

Abstract
Smad7 is a negative regulator of TGFβ superfamily signaling. Using a three-component triple transgenic system, expression of the inhibitory Smad7 was induced via doxycycline within the NCC lineages at pre- and post-migratory stages. Consistent with its role in negatively regulating both TGFβ and BMP signaling in vitro, induction of Smad7 within the NCC significantly suppressed phosphorylation levels of both Smad1/5/8 and Smad2/3 in vivo, resulting in subsequent loss of NCC-derived craniofacial, pharyngeal and cardiac OFT cushion cells. At the cellular level, increased cell death was observed in pharyngeal arches. However, cell proliferation and NCC-derived smooth muscle differentiation were unaltered. NCC lineage mapping demonstrated that cardiac NCC emigration and initial migration were not affected, but subsequent colonization of the OFT was significantly reduced. Induction of Smad7 in post-migratory NCC resulted in interventricular septal chamber septation defects, suggesting that TGFβ superfamily signaling is also essential for cardiac NCC at post-migratory stages to govern normal cardiac development. Taken together, the data illustrate that tightly regulated TGFβ superfamily signaling plays an essential role during craniofacial and cardiac NCC colonization and cell survival in vivo.
Keywords: mouse embryo, Smad7, neural crest, endocardial cushions, heart development, facial cleft, reverse tetracycline transactivator and tetracycline-responsive element-driven transgenes
Introduction
Neural crest cells (NCC) are a transient population created during higher vertebrate early embryonic development. During neurulation, NCC undergo successive epithelial-to-mesenchymal transformation (EMT) along a cranialcaudal embryonic axis (Serbedzija et al., 1992), delaminate and migrate down defined pathways to contribute to the formation of a wide variety of target tissues; including neurons and glia of the peripheral nervous system, melanocytes, smooth muscle cells and most craniofacial cartilages and bones (Nichols, 1981; Le Douarin and Kalcheim, 1999; Chai et al., 2000). NCC also contribute crucial cell populations to several thoracic tissues, including the developing aortic arch arteries (AAA) and outflow tract septum (OFT) of the heart (Kirby and Waldo, 1995; Jiang et al., 2000; Gittenberger-de Groot et al., 2005; Snider et al., 2007). Despite the diversity of NCC fates, they are divided broadly into cranial, cardiac and trunk NCC populations based on their site of origin and ability to colonize adjacent tissues and organs (Osumi-Yamashita et al., 1994; Le Douarin, 1999; Kirby and Waldo, 1995; Kanzler et al., 2000; Helms et al., 2005). This loose anatomical classification of the NCC subpopulations reflects the fact that the molecular mechanisms underlying the determination, pathway-dependent control and morphogenesis of the in utero NCC types remains far from clear.
TGFβ superfamily members include Activins, BMP and TGFβ cytokines. Ligands of the TGFβ superfamily are obligatory growth factors for early embryogenesis and heart morphogenesis, and play diverse biological roles during cell proliferation, differentiation, apoptosis and many other tissue remodeling processes. All TGFß family members mediate biological function by binding to a receptor within the cell membrane. BMP signaling (particularly via BMP2, BMP4 ligands and Alk2, Alk3, Alk4 receptors) has been implicated in promoting NCC induction, maintenance, migration and differentiation in several different model organisms (Aybar and Mayor, 2002; Knecht and Bronner-Fraser, 2002; Kaartinen et al., 2004; Stottmann et al., 2004; Dudas et al., 2004; Morikawa et al., 2009). Similarly, there is accumulating evidence that TGFβ signaling in NCC is critical, as NCC-restricted deletion of type I (Alk5) and type II receptors results in a spectrum of defects in the craniofacial, pharyngeal and cardiac regions (Choudhary et al., 2006; Wurdak et al., 2005; Ito et al., 2003; Wang et al., 2006). Signaling is initiated by the binding of ligands to the type II receptor, which then recruits and activates the type I receptor. Activated type I receptor then phosphorylates regulatory (R)-Smads (Smad1, 2, 3, 5 or 8), which form a complex with the co-Smad, Smad4. The complex then translocates into nucleus to regulate gene transcription (reviewed by Massagué et al., 2005, 2008; Itoh and ten Dijke, 2007; Moustakas and Heldin, 2009). Previous studies have shown that Wnt1-Cre mediated conditional knockout of Smad4 within the NCC results in craniofacial, pharyngeal and cardiac malformations (Jia et al., 2007; Ko et al., 2007; Nie et al., 2008; Büchmann-Møller et al., 2009). Overall, TGFβ superfamily signaling has been strongly implicated in NCC development, but its detailed in utero molecular mechanisms are still poorly understood due to the large number of family members with wide range of overlapping functions, genetic compensation, promiscuous receptor-ligand associations and functional redundancy. Due to Smad7's unique role in repressing both BMP and TGFβ signaling and its complementary role to that of Smad4 within TGFβ superfamily signaling, we hypothesized that Wnt1-Cre mediated Smad7 over-expression would phenocopy Wnt1-Cre mediated loss of Smad4 function.
To test this assumption and examine further the spatiotemporal role of TGFβ signaling during NCC morphogenesis and the temporal consequence of TGFβ superfamily inhibition during development, we generated a three-component triple transgenic Smad7 expression mouse model (herein termed trigenic) based on Cre/loxP recombination (to induce spatially-restricted Smad7) and doxycycline-inducible control elements (to temporally regulate Smad7). Since Smad7 has already been demonstrated to negatively regulate both TGFβ and BMP signaling in vivo in mammalian adult tissues, B cells, retinal pigment epithelium and Xenopus explant assays (Hayashi et al., 1997; Nakao et al., 1997; Casellas and Brivanlou 1998; Ishisaki et al., 1999; Saika et al., 2007), we are using its spatiotemporal induction as a tool to attenuate TGFβ in utero within restricted cell lineages and during defined developmental temporal windows. Results reported here show that pre-migratory Smad7 induction attenuates both TGFβ and BMP signaling by suppressing R-Smad phosphorylation, resulting in elevated NCC death, diminished NCC colonization of craniofacial, pharyngeal, cardiac tissues, dysregulated epithelial-mesenchymal interactions and in utero lethality. Additionally, Smad7 induction in post-migratory cardiac NCC results in isolated interventricular septal defects and neonatal lethality. This work demonstrates that tightly regulated TGFβ superfamily signaling plays an essential role during both craniofacial and cardiac NCC colonization and cell survival in vivo.
Materials & Methods
Conditional neural crest-specific trigenic mice
In the construct used to generate (tetO)-Smad7 transgenic mice, myc-tagged Smad7cDNA (Kuang et al., 2006) was inserted between a (tetO)7×CMV minimal promoter and the bovine growth factor polyadenylation signal sequence (bGHpA) within the pUHD10-3 vector (generously provided by Dr. Andras Nagy, Samuel Lunenfeld Research Institute; Belteki et al., 2005). Following diagnostic restriction digest verification and sequencing, the linearized construct was given to the IU Transgenic Core Facility for microinjected into inbred C3HeB/FeJ zygotes to obtain transgenic founders as described (Snider et al., 2009). Forward primer 5'-ATCCACGCTGTTTTGACCTC-3' and reverse primer 5'-GAGCGCAGATCGTTTGGT-3' were used for genotyping tetO-Smad7 transgenic offspring via PCR using mouse genomic DNA from tail using established protocols (Snider et al., 2008). Three independent lines were generated and all three were viable up to two years of age. In order to generate triple transgenic doxycycline-inducible mice, tetO-Smad7 mice were intercrossed with both the reverse tetracycline transactivator Rosa26rtTA-EGFP (R26rtTA-EGFP) (JaxLab stock #005670) and Wnt1-Cre (Jiang et al., 2000) mice. For the lineage mapping studies, Rosa26 reporter (R26r) mice (JaxLab stock #003474) were intercrossed with the trigenics. Genotyping was carried out using PCR primers specific for each transgene (http://www.jax.org; Jiang et al., 2000). All mice were maintained on mixed genetic backgrounds and age-matched littermates were used as appropriate controls. To induce Smad7 transgene expression in trigenic embryos, pregnant females were given doxycycline administered in green dyed food pellets at a concentration of 200mg/kg (Bio-Serv; Frenchtown, NJ) for specified time periods. Mice were maintained under specific-pathogen-free conditions with a 12 hours light/dark cycle. The animal use protocols were approved by the Institutional Animal Care and Use Committee at IUPUI (study #3301).
RNA and Protein Analysis
In order to verify Smad7 cDNA induction by RT-PCR, cDNA was synthesized from RNA isolated from individual control (trigenic mice without doxycycline food) and mutant (trigenic mice feed doxycycline food) embryonic day 10.5 (E10.5) whole embryos (n = 4 embryos of each treatment) using a Superscript-II kit (Invitrogen) with 5μg RNA and oligo(dT) primer. cDNAs were amplified with specific Smad7 primers (30 cycles; forward primer 5'-GCATTCCTCGGAAGTCAAGA-3' and reverse primer 5'-TTGTTGTCCGAATTGAGCTG-3') and normalized with GAPDH (16 cycles; Glyceraldehyde-3-phosphate dehydrogenase) as described previously (Rios et al., 2005). In order to verify myc-tagged Smad7 protein induction and suppressive effects upon TGFβ superfamily signaling via Western blotting, doxycycline-fed E10.5 embryos were homogenized on ice in RIPA buffer with 1% phosphatase inhibitor mixture (Sigma). Proteins were blotted onto PVDF membrane and the following antibodies used; Smad1 (1:2,000 dilution Santa Cruz Biotech, sc-81378), phospho-Smad1/5/8 (1:1,000 dilution Cell signaling, 9511), Smad2/3 (1:2,000 dilution Santa Cruz Biotech, sc-8332), phospho-Smad2 (1:1,000 dilution Cell signaling, 3101), Myc (1:5,000 dilution Santa Cruz Biotech, A-14), αTubulin (1:10,000 dilution Sigma, T-5168). Primary antibody binding was visualized by HRP-conjugated secondary antibodies and enhanced chemiluminescence (Amersham, GE Healthcare Biosciences). Signal intensity was quantified from at least 3 samples by Image J (http://rsweb.nih.gov/ij/) and the combined data graphically displayed.
Histological Analysis, X-Gal Staining and Immunohistochemistry
Tissue isolation, 4% paraformaldehyde fixation, processing, paraffin embedding, H&E staining, and wholemount detection of R26r indicator β-galactosidase activity were performed as described (Rios et al., 2005; Snider et al., 2008, 2009). Sections (n= 3 individual embryos of each genotype) were cut at 10μm thickness and counterstained with Eosin. As our mice lines are not isogenic, doxycycline-fed non-trigenic single and double transgenic age-matched littermates were used as negative controls.
Immunostaining was carried out using ABC kit (Vectorstain) with DAB and hydrogen peroxide chromogens as described previously (Zhou et al., 2008). The following primary antibodies were used to assess neural crest differentiation and TGFβ superfamily signaling: α-smooth muscle actin (1:5,000 dilution αSMA; Sigma), phospho-Smad1/5/8 (1:750 dilution Cell Signaling, 9511), phospho-Smad2 (1:1,500 dilution Cell Signaling, 3101). Negative controls were obtained by substituting the primary antibody with serum at 1:150 dilution and positive staining within serial sections was examined using at least three individual embryos of each genotype at each developmental stage.
Apoptosis and cell proliferation
Apoptotic cells were detected in paraffin-embedded tissue sections using TdT-FragEL™ DNA Fragmentation Detection Kit (Calbiochem). Cell proliferation was immunodetected using Ki67 antibody (1:25 dilution DakoCytomation, CA). Both assays were performed on paraffin serial sections (n=4). Both trigenic and control sections were placed on the same slides, thus receiving identical processing and treatment. The total cell number and positively stained cell number were counted manually in defined areas of tissues under 40× magnification. Statistical analysis of cell counts in serial sections and comparison of mutant specimens with controls was performed using one-tailed Student's t test (P values were assigned, with 0.05 being significant).
In Situ Hybridization
Radioactive in situ hybridization for Ap2α Crabp1, Fgf8, Msx1, Msx2, Pax3 and Sox10 expression was performed as described (Conway et al., 1997; Conway et al., 2000; Snider et al., 2009). Both sense and antisense 35S-UTP-labeled probes were used, and specific signal was observed only with hybridization of the antisense probe, in serial sections within at least three independent embryos of each genotype. In order to quantitate expression differences, silver grains were counted/cell on serial sections and comparison of mutant specimens with controls was performed using one-tailed Student's t test.
Results
Generation of neural crest-restricted inducible Smad7 mouse line
In order to spatially restrict Smad7 over-expression to the NCC lineages, we used the Wnt1-Cre transgenic driver line (Fig. 1A) (Brault et al., 2001; Chai et al., 2000; Jiang et al., 2000; Stottmann et al., 2004; Snider et al., 2007). Temporal regulation of the onset of Smad7 within the NCC was achieved with the inducible reverse tetracycline transactivator (rtTA). The Rosa26rtTA-EGFP mice contain the rtTA with a nuclear localization signal targeted into the Rosa26 locus, and a downstream EGFP separated by an IRES sequence to ensure efficient EGFP translation (Fig.1A). The ROSA locus has been shown to be expressed in all cell types at all developmental and postnatal stages, and its expression is not subject to genetic or environmental changes (Zambrowicz et al., 1997). In the absence of its inducer doxycycline (a derivative of tetracycline), rtTA does not recognize its DNA binding sequence, tetO. On the other hand, addition of doxycycline allows rtTA to bind the tetO minimal promoter resulting in transcription of the tetO-Smad7 transgene (Fig. 1A). Furthermore, as the rtTA expression module is preceded by a floxed STOP expression cassette, rtTA expression is dependent upon Cre/loxP recombinase to remove the loxP-flanked STOP expression cassette from within the R26 locus. The R26rtTA-EGFP mice have previously been shown to drive robust expression of doxycycline-inducible tet-O controlled transgenes (Belteki et al., 2005). TetO-Smad7 transgenic mice (three separate lines) were generated by placing full length Smad7 cDNA under the control of heptamerized tetO promoter (Fig. 1A). As there are no commercially-available specific Smad7 antibodies available and in order to unequivocally detect transgenic Smad7 protein in vivo, we used a myc-tagged Smad7, as this cDNA has already been demonstrated to block TGFβ signaling in vivo in adult transgenic mice (Kuang et al., 2006). Utilizing our R26rtTA-EGFP/tetO-Smad7/Wnt1-Cre trigenic mice, we can control temporal induction of in utero NCC-restricted myc-Smad7 by feeding doxycycline-containing food to the pregnant mother (Fig. 1A); and in non-NCC lineages the tetO-Smad7 is silent.
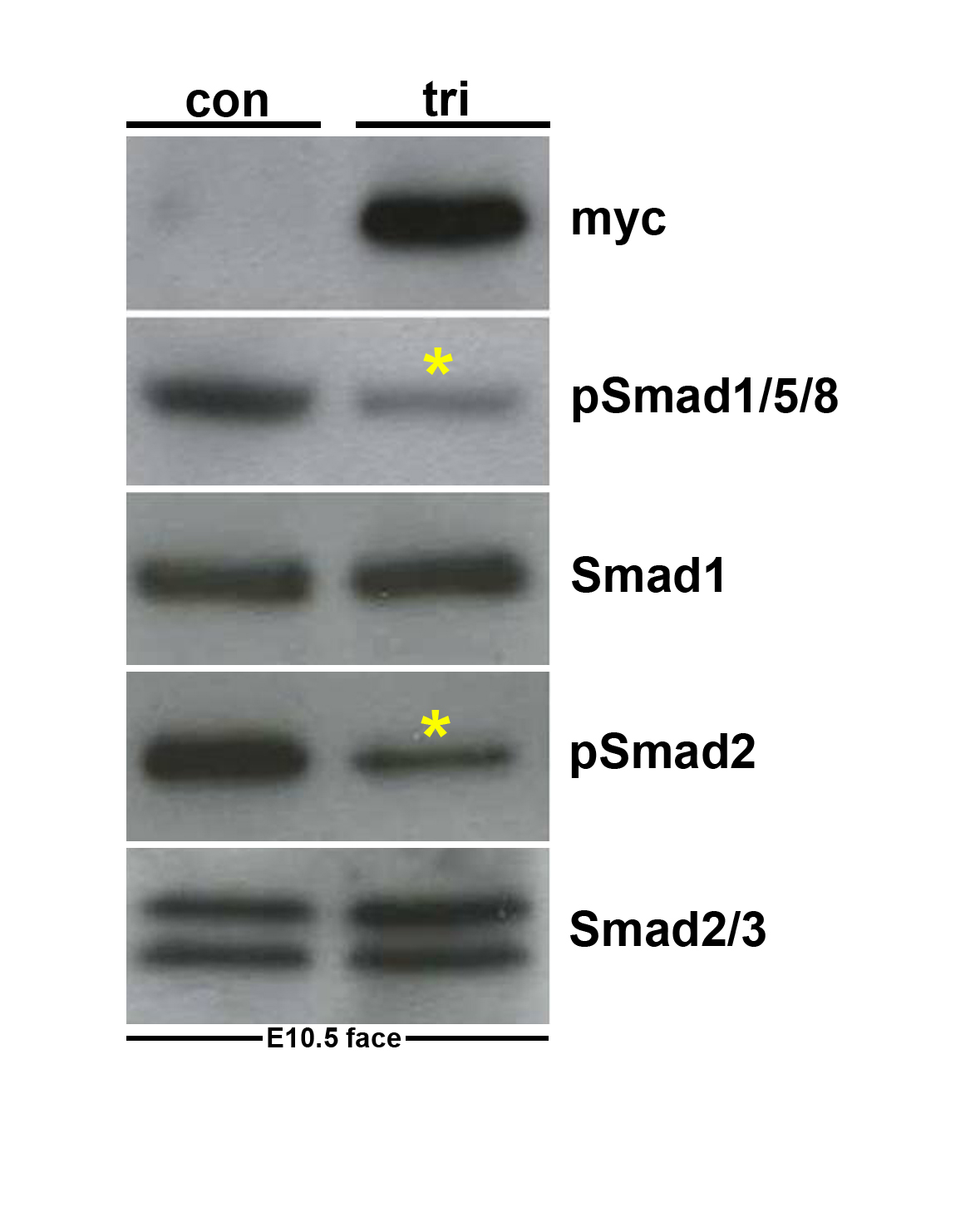
Figure 1. Evaluation of the trigenic Cre/loxP-dependent, tetracycline inducible transgenic system.

(A) Schematic illustrating operation of the trigenic system. The R26rtTA-EGFP knockin mice will only express the rtTA from the Rosa26 locus upon Cre-mediated recombination (Belteki et al., 2005). Wnt1-Cre transgenic mice have been shown to label almost all NCC, including the cardiac and craniofacial NCC lineages (Jiang et al., 2000; Chai et al., 2000). In the tetO-Smad7 transgenic mice, myc-Smad7 full length cDNA (myc tag is blue box 5' of Smad7 cDNA) is under the control of heptamerized tetOn promoter, but Smad7 is not expressed until both the transactivator (rtTA) and inducer (doxycycline) are present within the same cell. Although all three transgenes are individually silent within the compound trigenic mice, myc-tagged Smad7 can be specifically induced within Cre-positive neural crest lineages upon doxycycline feeding. Thus, the spatiotemporal timing of myc-tagged Smad7 expression is determined by the timing of doxycycline addition and positionally by the expression of Cre recombinase. (B) RT-PCR analysis reveals that Smad7 mRNA expression is upregulated ~10 fold in trigenic E10.5 embryos fed doxycycline at E7.5 compared to control embryo expression of endogenous Smad7 (n= 4 embryos of each genotype). Loading was normalized using GAPDH housekeeping control. (C,D) Western analysis verifies that myc-Smad7 is only expressed within duplicate doxycycline-fed duplicate trigenic E10.5 embryos fed doxycycline at E7.5, and is absent from duplicate control embryos (n=3 duplicates). The presence of myc-Smad7 suppressed both pSmad1/5/8 and pSmad2 levels relative to total Smad1 and pSmad2/3 levels when compared to age-matched control littermates, indicating over-expression of Smad7 attenuates both BMP and TGFβ signaling by ~55% (D). Loading was normalized using Tubulin housekeeping control. αTubulin signal was detected after 5 sec, while Myc and Smads was after 30 sec exposures. Abbreviations: tri, trigenic tetO-Smad7/Wnt1-Cre/RosartTA-EGFP embryos; con, control embryos.
Inducible over-expression of Smad7 in vivo
In order to verify the “silent but inducible” feature of our trigenic system, we placed each of the three individual TetO-Smad7 target mouse lines onto an R26rtTA-EGFP/Wnt1-Cre genetic background to generate R26rtTA-EGFP/tetO-Smad7/Wnt1-Cre trigenic mice. Male trigenic offspring were then crossed to homozygous female R26rtTA-EGFP mice (fed regular chow) and resultant trigenic embryos examined for myc-Smad7 protein and Smad7 mRNA overexpression. To simultaneously detect both endogenous and transgenic Smad7 mRNA, both forward and reverse primers located within the Smad7 cDNA transcript were employed. As expected, normal litter sizes (n=8 embryos/litter from 10 litters) and trigenic offspring were recovered at expected Mendelian ratios when harvested at E14.5 (data not shown). Additionally, neither Western analysis nor RT-PCR detected myc-Smad7 protein or elevated Smad7 mRNA expression in normal fed trigenic embryos (data not shown).
When the food was switched from regular to doxycycline-containing food, both myc-Smad7 protein induction and elevated Smad7 mRNA expression were observed specifically within trigenic embryos (Fig. 1B,C). Significantly, feeding pregnant females doxycycline at E10.5 gestation resulted in both Smad7 mRNA upregulation (8× fold) and myc-Smad7 induction in E11.5 trigenic whole embryos but not within control (remaining allelic single and double transgenic combinations) littermates (not shown). This rapid induction is consistent with other studies that have shown rtTA-driven transgene expression is detectable with adult lungs 6–12 hours after doxycycline (Perl et al., 2002). Additionally, the relative level of myc-Smad7 induction is comparable to the 3× fold elevation in aged skin and 4× fold elevation in some tumors (Han et al., 2006). Without doxycycline administration to the mother, these embryos develop normally and reach adulthood (not shown). As expected feeding pregnant females doxycycline from E7.5 gestation onwards also resulted in Smad7 mRNA upregulation (10× fold) in E10.5 trigenics relative to littermate controls (Fig. 1B). Similarly, myc is detected within trigenic E10.5 mutants only after being fed doxycycline (Fig. 1C). As similar levels of myc-Smad7 induction were observed using all three independent trigenic lines (not shown), results from just one line are presented to simplify data. Since Smad7 has been shown to negatively regulate both TGFβ and BMP signaling in vitro (Hayashi et al., 1997), we examined whether myc-Smad7 induction attenuated both phosphorylated Smad1/5/8 (pSmad1/5/8) and phosphorylated Smad2/3 (pSmad2/3) levels in vivo. Western analysis reveals that pSmad1/5/8 levels were reduced by 55.5% (P<0.037) and pSmad2 levels were reduced by 58.3% (P<0.021) in whole body doxycycline-fed trigenic E10.5 mutants (Fig. 1C,D). Similarly, when just NCC-enriched craniofacial and 1st pharyngeal arch tissues were used (Supplemental Fig. 1), the presence of myc-Smad7 suppressed both pSmad1/5/8 (by ~72%) and pSmad2 (by ~85%) levels relative to total Smad1 and Smad2/3 levels compared to age-matched control tissues. Combined, these data demonstrate that transgenic Smad7 expression is tightly controlled in this three-component genetic system, and that myc-Smad7 expression is induced rapidly in NCC-derived tissues by application of doxycycline to the system and the induced myc-Smad7 represses both TGFβ and BMP signaling in vivo. In addition, when doxycycline was administered to wild type pregnant females throughout gestation starting at E6, normal litter sizes (n=8 embryos/litter from 6 litters) were recovered postnatally, demonstrating that sustained doxycycline does not adversely affect in utero morphogenesis.
Over-expression of Smad7 in the neural crest impairs normal craniofacial and pharyngeal arch development
During craniofacial development, cranial NCC migrate ventrolaterally as they populate the craniofacial region. The proliferative activity of cranial NCC produces the frontonasal process and the discrete swellings that demarcate each branchial arch (Chai et al., 2000). In order to examine the early effects upon NCC morphogenesis, we induced myc-Smad7 transgene expression beginning E7.5 (early headfold pre-somitic stage), as this is prior to the onset of Wnt1-Cre expression (Stottmann et al., 2004) and NCC emigration from the neural tube (Nichols, 1981; Serbedzija et al., 1992; Kirby and Waldo, 1995; Le Douarin and Kalcheim, 1999; Snider et al., 2007). NCC lineage tracing experiments with Wnt1-Cre and the Rosa26 reporter (R26r; Soriano, 1999) show expression starts in the rostral hindbrain around the four somite (E8.0) stage and extends to the midbrain, forebrain and caudal hindbrain, and progresses to increasingly caudal cardiac and trunk levels by eight somite (E8.5) stages (Stottmann et al., 2004). Thus, Wnt1-Cre allows recombination of floxed STOP R26rtTA-EGFP rtTA expression within the NCC from very early stages of its development.
To examine the effects of Smad7 over-expression and to directly test the in vivo requirement of regulated TGFβ superfamily signaling during early NCC morphogenesis in utero, we fed pregnant trigenic females doxycycline from E7.5 onwards. Resultant embryos were harvested at E9 to birth. Initially, E9 trigenic embryos were indistinguishable from control littermates, but by E10 the trigenics were slightly smaller and exhibit subtle craniofacial and pharyngeal arch dysplasia defects (Fig. 2A–C). Specifically, the 1st pharyngeal arch was hypoplastic and the face/forebrain region was undersized. This became more evident at E11.5, as all the trigenic embryos exhibited significantly smaller craniofacial regions (n=7/7 trigenics exhibit craniofacial defects) and greatly reduced 1st, 2nd and 3rd pharyngeal arches (Fig. 2D–F). By E13.5, the trigenic embryos grossly lacked identifiable upper and lower jaws (Fig. 2G–I) but were otherwise viable until birth. However, none of the doxycycline-fed trigenic offspring were recovered at birth (n=7 litters). The non-trigenic littermates with remaining allelic combinations were phenotypically normal and serve as genetic background, age-matched and doxycycline-exposure controls. Histology revealed that the upper and lower jaws were indeed hypoplastic and the tongue was reduced in size, but Meckel's cartilage, which forms a template for mandible formation and is derived from cranial NCC (Chai et al., 2000) was still present (Fig. 3B). These data are consistent with the underdevelopment of medial nasal prominences and incomplete tongue formation observed in Wnt1-Cre;Smad4loxp/loxp embryos (Ko et al., 2007; Jia et al., 2007). Histology also revealed that 100% E13.5 trigenic mutants (n=5) lack the choroid plexus that extends into the fourth ventricle, even though the choroid plexus extending into the lateral ventricle was present (Fig. 3B). As X-Gal staining of Wnt1-Cre;R26r brains has shown that recombination of the R26r allele is confined to the CNS midbrain, hindbrain and cerebellum & choroid plexus in 4th vent/hindbrain (Dietrich et al., 2009) and that robust TGFβ signaling is present within the migrating cranial NCC, meninges, and choroid plexus (Wang et al., 1995); this selective loss of the choroid plexus that extends into the fourth ventricle was directly due to the restricted Smad7 overexpression within the NCC that contribute to this particular choroid plexus. As cranial NCC, located at the anterior neural tube are the first NCC population to migrate and populate the 1st, 2nd and 3rd pharyngeal arches and frontonasal mass, it is not surprising that these are the NCC lineage most visibly affected.
Figure 2. Early Smad7 induction within the neural crest lineage suppresses normal craniofacial and pharyngeal arch development.

(A–C) E10.0 control and trigenic (right embryo in A) whole embryos fed doxycycline at E7.5 appear grossly similar except smaller facial processes, but higher magnification reveals that the mandibular component of the E10.0 trigenic mutant 1st pharyngeal arch is undersized and the oropharyngeal region below the nasal process is enlarged (indicated by * in C) when compared to age-matched control (B). (D–F) E11.5 control and trigenic (right embryo in D) whole embryos. Note trigenic 1st and 2nd arches are dramatically underdeveloped and the facial processes are hypoplastic (F) when compared to controls (E), but the size of the trigenic heart appears is unaffected. (G–I) E13.5 control and trigenic (right embryo) whole embryos fed doxycycline at E7.5. Note in isolated heads (severed immediately below lower jaw), both the upper and lower jaws of the trigenic mutant are severely hypoplastic and largely absent (I) when compared to control littermates (H). Abbreviations: 1, first pharyngeal arch; 2, second pharyngeal arch; a, atrium; v, ventricle; lv, left ventricle; e, eye. Bar in A=0.1mm; B,C=0.05mm.
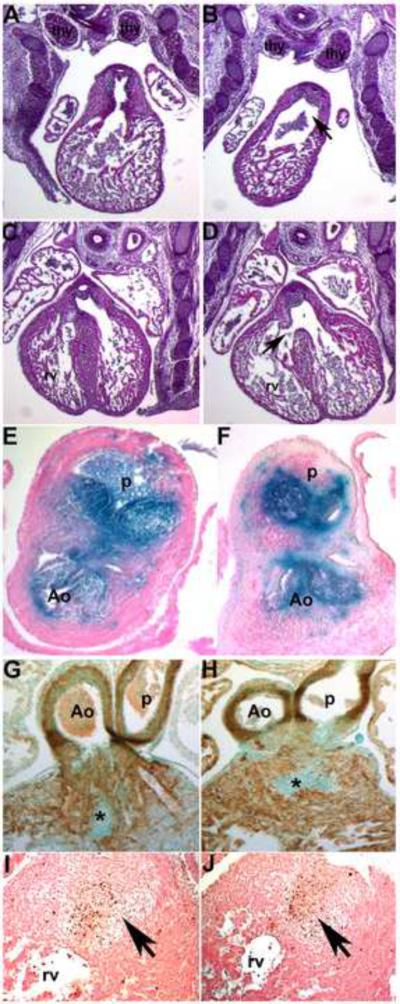
Figure 3. Histological examination of E13.5 Smad7 trigenic phenotypes fed doxycycline at E7.5.

(A,B) Control and trigenic littermate cranial regions sectioned sagittally and stained with H&E. Note the absent ventral extremity of the lower jaw and lip, hypoplastic tongue, absent primary palate, and absent upper jaw and lip in trigenic mutant (* in B). However, Meckel's cartilage is still present in the mutant (yellow arrowhead). Whilst the choroid plexus (cp) extending into the trigenic lateral ventricle is present, the choroid plexus differentiating from the roof of the fourth ventricle is absent (arrow in B). (C–H) Low power images of serial sagittal sections through control (C,E,G) and trigenic (D,F,H) cardiothoracic regions at the level of the OFT and high power images of the interventricular septum (G,H). While the control has separate ascending aorta and pulmonary trunk vessels (C,E), the trigenic mutant outflow tract has failed to septate and remains as a single outlet (PTA; E) and the right subclavian is retroesophageally located in the trigenic embryo (arrow in D). Additionally, the trigenic embryo exhibits accompanying interventricular septal defects (H). (I–L) Higher power images of transverse sections reveal that both the trigenic dorsal root ganglia (J) and thymus (L) are histologically normal when compared to control littermates (I,K), but are proportionately smaller in line with the overall reduced size of the trigenics. Abbreviations: t, tounge; nt, neural tube; PTA, Persistent Truncus Arteriosus; VSD, Ventricular Septum Defect; sep, septum; rv, right ventricle; lv, left ventricle; la, left ventricle; Ao, aortic trunk; dAo, descending aorta; p, pulmonary trunk; drg, dorsal root ganglia; thy, thymus; X, vagal X trunk. Bars in I,J=10μm.
Induced Smad7 over-expression impairs normal cardiac development
As trigenic mutants fed doxycycline from E7.5 onwards were not recovered at birth and Wnt1-Cre:R26r marked NCC have been shown to be essential for heart morphogenesis (reviewed by Kirby and Waldo, 1995; Snider et al., 2007), we examined the effects of NCC-restricted over-expression of Smad7 within the cardiovascular system. Cardiac NCC migrate through 3rd, 4th and 6th pharyngeal arches to colonize the OFT cushions, where they are required for septation of the truncus arteriosus into the aorta and pulmonary artery (Kirby and Waldo, 1995; Conway et al., 2003). Histology revealed that all trigenic mutants exhibit OFT defects (n=11/11 trigenics fed doxycycline at E7.5), specifically a single outlet forming persistent truncus arteriosus (PTA). Besides a failure of OFT separation, the right subclavian in trigenic embryos was retroesophageally located (Fig.3D) and the outlet valve leaflets were abnormally thickened (Fig. 3F), when compared to those seen in the control embryos (Fig. 3C,E). Additionally, trigenic embryos present accompanying membranous interventricular septal defects (VSD) (Fig. 3H). However, trigenic dorsal root ganglia and thymus appeared largely unaffected relative to the overall embryo size (Fig. 3J,L), notwithstanding having a NCC contribution (Jiang et al., 2000). Furthermore, trigenic dorsal root ganglia were normally colonized via NCC lineage (Fig. 4) and appropriately expressed Ap2α, Crapb1 and Sox10 mRNA (not shown).
Figure 4. NCC develop in Smad7 trigenic mutant embryos fed doxycycline at E7.5.

To lineage map both the trigenic and control NCC populations, R26r lacZ reporter mice were crossed with Smad7 trigenic to enable us to visualize NCC migration and colonization of the craniofacial and OFT regions. (A–C) E10 wholemount lacZ staining of trigenic and control littermates, revealed that initial NCC migration was largely unaffected within the trigenic (right embryo in A) cranial, cardiac and trunk regions when compared to control littermates. Robust lacZ expression is evident in trigenic frontonasal prominence, trigeminal nerve ganglia, hypoplastic 1st, 2nd and 3rd arches and within the facial nerve ganglia, and primordium of the 3rd pharyngeal arch. Similarly, lacZ-marked NCC are present within the cardiac 4/6th arch region and the DRGs (*) in trigenic mutants. (D–J) LacZ stained E11.5 trigenic and control littermate whole embryos (D); isolated control (E,F) and trigenic (G,H) hearts viewed frontally (E,G) and from the right (F,H); and higher power views of control (I) and trigenic (J) craniofacial regions. Note there is a deficiency of NCC-derived Schwann cells within SNS of trigenic forelimb (* in D), but trunk NCC migration is unaltered. (E–H) Higher power views of isolated hearts clearly show that Smad7 trigenic NCC reach the pharyngeal arches and aortic sac region, that a few mutant NCC can enter the OFT truncal region but that there are no lacZ stained NCC within the trigenic OFT conal region (* in H) when compared to controls (E,F). Similarly, although trigenic NCC do colonize the hypoplastic craniofacial regions, there are reduced numbers of lacZ positive cells, particularly evident within the frontal nasal process and 3rd, 4th and 6th pharyngeal arches (arrow in J). (K,L) Sections through OFT from distal to proximal in E11.5 control (K, K', K”) and trigenic embryos (L, L', L”). Histology confirms a lack of trigenic NCC colonizing the OFT, that there are fewer cells within the conal cushions and that the mutant NCC are not found in ectopic locations within the adjacent myocardial cuff (arrow in L') or overlying endothelium. (M–P) LacZ stained E13.5 control (M,O) and trigenic littermate whole embryos and isolated hearts viewed frontally. Note the absent lacZ NCC within the trigenic upper and lower craniofacial regions (arrow in N) and the absence of lacZ positive NCC within the trigenic OFT (arrow in P). Abbreviations: fl, forelimb; lv, left ventricle; a, atria; oft, outflow tract; ec, endocardial cushions; rv, right ventricle; ra, right atria; e, eye. Bars in I,J=0.2mm.
Neural crest lineage mapping reveals regional deficiencies
To verify Wnt1-Cre recombination efficiency in trigenics and to determine the origin of these defects, trigenic mice NCC were lineage mapped. Earlier studies have shown that elements of TGFβ superfamily signaling are required for normal craniofacial and cardiac NCC migration. For example, Wnt1-Cre conditional deletion of the canonical TGFβ factor signaling cofactor Smad4 (Jia et al., 2007; Ko et al., 2007; Nie et al., 2008; Büchmann-Møller et al., 2009) and transgenic overexpression of the BMP-antagonist Noggin (Kanzler et al., 2000) can result in NCC-deficiency. Analysis of β-galactosidase stained trigenic mutant embryos carrying the R26r reporter showed normal NCC migration and contribution to the craniofacial region and dorsal root ganglia, but reduced NCC colonization of the OFT itself. From E10.5–11.5, robust lacZ staining was evident in the frontonasal prominence, trigeminal nerve ganglia, 1st, 2nd pharyngeal arches along with facial nerve ganglia, and primordium of the 3rd pharyngeal arch in trigenic embryos (Fig. 4A,C,D), similar to that observed in controls (Fig. 4A,B,D). Intriguingly, this assay revealed that whereas the NCC populate the craniofacial and pharyngeal regions, the 1st, 2nd and facial regions failed to form normally and were hypoplastic in trigenics (Fig. 2). However, in E13.5 trigenic embryos there were subsequent lacZ-negative areas of frontonasal mesenchyme, suggesting a subsequent loss of lacZ-positive cells (Fig. 4N). This was confirmed via histology (not shown). Similar to the cranial NCC, migration of the lacZ-marked cardiac NCC was largely unaffected in E10.5 trigenic embryos (Fig. 4A–C), and trunk NCC contribution to the trigenic dorsal root ganglia was unchanged when compared to littermate controls (Fig. 4A,D). However, further analysis of cardiac NCC colonization of the heart shows that although a significant number of NCC have populated the pharyngeal arches and aortic sac, only a few trigenic NCC colonized the truncal region and there was a complete lack of colonization of the more proximal conal cushion region of the trigenic truncus arteriosus (Fig. 4G,H,L) when compared to control littermates (Fig. 4E,F,K). The foremost difference of cardiac NCC contribution to the trigenic OFT embryos could also be seen at later stages (Fig. 4P). The difference between the presence of fully penetrant OFT defects and dramatically reduced cardiac NCC colonization and unaffected NCC contribution to the dorsal root ganglia and normal cranial NCC migration patterns, suggests that the cardiac NCC population and morphogenesis of the OFT are especially sensitive to suppression of TGFβ superfamily signaling. Furthermore, lineage mapping established that Smad7-mediated suppression of TGFβ superfamily signaling principally inhibits later cardiac NCC colonization of the OFT, rather than early NCC emigration and migration from the neural tube.
Trigenic mutants exhibit normal proliferation but increased cell death
Such dramatic changes in the morphology of the frontonasal and OFT tissues could be accomplished via a change in the amount of cell death and/or cell proliferation. Cell proliferation assays, using Ki67 to mark cells in all active phases of the cell cycle (G1, S, G2, and mitosis) in sectioned E10.5 trigenic embryos fed doxycycline from E7.5 onwards, did not detect any decrease in facial, 1st pharyngeal arch mesenchyme or OFT cushion cell proliferation index (Fig. 5A–C). Total E10.5 1st pharyngeal arch proliferation in trigenic mutants was reduced (~675 Ki67 positive cells/arch/10μm section) when compared to age-matched control embryos (~1,003 Ki67 positive cells/arch/10μm section), due to the hypoplastic trigenic phenotype. This was specifically apparent at the base of the 1st trigenic arch (Fig. 5B), where the mesenchyme was sparse compared to control littermates. However, when proliferating cells were tallied (as a percentage of total cells in the 1st pharyngeal arch) to calculate a mitotic index, there was no significant difference by this measure (90.9% in controls versus 91.2% in trigenics). Thus, the defects in mutant embryos were unlikely to be caused by reduced NCC proliferation.
Figure 5. Elevated cell death of NCC in Smad7 trigenic mutants fed doxycycline at E7.5.

(A–C) Ki67 staining at E10.5 on sagittal sections reveals equivalent levels of cell proliferation (n=4) within the trigenic 1st (B) and 2nd (not shown) pharyngeal arches when compared to controls (A). (D–F) In contrast, TUNEL staining at E10.5 (n=4) demonstrates a notable increase in cell death in trigenic mutants within the mandibular component of the 1st arch (arrow in E) compared to controls (D). (G) TUNEL staining (brown) of lacZ-stained E11 R26r trigenic embryo (transverse section of right pharyngeal/OFT region lying on left hand side), demonstrates that apoptotic cells (arrows) are of NCC (blue) origin. Note that trigenic non-NCC derived core arch mesenchyme (indicated by yellow line), cephalic mesenchyme (cm) and body wall (bw) are largely devoid of apoptotic cells. However, as expected the pharyngeal pouch endoderm exhibits robust apoptosis (*). (H–K) α-SMA staining showed that NCC differentiation into smooth muscle within the E10.5 trigenic (I) aortic sac was comparable to controls (H), that α-SMA staining (brown) within the trigenic OFT cushions was reduced (*) and that transient cardiomyocyte expression of α-SMA was unaffected in both control and trigenic mutant hearts. Similarly, NCC-derived smooth muscle colonization of the control (J) and trigenic (K) 6th aortic arch arteries (arrows) was comparable at E13.5. Abbreviations: as, aortic sac; a, atria; h, heart; PTA, Persistent Truncus Arteriosus.
In contrast, using the TUNEL assay to mark apoptotic cells, dramatic increases in cell death in the E10.5 trigenic facial and pharyngeal arch mesenchyme and within migratory NCC lineages were observed (Fig. 5D–F). Considerable and aberrant cell death was localized to the trigenic facial primordia and 1st (Fig. 5E), 2nd and 3rd pharyngeal arches of mutant embryos, but not within the OFT cushions nor the cardiomyocytes of the heart or the neuroepitheium (not shown). The sparse mesenchyme at the base of the 1st trigenic arch still contained several apoptotic cells, but is adjacent to a region of very robust apoptosis (Fig. 5E). TUNEL staining of older E11 and E11.5 embryos revealed that the entire 1st arch mesenchyme was sparse and apoptotic (not shown), suggesting that apoptosis initiates at the base of the arches and progresses more ventrally as development proceeds. When TUNEL positive cells were tallied as a percentage of total cells in the 1st arch to calculate a cell death index, there was a significant increase in cell death (1.55% in controls versus 16.52% in trigenics;P<0.001). To test if these apoptotic cells were limited to NCC, we performed X-gal staining on E11.0 trigenic; R26r embryos to label NCC and their derivatives followed by TUNEL (Fig. 5G). As expected, many NCC were positively stained with the TUNEL signal. These data are consistent with the dramatic increases in cell death observed in Wnt1-Cre;Smad4loxp/loxp (Ko et al., 2007; Jia et al., 2007; Nie et al., 2008; Büchmann-Møller et al., 2009) and Wnt1-Cre;Alk5loxp/loxp embryos (Wang et al., 2006), as both Smad4 and Alk5 are required for survival of cardiac NCC. Similarly, Wnt1-Cre;Alk3loxp/loxp embryos exhibit normal NCC migration, but NCC subpopulations die immediately after colonization of their target tissues (Morikawa et al., 2009). Intriguingly, loss of BMP antagonists (Noggin and Chordin) result in excessive NCC emigration from the neural tube that results in elevated NCC-restricted apoptosis (Anderson et al., 2006). Combined, these data reveal that Smad7 suppression of TGFβ superfamily signaling within early NCC is required for normal survival of migratory NCC-derived mesenchyme and furthermore demonstrate that combined suppression of TGFβ and BMP pathways, deletion of either individual TGFβ or BMP pathway effectors or increased BMP signaling can all likewise distress NCC survival in vivo.
Aortic arch artery remodeling is unaffected
Given the frequent association of anomalous pharyngeal AAA remodeling and pathogenesis of OFT defects (Kirby and Waldo, 1995; Conway et al., 2000; Stalmans et al., 2003; Snider and Conway, 2007) we used histology and α-smooth muscle actin (αSMA) to determine whether AAA were properly formed in myc-Smad7 trigenic mutants fed doxycycline from E7.5 onwards (Fig. 5H–K). This analysis revealed the trigenic AAA were present and patent by E10.5, suggesting that induction of Smad7 in NCC does not disrupt initial arch artery formation. However, αSMA staining of the OFT cushions was reduced in E10.5 mutants, and as colonizing cardiac NCC express αSMA (Epstein et al., 2000), these results confirm the reduced Wnt1-Cre;R26r lineage mapping data in trigenic OFT cushions (Fig. 4). We further examined whether αSMA positive NCC-derived cells ensheath the remodeled trigenic AAA. Both the control and trigenic 4th and 6th AAA were intact and exhibit similar αSMA expression patterns (Fig. 5J,K). Despite suppression of TGFβ signaling within NCC lineage, we did not observe abnormal regression of AAAs, a defect which was reported in Wnt1-Cre;Alk2loxp/loxp and Wnt1-Cre;Alk5loxp/loxp embryos (Kaartinen et al., 2004; Wang et al., 2006)
Localized attenuated TGFβ and BMP signaling
To further resolve the molecular defects in trigenic mutants caused by NCC-restricted induction of Smad7, we examined the spatial expression of both pSmad1/5/8 and pSmad2 in trigenic and control embryos fed doxycycline from E7.5 onwards. Consistent with Western data (see Fig. 1C,D and Supplemental Fig. 1), pSmad1/5/8 and pSmad2 expression was reduced in E10.5 trigenic embryo NCC lineages compared to control littermates (Fig. 6). Significantly reduced numbers of pSmad1/5/8-positive and pSmad2-positive cells were detected in the mesenchyme and ectoderm-derived epithelium of the trigenic frontal facial region (Fig. 6B,J) and pharyngeal arches (Fig. 6D,L), and within the aortic sac mesenchyme (Fig. 6F,N), but not in the trigenic ventricular myocardium/epicardium/endothelium (Fig. 6H,P). Each of the mesenchymal sites of reduced pSmad1/5/8 and pSmad2 expression correlate with Wnt1-Cre;R26r lineage mapped NCC (Fig. 4; Chai et al., 2000; Jiang et al., 2000), indicating that both TGFβ and BMP signaling were significantly attenuated within NCC in vivo by over-expression of myc-Smad7. However, both normal (Chai et al., 2000) and Smad7 overexpressing ectoderm derived-epithelium (Fig. 4) are completely free of NCC progeny, suggesting that NCC migration and colonization were unaffected, suggesting a bystander effect. These immunohistochemical data are consistent with the significantly reduced pSmad levels detected via whole body and NCC-enriched Westerns, as we observe almost 100% suppression within NCC-specific tissues.
Figure 6. NCC-restricted Smad7 over-expression results in decreased TGFβ and BMP signaling.

(A–H) pSmad2 immunohistochemistry on E10.5 sagittal sections demonstrated that TGFβ signaling is reduced in both craniofacial and cardiovascular tissues colonized by NCC within trigenic (B,D,F) embryos fed doxycycline at E7.5 when compared to control littermates (A,C,E) but that pSmad2 expression is unaffected within control (G) and trigenic (H) ventricles. Note that pSmad2-positive cells (brown staining) are significantly reduced within the trigenic mesenchymal nasal process and the overlying epithelium (* in B), as well as within the 1st pharyngeal arch mesenchyme and overlying epithelium (* in D). Similarly, there is almost a complete absence of pSmad2-positive NCC around the trigenic aortic sac and within the truncal region of the OFT (F). (I–P) pSmad1/5/8 immunohistochemistry on E10.5 sagittal sections similarly demonstrated that Bmp signaling is attenuated in both craniofacial and cardiovascular tissues colonized by NCC within trigenic (J,L,N) embryos fed doxycycline at E7.5, when compared to control littermates (I,K,M) but that pSmad2 expression (brown staining) is unaffected within control (O) and trigenic (P) ventricles to which NCC do not contribute. Sections were counterstained with 0.05% toluidine blue. Abbreviations: mes, mesenchyme of 1st arch; as, aortic sac. Bars in B,G,J,O=10μm.
Altered gene expression in pharyngeal arches in mutant embryos
To investigate the signaling mechanism of Smad7 induction in regulating epithelial-mesenchymal interactions; we examined expression patterns of genes expressed in the early trigenic (fed doxycycline from E7.5 onwards) neural tube, in migratory NCC, and genes known to be critical for early mandible development. We first examined expression of different NCC markers including Pax3, Crapb1, Ap2α, Msx1 and Msx2 in E9 embryos by in situ hybridization analysis. Pax3 transcription factor is required for normal OFT development and is expressed within the early dorsal neural tube prior to NCC emigration and during initial migration from the neural tube (Conway et al., 1997; Epstein et al., 2000), while Msx2 transcription factor is co-expressed with Pax3 within the neural tube and is an immediate downstream effector of Pax3 in the cardiac NCC (Kwang et al., 2002). Msx2, a homeobox gene regulating BMP signaling, can act in concert with Msx1 to regulate cranial NCC differentiation (Han et al., 2007). Both Crabp1 and Ap2α transcription factor are retinoic acid responsive genes that are expressed in migrating NCC (Conway et al., 1997; Conway et al., 2000). We did not observe any obvious abnormal expression of these genes (not shown), suggesting that the initial formation of NCC was intact in trigenic embryos. These data are consistent with our Wnt1-Cre;R26r cell lineage analysis (see Fig. 4).
Next we evaluated expression of the Crapb1 and Ap2α NCC markers, as well as Fgf8 and Sox10 expression in E10.5–13.5 trigenic and control embryos fed doxycycline from E7.5 onwards (Fig. 7). In developmentally older embryos, Ap2α is still expressed within migratory NCC but is also expressed robustly within the ectoderm (Brewer et al., 2002). Fgf8 encodes a signaling growth factor molecule expressed in the epithelia of pharyngeal arches, and is required for normal growth of pharyngeal arches and remodeling of OFT (Frank et al., 2002). While the Sox10 transcription factor is essential for the development of NCC-derived glia and neurons of the peripheral nervous system (Sonnenberg-Riethmacher et al., 2001). Significantly, expression of Crabp1 (Fig. 7B) and Ap2α were visibly reduced (but not eliminated) from their expression domains within E10.5 trigenic migratory cardiac NCC, whereas expression of CrabpI was not noticeably affected within non-NCC derived body wall. Ap2α was also reduced within trigenic facial mesenchyme, but that most likely reflected apoptotic loss of cranial NCC. Intriguingly, while ectodermal Ap2α expression appeared comparable between control and trigenic littermates, trigenic 1st arch mesenchyme Ap2α expression was increased compared to control mesnchyme (Fig. 7C,D). Correspondingly, Fgf8 was specifically elevated within the trigenic 1st pharyngeal arch and facial ectodermal lineages (Fig. 7F). Counting of silver grains/cell revealed a ~4× increase in Fgf8 expression in trigenic (87 +/−11 grains/cell; n=3 embryos, five 10μm sections/embryo) vs. control (22 +/−8 grains/cell; n=3 embryos, five 10μ sections/embryo) 1st arch ectoderm. Consistent with the normal histology (see Fig. 3J) and correct NCC-contribution (see Fig. 4A,D); Ap2α, Crapb1 and Sox10 were each expressed normally in E10.5 and 13.5 trigenic dorsal root ganglia (not shown). Collectively, these results indicate that suppression of TGFβ superfamily signaling within the cranial and cardiac NCC, disrupted not only distinct NCC populations gene expression but also normal developmental processes in neighboring cell populations. Thus, bystander affects upon neighboring epithelial TGFβ superfamily signaling may augment the subsequent hypoplasia of these structures and contribute to the overall pathogenesis of the severe malformations in trigenic mutant embryos
Figure 7. Molecular marker analysis of E10.5 trigenic mutant phenotype.

Radioactive in situ hybridization detection of Crabp1 (A,B), Ap2α (C,D) and Fgf8 (E,F) mRNA in control (A,C,E) and trigenic mutants (B,D,F) fed doxycycline at E7.5. (A,B) Transverse sections through the OFT region of trigenic mutants reveal that Crabp1 expression is reduced within the pharyngeal arch region around the foregut (indicated by * in B) and along the NCC migration routes around the dorsal aorta, but not within the thoracic body wall (arrows). (C,D) Sagittal sections of trigenic mutants indicate that Ap2α expression is reduced in the trigenic nasal process (* in D) and that Ap2α is ectopically expressed within the mesenchyme of 1st arch (arrow) when compared to control littermate (C). (E,F) Sagittal sections of trigenic mutants also reveal that Fgf8 expression is upregulated within the epithelium overlying the nasal process (* in F) and 1st arch mesenchyme (arrow in F) but that trigenic Fgf8 expression within the neural epithelial layer of the optic cup is comparable with control expression levels. Abbreviations: da, dorsal aorta; oft, outflow tract; e, eye.
Over-expression of Smad7 in the neural crest following OFT colonization also results in abnormal heart development
To examine the effects of Smad7 over-expression within post-migratory NCC following colonization of their target tissues, we fed pregnant trigenic females doxycycline from E10 onwards. Our initial characterization of the trigenic system revealed that feeding pregnant females doxycycline at E10.5 gestation resulted in both Smad7 mRNA upregulation and myc-Smad7 induction by E11.5 in only trigenic but not control littermates (see Fig. 1). Grossly, E14.5 trigenic fetuses were indistinguishable from control littermates, but histology revealed that 100% of the E14.5 trigenic mutants exhibit membranous VSD (n=7/7 trigenics exhibit heart defects; Fig. 8D) that persisted until birth (n=3/3 trigenics). Additionally, although the trigenic OFT was divided and separate aorta and pulmonary arteries exit their respective right and left ventricles, there was a significantly dilated outflow lumen and possibly reduced endocardial cushion cells (Fig. 8B). Further examination of the dorsal root ganglia, thymus and craniofacial regions did not reveal any other histological defects (not shown). Wnt1-Cre;R26r lineage mapping revealed that regular numbers of NCC colonize the trigenic OFT and aorticopulmonary septum at the appropriate developmental stage, and that they survive until at least E13.5 (Fig. 8F) following post-migratory Smad7 overexpression. Additionally, histology revealed that the OFT was fully divided and that the trigenic valves were normal (Fig. 8F). Significantly, myocardialization of the cushions occurs (Fig. 8G,H) and there are similar levels of apoptosis (Fig. 8I,J) in both trigenic and wildtype littermates. This is significant as it has been proposed that the relatively small population of cardiac NCC cells that migrate into the proximal cushions are required to die to promote myocardialization (Poelmann et al., 2000).
Figure 8. Analysis of E14.5 Smad7 trigenic heart phenotypes fed doxycycline at E10.
(A,B) Control and trigenic littermate OFT regions sectioned transversely and stained with H&E. Note both the control (A) and trigenic (B) left and right lobes of the thymic rudiment are normally situated and unaffected by myc-Smad7 induction (n=4 trigenic and 5 control fetuses). However, the trigenic OFT truncal endocardial cushions are smaller and the lumen of the outlet of the right ventricle is abnormally dilated (arrow in B). (C,D) Additionally, the trigenic embryo exhibits an isolated VSD (arrow in D) but the control heart is septated normally (C). (E,F) To lineage map both the trigenic and control post-migratory NCC populations, R26rlacZ reporter mice were crossed with Smad7 trigenic mice. Transverse sections through the E13.5 OFT revealed similar patterns of cardiac NCC (blue) within the aorticopulmonary septum (not shown) and valves in both control (E) and trigenic (F) littermates. Note that trigenic cardiac NCC are still present and that the valves are normal. (G,H) α-SMA staining showed that myocardialization occurs normally in both E13.5 control (G) and trigenic (H) hearts. Note active myocardialization of endocardial cushions at base of the OFTs (indicated by *). (I,J) TUNEL staining (brown) revealed that both the patterns and level of apoptosis (indicated by arrows) are grossly similar in E13.5 control (I) and trigenic (J) OFTs. Abbreviations: thy, thymic lobes; rv, right ventricle.
Despite being the foremost CHD found in patients, isolated VSD in mice mutants are usually found in association with altered myocardial growth, OFT/AAA abnormalities and/or valvular defects (Conway et al., 2003). VSD pathogenesis is complex and multifactorial and may include underlying defects within the NCC, cardiomyocyte, endothelial, endocardial cushion, epicardial and/or cardiac fibroblast lineages; that can each alter left-right ventricular morphogenesis, chamber formation and septal positioning. Thus, the ability to induce isolated VSDs will provide a useful mouse model in which to begin to address the role of the NCC lineage in these CHDs.
Discussion
The physiological consequences of postnatal Smad7 dysregulation is demonstrated by the finding that SMAD7 is upregulated in a number of pathological conditions in patients; including development of malignancy (Boulay et al., 2003; Arnold et al., 2004), scleroderma (Asano et al., 2004), and chronic inflammatory bowel diseases (Monteleone et al., 2001). Smad7 has also recently been shown to be required for embryogenesis, as the majority of Smad7 knockout mouse embryos die in utero due to multiple defects in cardiovascular development, including VSD and OFT malformations (Chen et al., 2009). Indeed, endogenous Smad7 mRNA is transiently activated preimplantation, then is re-expressed widely post-gastrulation (Zwijsen et al., 2000). In E10 and E12.5 embryos, Smad7 mRNA is widespread outside the developing central nervous system; including the NCC-derived mandibular and medial nasal processes, developing pharyngeal arches, OFT cushions of the heart, adrenal primordium and trigeminal ganglion (Zwijsen et al., 2000; Luukko et al., 2001). Transgenic reporter and Cre/loxP lineage mapping using a partial Smad7 promoter have also confirmed the robust expression of Smad7 mRNA within the craniofacial, pharyngeal arch and cardiovascular system (Liu et al., 2007; Snider et al., 2009). However, the role of Smad7 within the NCC lineage and specifically the effects of mis-regulation of Smad7 and overexpression in utero remain unknown.
We hypothesized that in vivo misexpression of a potent TGFβ signaling inhibitor like Smad7, would generate severe NCC cell hypoplasia and resultant neurocrestopathies. Our data provide compelling genetic evidence demonstrating that forced expression of Smad7 within NCC is detrimental to the NCC subpopulations that colonize the OFT and craniofacial regions. Moreover, we showed that levels of both TGFβ and BMP phosphorylated R-Smads were reduced in the NCC expressing myc-Smad7. Since TGFβ superfamily signaling is known to increase production of phosphorylated forms of R-Smad proteins, our data provide good evidence that TGFβ signaling in vivo is impaired by myc-Smad7 induction in NCC. The spectrum of trigenic phenotypes fit well with previous studies reporting craniofacial, pharyngeal, cardiac defects and in utero lethality within NCC-restricted Wnt1-Cre;Smad4loxp/loxp embryo mutants (Ko et al., 2007; Jia et al., 2007; Nie et al., 2008; Büchmann-Møller et al., 2009). Thus, combined these data strongly suggest that altered NCC TGFβ superfamily signaling, either via targeted deletion of Smad4 or upregulation of Smad7 can both play pathogenic roles that result in neurocrestopathies. Although we observed an OFT defect and VSD in the trigenic embryos fed doxycycline from E7.5 onwards, it may not be the primary cause of the mid-gestation lethality, because an OFT defect or PTA is not necessarily lethal until birth (Conway et al., 1997; Kirby et al., 1995; Conway et al., 2003). Further experiments are required to determine the primary cause of embryonic lethality of the myc-Smad7 trigenic embryos.
Although members of the Wnt family have been shown to specify NCC from early dorsal neuroepithelial cells (Dorsky et al., 2000; García-Castro et al., 2002) and Wnt/ß-Catenin signal activation plays an instructive role in specification of NCC sensory fate (Lee et al., 2004); the molecular mechanisms underlying the determination, pathway-dependent control and morphogenesis of the diversity of NCC fates remains far from clear. Some alternative NCC fates have been shown to be instructively promoted by TGFβ superfamily members (Shah et al., 1996: Dorsky et al., 2000). For example, Bmp2 causes cloned NCC to form autonomic neurons and can also induce some smooth muscle differentiation, whilst TGFβ1 exclusively promotes smooth muscle differentiation (Shah et al., 1996). Surprisingly, although our lineage analysis and immunohistochemical assays revealed robust NCC death within trigenic embryos fed doxycycline from E7.5 onwards, subsequent NCC colonization of the trigenic dorsal root ganglia, trigeminal ganglion, thymus and αSMA-positive NCC-derived subpopulation that ensheath the remodeling trigenic AAA which give rise to the great vessels exiting the heart were all unaffected (Figs. 4,5). Thus, Smad7 induction appears to aberrantly affect specific subpopulations of NCC, independent of where they originate from, when they initiate migration or even how far they are required to migrate prior to target tissue colonization. Indeed, forced expression of Smad7 within pancreatic beta cells (Smart et al., 2006) and human umbilical cord blood cells (Chadwick et al., 2005) has similarly been shown to inhibit differentiated cell formation and suppression of TGFβ signaling alters cell fate decisions.
Craniofacial and cardiac defects thought to arise principally from perturbation of NCC morphogenesis are prominent features of DiGeorge syndrome (DGS)/velocardiofacial syndrome (VCFS), as well as Noonan syndrome (NS) (Moon et al., 2006; Nakamura et al., 2009). While NS is caused by a gain-of-function mutation in SHP2 (Nakamura et al., 2009) and Wnt1-Cre lineage restricted deletion of Shp2 (Nakamura et al., 2009) and subsequent Erk1/2 activation (Newbern et al., 2008) both result in a similar spectrum of cardiac and craniofacial defects as seen in the trigenics, DGS/VCFS is the result of a recurrent deletion in chromosome 22q11 (Kobrynski and Sullivan, 2007). DGS/VCFS is the most common micro-deletion syndrome in patients (Moon et al., 2006), but not all DGS/VCFS patients have the 22q11 deletion (Shaikh et al., 2007; Rope et al., 2009). Additionally, several modifiers of the DGS/VCFS have been identified in patients and modeled using mouse transgenics; including VEGF (Stalmans et al., 2003), FGF8 (Moon et al., 2006) and CHD7 (Randall et al., 2009). Although the microdeletion of 22q11 encompasses ~30 genes, it does not include any of the TGFβ superfamily receptors or ligands. Thus, gene products eliminated by 22q11 deletions may functionally interact with TGFβ signaling within the NCC lineage. For instance, MED15 has been shown to modulate TGFβ signaling within embryos (Kato et al., 2002), and both Tbx1 (Randall et al., 2009) and Crkl (Wurdak et al., 2005) can directly regulate TGFβ signaling within murine NCC via modulating R-Smad phosphorylation and altering the ability of R-Smads to bind Smad4 (Fulcoli et al., 2009). Furthermore, loss of Crkl disrupts Erk1/2 activation (Moon et al., 2006). These findings imply that the common developmental abnormalities observed in DGS/VCFS and NS patients may arise from perturbation of the same pathway. Although the reduced expression levels of Ap2α and Crabp1 mRNA are to be expected (Fig. 7) given the observed elevation of NCC death (Fig. 5), our molecular marker analysis did reveal an unexpected finding. One particularly significant discovery is that Fgf8 levels are elevated within the epithelia of the trigenic pharyngeal arches, as FGF8 is a known modifier of DGS/VCFS and Fgf8 signaling is mediated by Crkl (Moon et al., 2006). Functions of these genes have been implicated in mediating interactions between NCC and their neighboring cells in facial primordia and pharyngeal arches, and subsequent organogenesis (Noden and Trainor, 2005). Combined, these data indicate that suppression of TGFβ signaling within the cranial and cardiac NCC disrupts not only distinct NCC gene expression, but also normal developmental processes in neighboring cell populations. In order to find the common downstream effectors, detailed analysis of the resultant gene expression profile changes within isolated trigenic NCC subpopulations will be required to identify which NCC-restricted gene pathways are altered in response to suppressed TGFβ signaling.
The spectrum of cardiac defects caused by Smad7 induction in early NCC resembles those associated with cardiac NCC ablation in avian embryos, in which both anomalous AAA remodeling and lack of OFT septation with concomitant VSDs are observed (Hutson and Kirby, 2007). Cardiac NCC normally migrate to the OFT region via the 3rd, 4th and 6th pharyngeal arches to form the two-pronged cushions in the truncal region of the OFT where they contribute to the septation of aorta and pulmonary outlets (Gittenberger-de Groot et al., 2005). However, not all cardiac NCC colonize the OFT cushions, as a subpopulation remains in the caudal pharyngeal arches and differentiates in a smooth muscle layer enveloping the 4th and 6th arch arteries (Kirby et al., 1997; Jiang et al., 2000). Surprisingly, despite extensive NCC death and reduced Wnt1-Cre;R26r reporter expression within trigenic mutants that exhibit severe OFT septation and VSD defects, the AAAs have smooth muscle tunics (Fig. 4,5). This is consistent with the phenotypes reported in Wnt1-Cre;Smad4loxp/loxp and Wnt1-Cre;Tgfbr2loxp/loxp mutants (Ko et al., 2007; Jia et al., 2007; Nie et al., 2008; Büchmann-Møller et al., 2009; Choudhary et al., 2006), Thus, OFT septation defects appear to be a common feature of all mouse models in which TGFβ signaling is impaired in NCC, indicating a central role for TGFβ superfamily signaling within NCC during cardiac development. Although it is possible for embryos to exhibit OFT septation defects without AAA abnormalities and it is possible to have AAA defects without OFT septation defects (Kirby et al., 1997), it remains unclear why suppression of TGFβ signaling within the cardiac NCC subpopulation predominantly results in only OFT septation and VSD defects. It has been proposed that TGFβ signaling may not be required for specification of a smooth muscle fate, that this is a default pathway or that TGFβ signaling within cardiac NCC acts as a local morphogenic factor rather than as an instructural cue for cell lineage specification or smooth muscle differentiation (Choudhary et al., 2006). Our trigenic data would support these conclusions and additionally infer that TGFβ signaling in cardiac NCC within pharyngeal arches is required to provide the stimulus for subsequent NCC colonization of the OFT itself, septation and for survival of this colonizing OFT subpopulation. This conclusion is further supported by our significant finding that later induction of Smad7 in NCC that have already reached the pharyngeal arches and OFT also results in VSD (Fig. 8). Since Smad7 blocks multiple receptor-activated TGFβ signaling pathways, our work has not yet revealed roles of individual receptor-mediated TGFβ pathways that are responsible for maintaining these colonizing OFT NCC subpopulation.
Elucidating the molecular pathways and mechanisms regulating the fate of the cranial and cardiac NCC subpopulations is fundamental to our understanding of the pathogenesis of numerous congenital syndromes. In addition to stimulatory factors, migrating NCC encounter environments rich in inhibitors that may counteract the influences of signaling molecules such as Wnts, BMPs and TGFβ. Our Smad7 inhibitory NCC-restricted studies reinforce the notion that TGFβ signaling is dispensable for NCC formation and emigration but plays a critical role during craniofacial and cardiac NCC morphogenesis and colonization of target tissues. We observed a severe reduction of NCC pSmad2 and pSmad1/5/8 expression, resulting in suppression of the temporal and spatial specification of TGFβ signaling and subsequent loss of craniofacial and OFT cushion NCC in trigenic Smad7 embryos. Moreover, Smad7 overexpression within post-migratory cardiac NCC also resulted in cardiac defects, suggested that TGFβ signaling plays multiple roles at multiple times, even in the same NCC subpopulation. Although there is a paucity of data regarding the function of post-migratory NCC within the heart itself, it has been shown that NCC-specific N-cadherin, RhoA and Cx43 expression are each required for normal OFT morphogenesis, cell shape, alignment and cell-cell communication. Wnt1-Cre;N-cadherin mutant NCC are unable to elongate and align properly along the midline and remained rounded with limited contact with their neighbors within mutant OFTs (Luo et al., 2006). Activation of RhoA is required for NCC contact inhibition of locomotion in vivo to control NCC directional migration (Carmona-Fontaine et al., 2008); whilst Cx43 mediates modulation of polarized cell movement and the directional migration of cardiac NCC (Xu et al., 2009). Significantly, N-cadherin, RhoA and Cx43 are all known to be responsive to TGFβ-signaling (Massagué, 2008; Asazuma-Nakamura et al., 2006), thus these data suggest that further studies are required to understand the link between lineage commitment and the many changes in cushion cell shape, cell-matrix adhesion, and cell-cell adhesion that occur within the OFT itself and during interventricular septal morphogenesis, and how suppression of TGFβ superfamily signaling may aberrantly affect one or more of these processes. Perhaps inhibitors of TGFβ inhibitors are required to titrate or delay responses as NCC differentiate along particular pathways, and that Smad7 overexpression prevents endocardial cushion differentiation resulting in defective septation. Taken together, these results demonstrate that fine-tuning of TGFβ signaling via proteins including Smad7 is crucial for normal development. We speculate that this method for conditional myc-Smad7 expression may be useful for elucidating further TGFβ roles in other organs and tissues and addressing the biological significance of the concerted action between growth and transcription factors in regulating normal development and disease.
Supplementary Material
Supplemental Figure 1. Western analysis of NCC-enriched craniofacial tissues. Both E10.5 control (con) and Smad7 trigenic (tri) microdissected frontonasal regions (including 1st pharyngeal arch) fed doxycycline at E7.5, were examined via Western analysis. Note that myc-Smad7 is only expressed within pooled (n=4) doxycycline-fed trigenic E10.5 tissues, but is absent from pooled control (n=4) samples. Moreover, in tissues known to be mostly populated by NCC, densitometry revealed that the presence of myc-Smad7 suppressed both pSmad1/5/8 (by ~72%) and pSmad2 (by ~85%) levels relative to total Smad1 and pSmad2/3 levels when compared to age-matched controls. Abbreviations: tri, trigenic tetO-Smad7/Wnt1-Cre/RosartTA-EGFP embryos; con, control embryos.
{kind=link}
Acknowledgments
We thank Mica Gosnell, Goldie Lin and Jian Wang for histological, immunohistochemical and in situ hybridization support. We are also grateful to Dr. Andras Nagy (Samuel Lunenfeld Research Institute) for providing the pUHD10-3 vector; Dr. Yan Chen (Chinese Academy of Science) for the Smad7 cDNA vector and Dr. Henry Sucov (University of Southern California) for providing Wnt1-Cre mice. These studies were supported, in part, by NIH HL60714/HL92508 (SJC), HL85098 (AF and SJC), AHA Pre-Doc Fellowship (ST), Riley Children's Foundation and IU Department of Pediatrics/Cardiology support (SJC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson RM, Stottmann RW, Choi M, Klingensmith J. Endogenous bone morphogenetic protein antagonists regulate mammalian neural crest generation and survival. Dev Dyn. 2006;235(9):2507–20. doi: 10.1002/dvdy.20891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold NB, Ketterer K, Kleeff J, Friess H, Büchler MW, Korc M. Thioredoxin is downstream of Smad7 in a pathway that promotes growth and suppresses cisplatin-induced apoptosis in pancreatic cancer. Cancer Res. 2004;64(10):3599–606. doi: 10.1158/0008-5472.CAN-03-2999. [DOI] [PubMed] [Google Scholar]
- Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. Impaired Smad7-Smurf-mediated negative regulation of TGF-beta signaling in scleroderma fibroblasts. J Clin Invest. 2004;113(2):253–64. doi: 10.1172/JCI16269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aybar MJ, Mayor R. Early induction of neural crest cells: lessons learned from frog, fish and chick. Curr Opin Genet Dev. 2002;12(4):452–8. doi: 10.1016/s0959-437x(02)00325-8. [DOI] [PubMed] [Google Scholar]
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33(5):e51. doi: 10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L, Laffer U, Herrmann R, Rochlitz C. SMAD7 is a prognostic marker in patients with colorectal cancer. Int J Cancer. 2003;104(4):446–9. doi: 10.1002/ijc.10908. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128(8):1253–64. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Brewer S, Jiang X, Donaldson S, Williams T, Sucov HM. Requirement for AP2alpha in cardiac outflow tract morphogenesis. Mech. Dev. 2002;110:139–149. doi: 10.1016/s0925-4773(01)00579-2. [DOI] [PubMed] [Google Scholar]
- Büchmann-Møller S, Miescher I, John N, Krishnan J, Deng CX, Sommer L. Multiple lineage-specific roles of Smad4 during neural crest development. Dev Biol. 2009;330(2):329–38. doi: 10.1016/j.ydbio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Casellas R, Brivanlou AH. Xenopus Smad7 inhibits both the activin and BMP pathways and acts as a neural inducer. Dev Biol. 1998;198:1–12. doi: 10.1006/dbio.1998.8893. [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, Mayor R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456(7224):957–61. doi: 10.1038/nature07441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick K, Shojaei F, Gallacher L, Bhatia M. Smad7 alters cell fate decisions of human hematopoietic repopulating cells. Blood. 2005;105(5):1905–15. doi: 10.1182/blood-2004-03-0881. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chen H, Zheng D, Kuang C, Fang H, Zou B, Zhu W, Bu G, Jin T, Wang Z, Zhang X, Chen J, Field LJ, Rubart M, Shou W, Chen Y. Smad7 is required for the development and function of the heart. J Biol Chem. 2009;284(1):292–300. doi: 10.1074/jbc.M807233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Stottmann RW, Yang YP, Meyers EN, Klingensmith J. The bone morphogenetic protein antagonist noggin regulates mammalian cardiac morphogenesis. Circ Res. 2007;100(2):220–8. doi: 10.1161/01.RES.0000257780.60484.6a. [DOI] [PubMed] [Google Scholar]
- Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–429. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the (Sp2H) mutant. Development. 1997;124:505–514. doi: 10.1242/dev.124.2.505. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Abnormal neural crest stem cell expansion is responsible for the conotruncal heart defects within the Splotch (Sp2H) mouse mutant. Cardiovascular Res. 2000;47:314–328. doi: 10.1016/s0008-6363(00)00098-5. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35(1):1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- Dietrich P, Shanmugasundaram R, Shuyu E, Dragatsis I. Congenital hydrocephalus associated with abnormal subcommissural organ in mice lacking huntingtin in Wnt1 cell lineages. Hum Mol Genet. 2009;18(1):142–50. doi: 10.1093/hmg/ddn324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, Raible DW. Environmental signals and cell fate specification in premigratory neural crest. Bioessays. 2000;22(8):708–16. doi: 10.1002/1521-1878(200008)22:8<708::AID-BIES4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev. 2004;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, Lo CW. Migration of cardiac neural crest cells in Splotch embryos. Development. 2000;127(9):1869–78. doi: 10.1242/dev.127.9.1869. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129:4591–603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcoli FG, Huynh T, Scambler PJ, Baldini A. Tbx1 regulates the BMP-Smad1 pathway in a transcription independent manner. PLoS One. 2009;4(6):e6049. doi: 10.1371/journal.pone.0006049. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- García-Castro MI, Marcelle C, Bronner-Fraser M. Ectodermal Wnt function as a neural crest inducer. Science. 2002;297(5582):848–51. doi: 10.1126/science.1070824. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Bartelings MM, Deruiter MC, Poelmann RE. Basics of cardiac development for the understanding of congenital heart malformations. Pediatr Res. 2005;57:169–76. doi: 10.1203/01.PDR.0000148710.69159.61. [DOI] [PubMed] [Google Scholar]
- Han G, Li AG, Liang YY, Owens P, He W, Lu S, Yoshimatsu Y, Wang D, Ten Dijke P, Lin X, Wang XJ. Smad7-induced beta-catenin degradation alters epidermal appendage development. Dev Cell. 2006;11(3):301–12. doi: 10.1016/j.devcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Han J, Ishii M, Bringas P, Jr, Maas RL, Maxson RE, Jr, Chai Y. Concerted action of Msx1 and Msx2 in regulating cranial neural crest cell differentiation during frontal bone development. Mech Dev. 2007;124(9–10):729–45. doi: 10.1016/j.mod.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Lagna G, Massagué J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12(2):186–97. doi: 10.1101/gad.12.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA, Topper JN, Gimbrone MA, Jr., Wrana JL, Falb D. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–1173. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- Helms JA, Cordero D, Tapadia MD. New insights into craniofacial morphogenesis. Development. 2005;132:851–861. doi: 10.1242/dev.01705. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18(1):101–10. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishisaki A, Yamato K, Hashimoto S, Nakao A, Tamaki K, Nonaka K, ten Dijke P, Sugino H, Nishihara T. Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J Biol Chem. 1999;274:13637–13642. doi: 10.1074/jbc.274.19.13637. [DOI] [PubMed] [Google Scholar]
- Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Jr., Nakajima A, Shuler CF, Moses HL, Chai Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development. 2003;130:5269–5280. doi: 10.1242/dev.00708. [DOI] [PubMed] [Google Scholar]
- Itoh S, ten Dijke P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 2007;19:176–184. doi: 10.1016/j.ceb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Jia Q, McDill BW, Li SZ, Deng C, Chang CP, Chen F. Smad signaling in the neural crest regulates cardiac outflow tract remodeling through cell autonomous and non-cell autonomous effects. Dev Biol. 2007;311:172–184. doi: 10.1016/j.ydbio.2007.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- Kanzler B, Foreman RK, Labosky PA, Mallo M. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development. 2000;127:1095–1104. doi: 10.1242/dev.127.5.1095. [DOI] [PubMed] [Google Scholar]
- Kato Y, Habas R, Katsuyama Y, Näär AM, He X. A component of the ARC/Mediator complex required for TGF beta/Nodal signalling. Nature. 2002;418(6898):641–6. doi: 10.1038/nature00969. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Waldo KL. Neural crest and cardiovascular patterning. Circ. Res. 1995;77:211–215. doi: 10.1161/01.res.77.2.211. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Hunt P, Wallis K, Thorogood P. Abnormal patterning of the aortic arch arteries does not evoke cardiac malformations. Dev Dyn. 1997;208(1):34–47. doi: 10.1002/(SICI)1097-0177(199701)208:1<34::AID-AJA4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nat Rev Genet. 2002;3(6):453–61. doi: 10.1038/nrg819. [DOI] [PubMed] [Google Scholar]
- Ko SO, Chung IH, Xu X, Oka S, Zhao H, Cho ES, Deng C, Chai Y. Smad4 is required to regulate the fate of cranial neural crest cells. Dev Biol. 2007;312(1):435–47. doi: 10.1016/j.ydbio.2007.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–52. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- Kuang C, Xiao Y, Liu X, Stringfield TM, Zhang S, Wang Z, Chen Y. In vivo disruption of TGF-beta signaling by Smad7 leads to premalignant ductal lesions in the pancreas. PNAS USA. 2006;103:1858–1863. doi: 10.1073/pnas.0508977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, Maas RL, Maxson RE., Jr Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129(2):527–38. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Kalcheim C. The Neural Crest. Cambridge University Press; New York: 1999. [Google Scholar]
- Lee HY, Kléber M, Hari L, Brault V, Suter U, Taketo MM, Kemler R, Sommer L. Instructive role of Wnt/beta-catenin in sensory fate specification in neural crest stem cells. Science. 2004;303(5660):1020–3. doi: 10.1126/science.1091611. [DOI] [PubMed] [Google Scholar]
- Liu X, Chen Q, Kuang C, Zhang M, Ruan Y, Xu ZC, Wang Z, Chen Y. A 4.3kb Smad7 promoter is able to specify gene expression during mouse development. Biochim Biophys Acta. 2007;1769:149–152. doi: 10.1016/j.bbaexp.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, High FA, Epstein JA, Radice GL. N-cadherin is required for neural crest remodeling of the cardiac outflow tract. Dev Biol. 2006;299(2):517–28. doi: 10.1016/j.ydbio.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukko K, Ylikorkala A, Mäkelä TP. Developmentally regulated expression of Smad3, Smad4, Smad6, and Smad7 involved in TGF-beta signaling. Mech Dev. 2001;101(1–2):209–12. doi: 10.1016/s0925-4773(00)00556-6. [DOI] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108(4):601–9. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa Y, Zehir A, Maska E, Deng C, Schneider MD, Mishina Y, Cserjesi P. BMP signaling regulates sympathetic nervous system development through Smad4-dependent and -independent pathways. Development. 2009;136(21):3575–84. doi: 10.1242/dev.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Guris DL, Seo JH, Li L, Hammond J, Talbot A, Imamoto A. Crkl deficiency disrupts Fgf8 signaling in a mouse model of 22q11 deletion syndromes. Dev Cell. 2006;10(1):71–80. doi: 10.1016/j.devcel.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136(22):3699–714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Gulick J, Pratt R, Robbins J. Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. PNAS USA. 2009;106(36):15436–41. doi: 10.1073/pnas.0903302106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, Cherosky N, Karlo JC, O'Loughlin B, Wikenheiser J, Gargesha M, Doughman YQ, Charron J, Ginty DD, Watanabe M, Saitta SC, Snider WD, Landreth GE. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. PNAS USA. 2008;105(44):17115–20. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols DH. Neural crest formation in the head of the mouse embryo as observed using a new histological technique. J. Embryol. Exp. Morphol. 1981;64:105–120. [PubMed] [Google Scholar]
- Nie X, Deng CX, Wang Q, Jiao K. Disruption of Smad4 in neural crest cells leads to mid-gestation death with pharyngeal arch, craniofacial and cardiac defects. Dev Biol. 2008;316:417–430. doi: 10.1016/j.ydbio.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osumi-Yamashita N, Ninomiya Y, Doi H, Eto K. The contribution of both forebrain and midbrain crest cells to the mesenchyme in the frontonasal mass of mouse embryos. Dev Biol. 1994;164:409–419. doi: 10.1006/dbio.1994.1211. [DOI] [PubMed] [Google Scholar]
- Perl AK, Tichelaar JW, Whitsett JA. Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res. 2002;11(1):21–9. doi: 10.1023/a:1013986627504. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Molin D, Wisse LJ, Gittenberger-de Groot AC. Apoptosis in cardiac development. Cell Tissue Res. 2000;301(1):43–52. doi: 10.1007/s004410000227. [DOI] [PubMed] [Google Scholar]
- Randall V, McCue K, Roberts C, Kyriakopoulou V, Beddow S, Barrett AN, Vitelli F, Prescott K, Shaw-Smith C, Devriendt K, Bosman E, Steffes G, Steel KP, Simrick S, Basson MA, Illingworth E, Scambler PJ. Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J Clin Invest. 2009;119(11):3301–10. doi: 10.1172/JCI37561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios H, Koushik SV, Wang H, Wang J, Zhou HM, Lindsley A, Rogers R, Chen Z, Maeda M, Kruzynska-Frejtag A, Feng JQ, Conway SJ. Periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol Cell Biol. 2005;25(24):11131–44. doi: 10.1128/MCB.25.24.11131-11144.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rope AF, Cragun DL, Saal HM, Hopkin RJ. DiGeorge anomaly in the absence of chromosome 22q11.2 deletion. J Pediatr. 2009;155(4):560–5. doi: 10.1016/j.jpeds.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Saika S, Yamanaka O, Nishikawa-Ishida I, Kitano A, Flanders KC, Okada Y, Ohnishi Y, Nakajima Y, Ikeda K. Effect of Smad7 gene overexpression on transforming growth factor beta-induced retinal pigment fibrosis in a proliferative vitreoretinopathy mouse model. Arch. Ophthal. 2007;125:647–654. doi: 10.1001/archopht.125.5.647. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ito Y, Bringas P, Jr, Chou S, Urata MM, Slavkin H, Chai Y. TGFbeta-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development. 2006;133(2):371–81. doi: 10.1242/dev.02200. [DOI] [PubMed] [Google Scholar]
- Serbedzija GN, Bronner-Fraser M, Fraser SE. Vital dye analysis of cranial neural crest cell migration in the mouse embryo. Development. 1992;116:297–307. doi: 10.1242/dev.116.2.297. [DOI] [PubMed] [Google Scholar]
- Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell. 1996;85:331–343. doi: 10.1016/s0092-8674(00)81112-5. [DOI] [PubMed] [Google Scholar]
- Shaikh TH, O'Connor RJ, Pierpont ME, McGrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC. Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res. 2007;17(4):482–91. doi: 10.1101/gr.5986507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart NG, Apelqvist AA, Gu X, Harmon EB, Topper JN, MacDonald RJ, Kim SK. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible (maybe future studies shown if only diabetes mellitus. PLoS Biol. 2006;4(2):e39. doi: 10.1371/journal.pbio.0040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Conway SJ. Developmental Biology: The power of the blood. Nature. 2007;450:180–181. doi: 10.1038/450180a. [DOI] [PubMed] [Google Scholar]
- Snider P, Olaopa M, Firulli AB, Conway SJ. Cardiovascular development and the colonizing cardiac neural crest lineage. ScientificWorldJ. 2007;7:1090–1113. doi: 10.1100/tsw.2007.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Fix JL, Rogers R, Peabody-Dowling G, Ingram D, Lilly B, Conway SJ. Generation and characterization of Csrp1 enhancer-driven tissue-restricted cre recombinase mice. Genesis. 2008;46:167–176. doi: 10.1002/dvg.20379. [DOI] [PubMed] [Google Scholar]
- Snider P, Tang S, Lin G, Wang J, Conway SJ. Generation of Smad7(-Cre) recombinase mice: A useful tool for the study of epithelial-mesenchymal transformation within the embryonic heart. Genesis. 2009;47(7):469–75. doi: 10.1002/dvg.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg-Riethmacher E, Miehe M, Stolt CC, Goerich DE, Wegner M, Riethmacher D. Development and degeneration of dorsal root ganglia in the absence of the HMG-domain transcription factor Sox10. Mech Dev. 2001;109(2):253–65. doi: 10.1016/s0925-4773(01)00547-0. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stalmans I, Lambrechts D, Desmet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, Molin D, Hellings P, Boetel T, Haardt M, Compernolle V, Dewerchin M, Emanuel B, Gittenberger-de Groot A, Esguerra C, Scambler P, Morrow B, Driscol D, Moons L, Carmeliet G, Behn-Krappa A, Devriendt K, Collen D, Conway SJ, Carmeliet P. VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nature Medicine. 2003;9:173–182. doi: 10.1038/nm819. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–2218. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YQ, Sizeland A, Wang XF, Sassoon D. Restricted expression of type-II TGF beta receptor in murine embryonic development suggests a central role in tissue modeling and CNS patterning. Mech Dev. 1995;52(2–3):275–89. doi: 10.1016/0925-4773(95)00408-s. [DOI] [PubMed] [Google Scholar]
- Wang J, Nagy A, Larsson J, Dudas M, Sucov HM, Kaartinen V. Defective ALK5 signaling in the neural crest leads to increased postmigratory neural crest cell apoptosis and severe outflow tract defects. BMC Dev Biol. 2006;6:51. doi: 10.1186/1471-213X-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdak H, Ittner LM, Lang KS, Leveen P, Suter U, Fischer JA, Karlsson S, Born W, Sommer L. Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005;19:530–535. doi: 10.1101/gad.317405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Francis R, Wei CJ, Linask KL, Lo CW. Connexin 43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development. 2006;133(18):3629–39. doi: 10.1242/dev.02543. [DOI] [PubMed] [Google Scholar]
- Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. PNAS USA. 1997;94(8):3789–94. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HM, Wang J, Rogers R, Conway SJ. Lineage-specific responses to reduced embryonic Pax3 expression levels. Dev Biol. 2008;315(2):369–82. doi: 10.1016/j.ydbio.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen A, van Rooijen MA, Goumans MJ, Dewulf N, Bosman EA, ten Dijke P, Mummery CL, Huylebroeck D. Expression of the inhibitory Smad7 in early mouse development and upregulation during embryonic vasculogenesis. Dev Dyn. 2000;218(4):663–70. doi: 10.1002/1097-0177(200008)218:4<663::AID-DVDY1020>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Western analysis of NCC-enriched craniofacial tissues. Both E10.5 control (con) and Smad7 trigenic (tri) microdissected frontonasal regions (including 1st pharyngeal arch) fed doxycycline at E7.5, were examined via Western analysis. Note that myc-Smad7 is only expressed within pooled (n=4) doxycycline-fed trigenic E10.5 tissues, but is absent from pooled control (n=4) samples. Moreover, in tissues known to be mostly populated by NCC, densitometry revealed that the presence of myc-Smad7 suppressed both pSmad1/5/8 (by ~72%) and pSmad2 (by ~85%) levels relative to total Smad1 and pSmad2/3 levels when compared to age-matched controls. Abbreviations: tri, trigenic tetO-Smad7/Wnt1-Cre/RosartTA-EGFP embryos; con, control embryos.