

Abstract
Mouse genetic studies have defined a set of signaling molecules and transcription factors that are necessary to induce the forebrain. Here we describe an ENU-induced mouse mutation, nearly headless (nehe), that was identified based on the specific absence of most of the forebrain at midgestation. Positional cloning and genetic analysis show that, unlike other mouse mutants that disrupt specification of the forebrain, the nehe mutation disrupts mitochondrial metabolism. nehe is a hypomorphic allele of Lipoic acid Synthetase (Lias), the enzyme that catalyzes the synthesis of lipoic acid, an essential cofactor for several mitochondrial multienzyme complexes required for oxidative metabolism. The defect in forebrain development in nehe mutants is apparent as soon as the forebrain is specified, without a concomitant increase in apoptosis. Two tissues required for forebrain specification, the anterior visceral endoderm and the anterior definitive endoderm, develop normally in nehe mutants. However, a third head organizer tissue, the prechordal plate, fails to express markers of cell type determination and shows abnormal morphology in the mutants. We find that the level of phosphorylated (active) AMPK, a cellular energy sensor that affects cell polarity, is up-regulated in nehe mutants at the time when the prechordal plate is normally specified. The results suggest that the nehe phenotype arises because high levels of energy production are required for the specialized morphogenetic movements that generate the prechordal plate, which is required for normal development of the mammalian forebrain. We suggest that a requirement for high levels of ATP for early forebrain patterning may contribute to certain human microcephaly syndromes.
Introduction
Defects in formation of the human brain are common, devastating birth defects. Neural tube closure defects and microcephaly both occur at frequencies of approximately one in 1000 live births (Detrait et al., 2005; Zamorano and Chuaqui, 1979; Krauss et al., 2003). Both genetic and environmental factors contribute to these conditions. Dozens of genes have been identified that affect these aspects of neural development; in addition, dietary deficiencies, maternal diabetes, and maternal use of alcohol or tobacco increase the frequency of neural tube defects and microcephaly (Krauss et al., 2003). Despite the complex interactions between genes and environment that lead to these birth defects, studies of human genetic conditions that affect brain morphology and mouse genetic studies have made inroads into understanding the molecular bases of these processes (Copp et al., 2003; Frances et al., 2006).
In the mouse, genetic and embryological studies have defined sequential inductive interactions that are required early in development for specification of the forebrain. In the first inductive interaction, a population of extraembryonic cells, the anterior visceral endoderm (AVE), signals to the adjacent embryonic cells to initiate forebrain specification. Mechanical removal of the AVE at the early-to-late streak stages results in a loss or reduction of markers of the developing forebrain (Thomas and Beddington, 1996). The AVE cells express inhibitors of Wnt and Nodal signaling (Belo et al., 1997; Meno et al., 1998; Zakin et al., 2000), and the evidence indicates that these inhibitors promote forebrain development in the adjacent neural tissue (Niehrs 2004). The genes encoding the transcription factors Lhx1 and Otx2 are expressed in the AVE and chimeric embryos that lack the Lhx1 or Otx2 genes in extraembryonic tissues show anterior truncations of the brain, indicating that these genes are required for this activity of the AVE (Rhinn et al., 1998; Shawlot et al., 1999; Varlet et al., 1997). At a slightly later stage (the zero-to-late allantoic bud stage), two derivatives of the anterior primitive streak (APS), the anterior definitive endoderm (ADE) and the prechordal plate (PCP, the anterior-most part of the axial mesendoderm), lie beneath the presumptive forebrain. Experimental removal of the ADE and PCP causes the loss of markers of the developing forebrain (Camus et al., 2000). Inhibitors of BMP and Wnt signaling are expressed in the ADE and PCP, and are thought to allow expression of forebrain markers in the overlying neural plate. The transcription factors Lhx1 and Foxa2 are expressed in the axial mesendoderm and embryos that lack Foxa2 or Lhx1 specifically in embryonic tissues lack a normal forebrain (Hallonet et al., 2002). Although both the AVE and APS are required for forebrain specification, neither the AVE nor the derivatives of the anterior primitive streak are sufficient to induce anterior neural tissues after transplantation (Tam and Steiner, 1999), but grafts that include both AVE and APS can induce anterior neural marker expression at ectopic positions (Tam and Steiner, 1999). These findings indicate that specification of the forebrain requires synergistic, sequential signaling activities from the AVE and the anterior midline tissues of the embryo.
In a genetic screen for mutations that affect the body plan of the mouse embryo, we identified a mutation, nearly headless (nehe), that led to the absence of most of the forebrain. In contrast to the signaling components and transcription factors that have previously been shown to play roles in development of the mouse forebrain, we find that nehe disrupts a gene required for normal mitochondrial metabolism. The nehe mutation disrupts lipoic acid synthetase (Lias), which catalyzes the synthesis of lipoic acid (Marquet et al., 2001; Miller et al., 2000), a disulfide-containing short-chain fatty acid that is an essential cofactor for mitochondrial multienzyme complexes required for oxidative metabolism, including the pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes (Reed and Hackert, 1990). We find that initial specification of the forebrain fails in nehe mutants due to a defect in specification of the prechordal plate, a non-neural tissue required for the induction of the forebrain. These findings define a specific requirement for mitochondrial metabolism in the initial specification of the mammalian forebrain.
Materials and Methods
Mouse strains and genotyping
Liasnehe was identified in a screen for recessive ethylnitrosourea-induced mutations (García-García et al., 2005). Liasnehe mutant mice were genotyped based on linkage to flanking SSLP markers (D5SKI96 and D5SKI75 http://mouse.ski.mskcc.org/). Phenotypic analysis was carried out in congenic C3HeB/FeJ animals. Lias null mice (Liastm1Mae) have been described previously (Yi and Maeda, 2005).
Genetic mapping and cloning of nehe
The nehe mutation was induced in C57BL/6J and was mapped by backcrossing to C3HeB/FeJ mice (Kasarskis et al., 1998). The nehe mutation was mapped to a 1.2 Mb region on chromosome 5 between the SSLP markers D5Mit197 and D5SKI36 (http://mouse.ski.mskcc.org). Candidate genes in the critical interval were sequenced based on physical map information from Ensembl database (http://www.ensembl.org/). Complementary cDNAs of candidate genes were generated from nehe mutant embryos and C57BL/6J control embryos using Superscript OneStep (Invitrogen) and the RT-PCR products were sequenced. The sequence of RT-PCR products amplified from nehe embryos showed a deletion of exon six in Lipoic acid synthetase (Lias). The genomic sequence of nehe embryonic DNA carried a T to C change at the splice donor site downstream of exon six, changing the GT in the intron to GC.
Phenotypic analysis and embryo staging
In situ hybridization, immunofluorescence and scanning electron microscopy (SEM) were done as previously described (Eggenschwiler and Anderson, 2000; Huangfu and Anderson, 2005). Because of the developmental delay of nehe embryos, wild-type controls were stage-matched rather than littermates (Downs and Davies, 1993). At early somite stages, the nehe embryos were smaller than somite-matched wild-type embryos; we therefore staged mutant embryos between late streak and early somite stages not by their size but by morphological landmarks such as the shape of the node and midline. Whole-mount embryos were imaged using a Zeiss Axiocam HRC digital camera on a Leica DM1RE2 inverted confocal microscope. Sections were imaged on a Leica MZFLIII microscope. For SEM, the embryos were viewed with a LEO1550 Field Emission Scanning Electron Microscope at 2.5 kv. Confocal datasets were analyzed using the Volocity software package (Improvision); 3D reconstructions were created using the Volocity software package (Improvision) and Amira package (Mercury Computer Systems). The mitotic index was defined as the ratio of phospho-histone-H3-positive cells to DAPI-positive cells in the sections.
Western blotting
Embryos were lysed in RIPA buffer with a protease inhibitor cocktail (Roche). Total protein concentration was determined by BCA Protein Assay Kit (Thermo Scientific). Signals were detected with ECL Western Blotting Detection Reagents (Amersham). The levels of lipoic acid-modified proteins, p-AMPK and cyclin E on Westerns were quantified using Photoshop.
Antibodies and reagents for immunofluorescent staining
Commercial antibodies used were: anti-lipoic acid (Calbiochem), anti-phosphohistone H3 (Upstate), anti-cleaved caspase 3 (Cell Signaling), anti-AMPKα (Cell Signaling), anti-phosphoAMPKα (Cell Signaling), anti-Cyclin E (Santa Cruz Biotechnology) and anti-γ-tubulin (Sigma). Fluorescent secondary antibodies were from Jackson Laboratories and Molecular Probes. Filamentous actin was detected with rhodamine-conjugated phalloidin (Molecular Probes) and nuclei were visualized with DAPI (Sigma).
Results
The nearly headless phenotype
The nearly headless (nehe; formerly called 7A5) mutation was identified in an ENU mutagenesis screen designed to identify recessive mutations that affect patterning and morphogenesis in the midgestation mouse embryo (García-García et al., 2005) (Fig. 1). The mutant phenotype was identified at e9.5 based on the small head and open anterior neural tube of homozygous embryos. The mutants survived until e12.5 when the embryos showed a severe anterior truncation of the brain, in which the telencephalon was completely absent. At that stage, the heart was swollen, which suggested that death was due to cardiovascular defects. In addition to these morphological defects, nehe embryos were delayed in development relative to their littermates; a delay of 10–12h was seen beginning at the head-fold stage and mutants remained 10–12h delayed relative to littermates at all stages thereafter.
Figure 1. The nehe mutant phenotype.
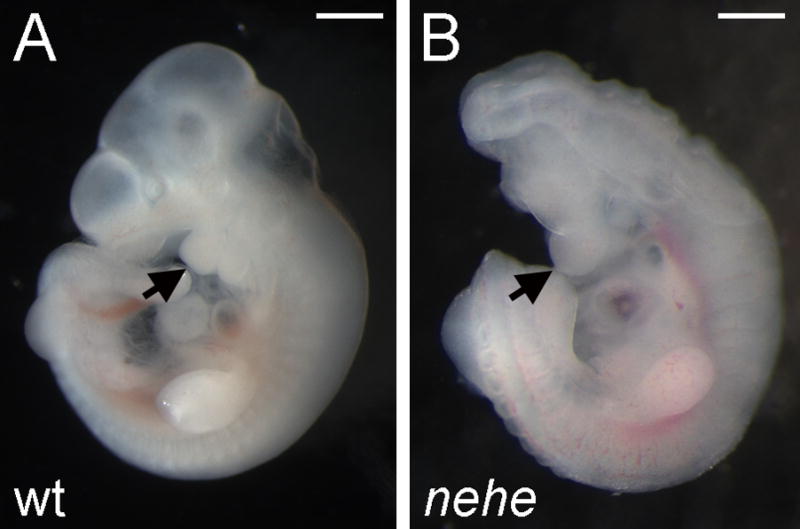
Wild-type (A) and nehe mutant (B) embryos, dissected at embryonic day 10.5 (e10.5) and e11.0, respectively. The nehe mutant shows the anterior truncation and open neural tube characteristic of this genotype. Arrows point to the first branchial arch and highlight the loss of tissue anterior to this structure in the mutant. Scale bars: 0.5mm.
To define the brain regions that were affected in e10.5 nehe mutants, we analyzed the expression of genes that mark specific regions of the developing brain by in situ hybridization. As in wild type, Fgf8, which marks the midbrain-hindbrain junction, was expressed in a stripe in the e10.5 nehe brain (Fig. 2A, B). En2, which is expressed in a broader region flanking the midbrain-hindbrain junction in wild-type embryos, was expressed at the anterior tip of the nehe brain (Fig. 2C, D), indicating that tissues anterior to the midbrain were absent in nehe embryos. Shh, which marks the ventral midline of the neural tube was expressed, but only a small amount of tissue dorsal to the Shh expression domain was detected at the anterior of nehe embryos (Fig. 2E, F).
Figure 2. Loss of forebrain domains in nehe embryos.
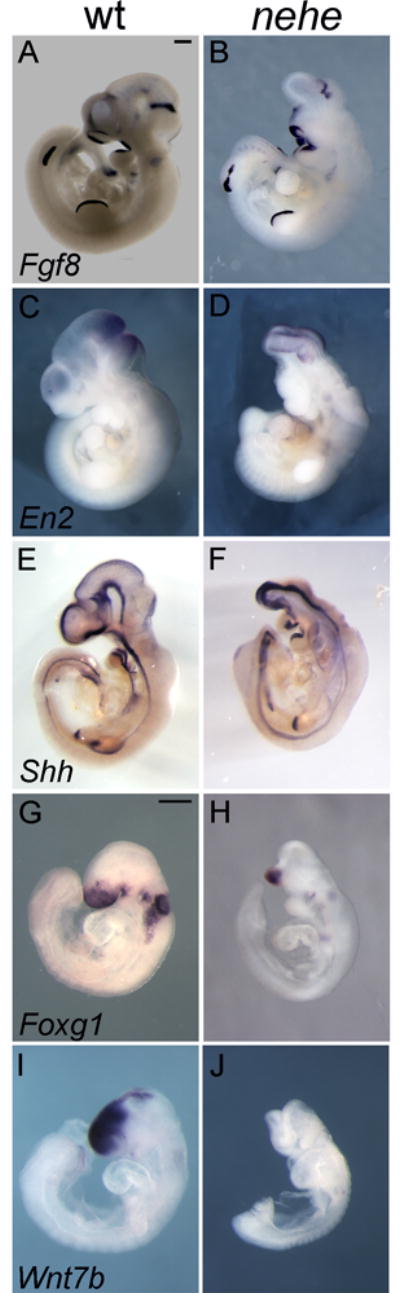
(A–F) Whole mount in situ hybridizations on e10.5 wild-type and somite-matched nehe embryos. Fgf8 is expressed in a stripe at the mid-hindbrain junction (arrows) in wild-type (A) and mutant (B) embryos (n=5). The discontinuity in the staining in the hindlimb is the result of a tear in this embryo and was not seen in other cases. En2 is expressed in a broader domain that reaches to the middle of the wild-type midbrain (C) and is expressed in the most anterior region of the mutant brain (D) (n=4). Shh is expressed at the ventral midline of both wild-type and mutant embryos (E, F), but there is little or no tissue dorsal to the Shh domain in the nehe forebrain (n=5). (G–J) e9.5 wild-type and somite-matched nehe embryos. Foxg1 is expressed in the forebrain of wild-type (G), but is limited to a small ventral domain in nehe (H) (n=5). Wnt7b is expressed in the dorsal forebrain of wild type (I), but is completely absent in mutant embryos (J) (n=5).
A similar loss of forebrain structures was apparent one day earlier, at e9.5. Foxg1, a marker of the telencephalon, was expressed in only a small, ventral region of the nehe neural tissue (Fig. 2G, H), suggesting that the dorsal forebrain tissue was reduced in mutant embryos. The loss of the dorsal forebrain in the mutants was confirmed by the lack of expression of Wnt7b, a marker of the dorsal forebrain (Fig. 2I, J).
To determine whether the lack of anterior neural tissue was due to a failure to specify anterior neural tissue or to a later loss of neural cells, we examined the expression of the earliest markers of the forebrain region. At the four somite stage (e8.5), Otx2 is expressed in the presumptive forebrain and midbrain of wild-type embryos (Ang et al., 1994), but Otx2 was expressed at high levels only on the midline of nehe embryos and could barely be detected in the adjacent anterior neural plate (Fig. 3A, B). At the headfold stage (e7.75), Otx2 is expressed in the presumptive anterior neural plate of wild-type embryos (Ang et al., 1994), but its expression was less intense and did not extend all the way to the anterior of nehe embryos (Fig. 3C, D). Similarly, Hesx1 is normally expressed in the anterior neural plate of wild-type embryos at e7.5 (Lowe et al., 2001), but its expression does not extend to the rostral tip of the nehe mutant neurectoderm (Fig. 3E, F). Thus from the earliest stage when cells have anterior neural plate identity, the domain of forebrain progenitors in nehe embryos was dramatically reduced in size.
Figure 3. Loss of anterior neural progenitors in early nehe embryos.
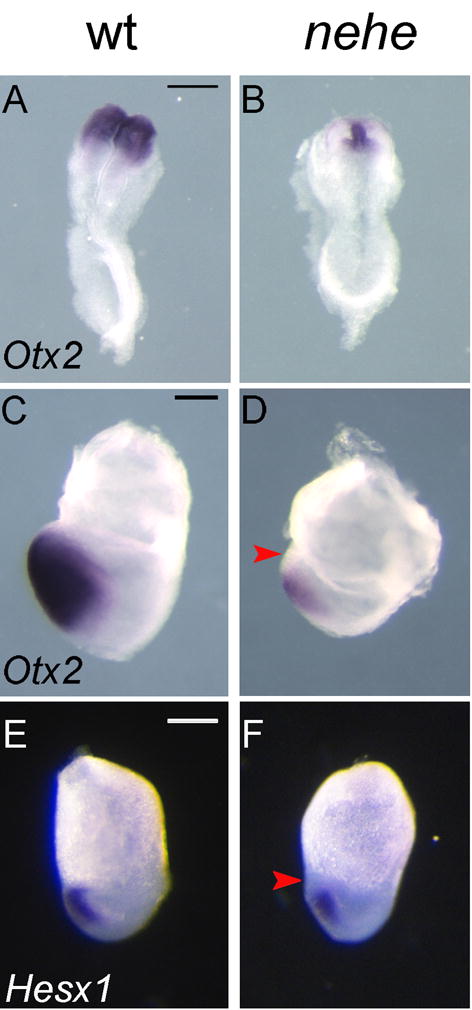
Otx2 is expressed throughout the headfolds of wild-type e8.5 embryos (A) but is detected only at the midline of stage-matched mutant embryos (B) (n=4). The embryos in (A) and (B) are both at the four somite stage. At the headfold stage (e7.75), Otx2 is expressed in the neurectoderm to the anterior end of the embryo, all the way to the embryonic/extraembryonic boundary (C), but Otx2 is more weakly expressed in stage-matched nehe embryos and does not extend as far anteriorly (red arrow) (D) (n=2). The embryos in (C) and (D) are stage matched based on the length of the allantois (black arrows). Hesx1 is expressed in the anterior neurectoderm of wild-type embryos (E), but is not expressed in the most anterior neurectoderm of nehe mutants (red arrow) (F) (n=4). Scale bars: 0.25mm.
The nearly headless mutation disrupts Lipoic Acid Synthetase (Lias)
Meiotic recombination mapping localized the nehe mutation to a 1.2 Mb region on chromosome 5 (Materials and Methods). We sequenced RT-PCR products amplified from mutant embryonic RNA for all eight genes in this interval. The only change detected was the deletion of exon six in the Lipoic acid synthetase (Lias) transcript in nehe embryos, which suggested aberrant splicing of the mutant Lias transcript. The genomic sequence revealed a T to C substitution at the splice donor site downstream of the sixth exon in nehe genomic DNA. The aberrant splicing event would truncate the open reading frame after amino acid 182 (out of 373 total amino acids) and delete half of the radical SAM domain required for catalytic activity (Fig. 4A; Layer et al., 2004; Morikawa et al., 2001). In addition to the unspliced transcript, we were able to detect a lower abundance RT-PCR product of the size of wild type Lias transcript (data not shown), suggesting that some wild-type Lias transcript was made in the mutant embryos.
Figure 4. The nehe phenotype is caused by a hypomorphic mutation in Lipoic acid synthetase.

(A) The nehe mutation disrupts a splice site in Lias; translation of the mutant transcript would add 17 amino acids from the fifth intron to the open reading frame before a stop codon. The truncated protein made by the nehe allele lacks most of the radical SAM catalytic domain. (B) Liasnehe fails to complement the targeted null allele of Lias. Lias−/− embryos arrest at e7.5, with a poorly developed embryonic region and an allantois (arrow). In contrast, both Liasnehe homozygotes and Liasnehe/Liasnull embryos survive to later stages and show an anterior truncation and open anterior neural tube at e9.5. (C) Western blot analysis with an antibody that recognizes lipoic acid in e9.5 embryo extracts, compared to γ-tubulin levels. The horizontal bars next to wild-type lane represent 75 and 50 kD size markers. The two major substrates of lipoic acid are the E2 of KGDH, dihydrolipoamide succinyltransferase (Dlst), (predicted MW = 49 kDa) and the E2 of PGH, dihydrolipoamide S-acetyl transferase (Dlat) (predicted MW = 68 kDa). The levels of lipoic-acid modified proteins are greatly reduced in nehe embryos. (D) Immunofluorescent staining to detect Lipoic acid in e7.5 embryos shows that Lipoic acid-modified proteins are broadly distributed in wild-type embryos, but not detectable in nehe mutants.
Embryos homozygous for a null allele of Lias arrest at e7.5 (Yi and Maeda, 2005). In a complementation test, we found that nehe/Liasnull embryos showed same anterior truncation seen in nehe homozygotes (Fig. 4B). The only difference between the phenotypes of the nehe homozygotes and nehe/Liasnull embryos was that the developmental delay was exaggerated in nehe/Liasnull embryos, so that e10.5 mutants were approximately 24h, rather than 12h, delayed relative to littermates. Because the nehe mutation failed to complement the null Lias allele, we concluded that the nehe phenotype was due to partial loss of function of the Lias gene.
Lias catalyzes the synthesis of lipoic acid, an essential cofactor for several multienzyme complexes required for oxidative metabolism (Marquet et al., 2001; Miller et al., 2000). Lipoic acid is covalently attached to the E2 subunits of the pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (KGDH) complexes and can be detected as abundant covalent conjugates in mitochondrial protein extracts (Reed and Hackert, 1990). To test whether the nehe mutation affected lipoic acid production, we examined the proteins covalently modified by lipoic acid in mutant embryo extracts using an antibody that recognizes lipoic acid. Two prominent proteins were detected on Western blots of wild-type embryos; these proteins were the size of the E2 subunits of PDH and KGDH, the most abundant lipoic acid modified proteins (Reed and Hackert, 1990). In contrast, the level of the lipoic acid-modified proteins was reduced by more than five-fold in nehe embryos (Fig. 4C); the presence of some lipoic-acid modified protein was consistent with the RT-PCR results that detected some full-length transcript and confirmed that nehe is a partial loss-of-function allele. It has been shown previously that Lias is localized specifically to mitochondria of all species examined, including mammalian cells (Morikawa et al., 2001; Yasuno and Wada, 1998; Yasuno and Wada, 2002). As expected, immunofluorescent staining for lipoic acid in sections of wild-type headfold-stage embryos showed that lipoic-acid modified proteins were present in cytoplasmic puncta that presumably corresponded to mitochondria in all embryonic cell types of wild-type embryos, but these puncta could not be detected in nehe embryos (Fig. 4D).
Reduced proliferation in early nehe embryos does not account for the loss of the forebrain
At the late streak stage, wild-type and nehe embryos were indistinguishable in size or morphology. However, as described above, nehe embryos were delayed in development by about 12h relative to their littermates from the 6-somite stage onward. We therefore examined apoptosis and proliferation in the mutant embryos between the late streak and the 2–4 somite stages. Based on staining for activated Caspase-3, there were only very small numbers of apoptotic cells in either wild-type or mutant embryos in late streak to early somite stage embryos (data not shown). Elevated levels of cell death were observed in the brain and other tissues in the mutants, but only after e9.5 (data not shown).
We assayed the number of number of mitotic cells by immunofluorescent detection of phospho-histone H3 in wild-type and mutant embryos (Fig. 5A–F). At the late streak stage (e7.5), the mitotic index was the same in wild-type and mutant embryos (Fig. 5G). However, at the headfold stage (e7.75 in wild type), the mitotic index of nehe embryos was about one-third that of wild type (3% vs 10% PHH3+). At the 2–4 somite stage (e8.5 in wild type), the mitotic index of nehe embryos had recovered, so that it was again similar to that of wild type (8% vs 9% PHH3+). These data suggest that there is a critical developmental time window at the headfold stage that requires a higher level of Lias activity for normal cell proliferation.
Figure 5. Reduced proliferation in headfold nehe embryos.
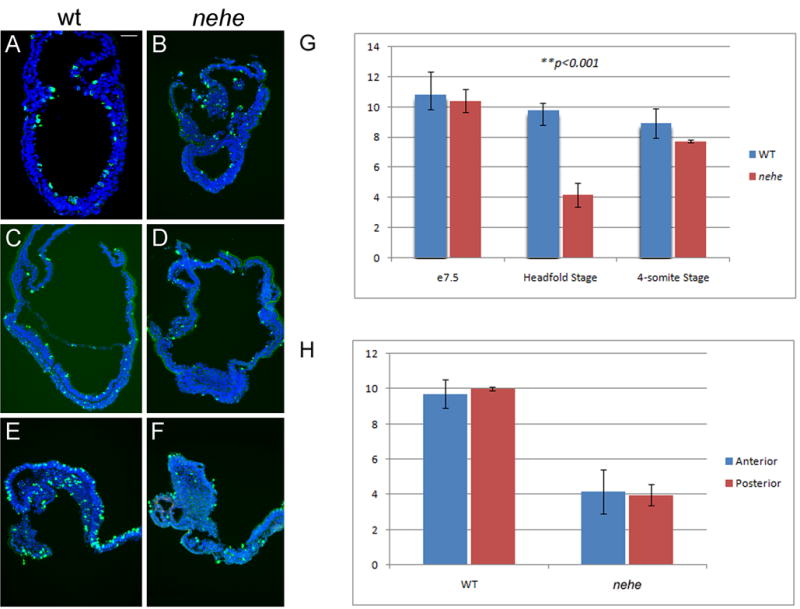
(A–F) Immunofluorescent staining with antibodies that recognize phospho-histone H3 (green) detects mitotic cells in wild-type (A, C, E) and nehe (B, D, F) embryos at e7.5 (A, B), headfold (C, D) and 4–6 somite (E, F) stages. Blue = DAPI staining. Anterior to the left. Scale bars: 100 μm in A–F. Note the strong reduction in the number of mitotic cells in the entire embryonic region of headfold-stage nehe embryos. (G) Phosphohistone H3-positive and total (DAPI-stained) nuclei were counted in sections from wild-type and mutant embryos at the three developmental stages. Embryonic and extraembryonic regions were counted separately. The only significant difference between wild type and mutant was in the embryonic region at the headfold stage, when the number of mitotic cells in the mutant was three-fold lower than in wild-type. These two values are different with p = 0.0001. At the same stage, there was no significant difference between the mitotic indices in the extraembryonic regions of wild type and mutant embryos. (H) The same sections from the headfold stage embryos analyzed in (G) were divided into two halves, anterior and posterior to the node, and phospho-histone H3 and DAPI-positive cells were counted. There was no significant difference in the mitotic index in the anterior versus posterior half of either wild-type or mutant embryos.
In mammalian cell cycle, the availability of essential nutrients in environment is monitored by the G1-S checkpoint. Previous studies have suggested that AMP-activated protein kinase (AMPK) can function as a sensor for cellular energy levels (Jones et al., 2005) and elevated AMPK activity can trigger cell cycle arrest by downregulation of cyclin E (Mandal et al., 2005). Consistent with a decrease in ATP levels, we found that the level of phosphorylated (active) AMPK was ~3-fold (2.6±0.1) higher in mutant headfold-stage embryos than in stage-matched controls (Fig. 6A). We also found that the level of cyclin E in headfold nehe embryos was ~3-fold lower (0.38±0.04) than the level in stage-matched wild-type embryos (Fig. 6B), consistent with the elevation of AMPK activity in the mutants. We conclude that decreased levels of Lias lead to decreased energy production at the headfold stage, which activates AMPK and partially blocks progression through the cell cycle. This would be sufficient to account for the delay in the development of mutant embryos that begins at this stage.
Figure 6. AMPK activation and decreased cyclin E in nehe embryos.
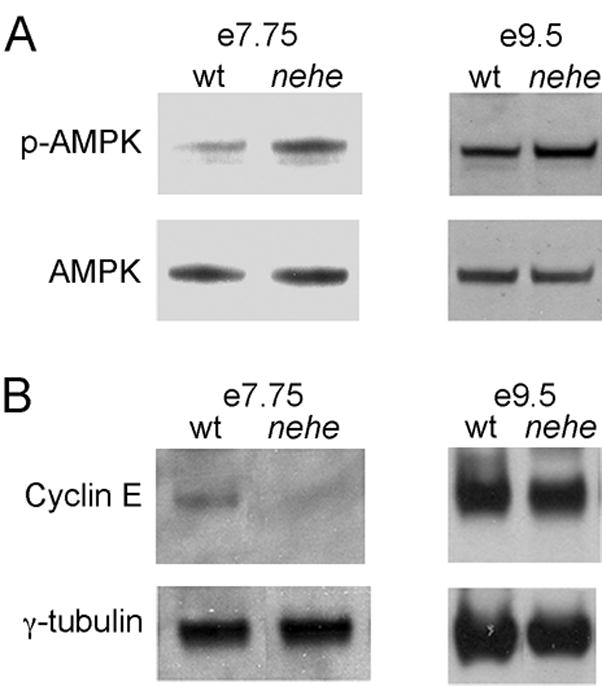
(A) Western blot analysis of phospho-AMPK in embryo extracts. Representative experiments are shown. Three independent samples for mutants and seven independent samples for wild type were analyzed; averaged among the experiments, the mutants had a 2.6+0.1-fold higher level of p-AMPK/total AMPK relative to wild type. At e9.5, three independent mutant protein extracts and seven independent wild-type extracts were analyzed, on average the p-AMPK/total AMPK ration in mutant embryos was 1.2±0.1-fold higher than in stage-matched controls. (B) Western blot analysis of cyclin E in headfold and e8.5 embryos; representative experiments are shown. Three independent mutant and five wild type samples were analyzed at headfold stage; the level of cyclin E in mutants (relative to the γtubulin loading control) was 0.38±0.04 the value in wild type. At e9.5, three mutant and five wild type extracts were analyzed; the level of cyclin E in mutants at this stage was equivalent to the amount in wild type (1.0±0.1). γtubulin was used as a loading control. Each lane in parts (A) and (B) represents protein extracted from 4–8 embryos.
Because nehe mutant embryos resumed the normal rate of after the early somite stage (although they never caught up with wild-type littermates), we hypothesized that compensatory mechanisms might restore ATP levels at later stages. To test this hypothesis, we assayed for phospho-AMPK level at e9.5, a stage when proliferation was normal in mutant embryos. In contrast to the three-fold higher level of p-AMPK at early stages, p-AMPK was only 20% (1.2±0.1) higher in e9.5 mutant embryos than in stage-matched controls (Figure 6A). Consistent with the more normal levels of p-AMPK, the amount of cyclin E in e9.5 nehe embryos was approximately the same level (1.0±0.1) present in stage-matched wild-type embryos (Figure 6B). Thus compensatory mechanisms that normalize the level of cellular ATP must be activated after the early somite stage, so that the rate development is no longer as sensitive to the reduction of Lias activity.
nehe embryos showed a smaller domain of forebrain progenitors by the early somite stage (Fig. 3), suggesting that the early proliferation defect could be the cause of the anterior truncation phenotype. However we found that the reduction in proliferation rate was evenly distributed across the mutant embryos at headfold stage; in particular there was no greater disruption in proliferation in the anterior neuroectoderm than in the rest of the embryo, arguing against the possibility that differential rates of proliferation accounted for the small forebrain of nehe embryos (Fig. 5H).
Defective specification of the prechordal plate precedes the loss of anterior neural fates in nehe embryos
Because defects in proliferation did not appear to account for the nehe anterior truncation phenotype, we analyzed the inductive interactions required for specification of the anterior forebrain in the mouse embryo. The first signal comes from the AVE, which initiates anterior specification of the adjacent epiblast at the onset of gastrulation (e6.5). The AVE is marked by Hhex and Cer1 expression at e6.5 (Thomas et al., 1998; Shawlot et al., 1998). Both the Hhex and Cer1 expression appeared normal in nehe embryos (Fig. 7A–D), indicating that a normal AVE was present in nehe mutants. The anterior neural fate is labile until the AVE signal is reinforced by signals from the ADE and prechordal plate (also called the anterior mesendoderm), two derivatives of the anterior primitive streak (Robb and Tam, 2004). Expression of Cer1, which marks the ADE (Shawlot et al., 1998), appeared normal in nehe embryos (Fig. 7E, F). Markers of the posterior axial mesendoderm such as Chordin (Bachiller et al., 2000) were also expressed normally in the mutant embryos (Fig 7E, F). Thus three of the tissues required for specification of the forebrain, the AVE, ADE and posterior axial mesendoderm, were specified correctly in nehe embryos.
Figure 7. The AVE, ADE and posterior AME are normal in nehe embryos.
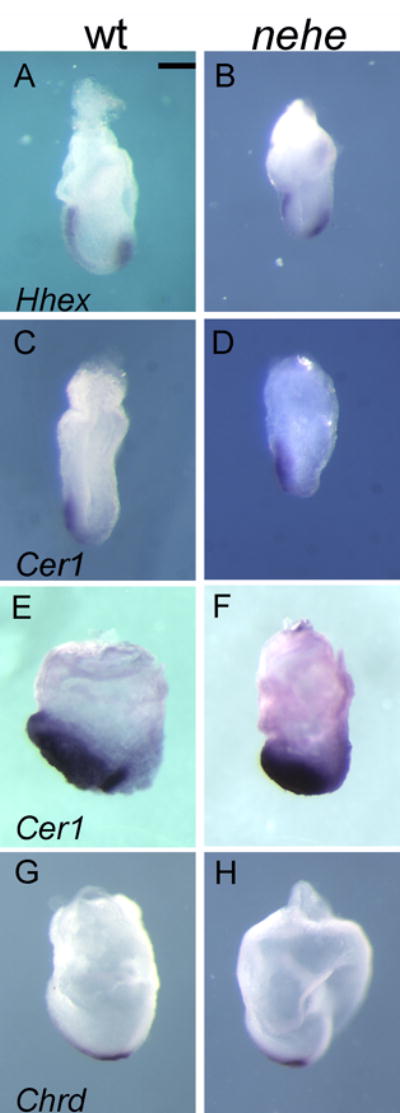
(A, B) The AVE marker Hhex is expressed normally in e7.5 mutant embryos (n=4). (C, D) Cer1, which marks the AVE, is expressed normally in mutant embryos (n=2). (E, F) Cer1, an early marker of the definitive endoderm, is expressed normally in stage-matched mutant embryos (n=5). (G,H) Chordin is expressed normally in the posterior axial mesendoderm in stage-matched mutant embryos (n=5). Scale bars: 0.25mm.
In contrast, the prechordal plate (the anterior axial mesendoderm) was abnormal in nehe embryos. Foxa2 was expressed in posterior axial midline, but was barely detectable in the anterior midline of nehe embryos, compared to stage-matched wild types (Fig 8A, B; Supp. Fig. 1). To confirm that there was a defect in the prechordal plate, we examined the expression of Gsc and Dkk1, which are expressed specifically in most anterior domain of the mesendoderm in wild-type embryos at the early headfold stage (Blum et al., 1992; Mukhopadhyay et al., 2001). Like Foxa2, expression of both Gsc and Dkk1 was reduced or absent in the anterior midline of nehe embryos (Fig 8C–F; Supp. Fig. 1).
Figure 8. Loss of anterior neural fates and defective specification of the prechordal plate in nehe embryos.
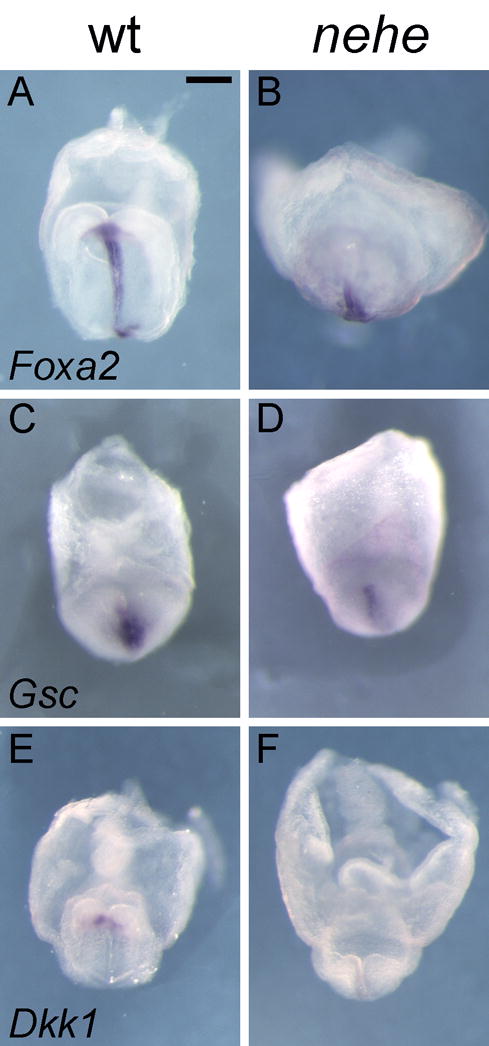
(A–B) Foxa2 is expressed normally in the posterior midline but is barely detectable in the anterior midline of stage-matched nehe embryos (n=3), compared to stage-matched wild types. (C–F) The anterior mesendoderm markers Gsc (C, D) (n=6) and Dkk1 (E, F) (n=4) are barely detectable at the anterior mesendoderm of the mutant embryos, compared to stage-matched wild types. Scale bars: 0.25mm in A–H.
The tissues that induce the forebrain have a characteristic morphology at the time of their interaction with the neural plate. The cells of the axial midline and prechordal plate are characterized by small apical surfaces (Sulik et al., 1994; Lee and Anderson, 2008). By the late headfold stage, the cells of the notochordal plate have aligned in a narrow stripe on the midline and the prechordal plate cells form a round cluster at the anterior of the midline. This morphology was apparent by both scanning electron microscopy (SEM) and immunofluorescent images of whole mount early bud to early headfold stage wild-type embryos (Fig. 9A, C; Supp Fig. 2). In contrast, cells with small apical surfaces at the anterior of the midline of stage-matched nehe embryos were intermixed with cells with large apical surface and the rosette-like arrangement of cells characteristic of the prechordal plate was not present (Fig. 9B, D; Supp. Fig. 2). The abnormal morphology of cells at the anterior midline, together with the failure of expression of the prechordal plate markers, demonstrates that a normal prechordal plate failed to form in nehe embryos. The failure to form the normal prechordal plate is sufficient to account for the failure of forebrain specification seen in the mutants.
Figure 9. The morphology of the prechordal plate is disrupted in nehe embryos.
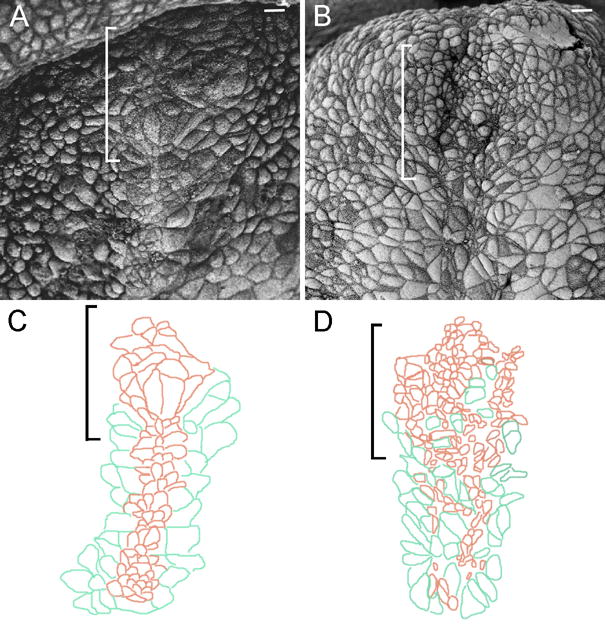
Scanning electron micrographs of stage-matched headfold wild-type (A) and mutant (B) embryos. (C, D) Camera lucida drawings tracings (Photoshop) of the cell boundaries in parts (A) and (B) highlight the morphology of the anterior midline cells. Anterior is up; brackets indicate position of the prechordal plate; scale bars: 10 μm. The prechordal plate lies anterior to the axial mesendoderm. Cells of the axial mesendoderm organize into a 3–4 cell-wide stripe of cells with small apical surfaces on the midline (brown in C) and are flanked by endoderm cells with larger apical surfaces (green). The cells of the wild-type prechordal plate anterior to the axial mesendoderm form a rosette-like structure (A; most anterior brown cells in C). In the mutant (B, D), cells with small (brown) and large (green) apical surfaces are not well sorted and no rosette-like organization at the anterior end of the midline is present. Embryos are at the zero somite stage.
Discussion
We identified the nehe mutant based the specific absence of forebrain structures at midgestation, a phenotype similar to that seen in mutants that lack the Lhx1 and Otx2 transcription factors (Shawlot and Behringer, 1995; Acampora et al., 1995) or the Wnt inhibitor Dkk1 (Mukhopadhyay et al., 2001). The identification of nehe as an allele of Lias adds a new dimension, regulation of energy metabolism, to the processes necessary for forebrain specification.
Lipoic acid synthetase is required for forebrain development
Genetic mapping and complementation experiments demonstrated that the nehe phenotype is caused by a partial loss-of-function mutation in the Lipoic acid synthetase gene. Lipoic acid, which is found only in mitochondria (Morikawa et al., 2001; Yasuno and Wada, 1998; Yasuno and Wada, 2002), acts as a covalently-bound cofactor for several mitochondrial enzymes. The vast majority of cellular lipoic acid in the early mouse embryo is bound to two components of the citric acid cycle, the pyruvate dehydrogenase complex and the α-ketoglutarate dehydrogenase complex (Fig. 4B). The pyruvate dehydrogenase complex is composed of three enzymes (E1, E2, and E3); lipoic acid is the cofactor for the E2, dihydrolipoyl acetyltransferase (Perham, 2000). Null alleles of mouse Lipoic acid synthetase or the gene encoding the E3 enzyme of the complex, dihydrolipoamide dehydrogenase (Dld) both cause lethality at about the late streak stage (e7.5) (Yi and Maeda, 2005; Johnson et al., 1997). Although Lias is also important for the catabolism of several amino acids and exogenous lipoic acid has antioxidant activity, the similarity of the Lias and Dld phenotypes argues that the major function of Lias in the early mouse embryo is to promote the activity of the citric acid cycle.
The Liasnehe mutation is associated with a nucleotide substitution in a splice site that blocks most normal splicing of the transcript. However, RT-PCR of mutant mRNA reveals that there is some wild-type length Lias transcript present in the mutants, which accounts for the low residual level of Lias protein present in nehe mutant embryos. Liasnehe homozygotes and Liasnehe/Liasnull embryos show identical anterior truncation phenotypes, however Liasnehe/Liasnull embryos show a longer developmental delay than the Liasnehe homozygotes, so that they are ~24h delayed relative to their littermates. We therefore infer that nehe is a hypomorphic allele and that either one or two copies of this mutant allele is sufficient to allow survival past e7.5 but insufficient to promote forebrain development.
nehe embryos lack the forebrain due to a failure of normal prechordal plate specification
The specific absence of the dorsal forebrain in nehe mutants resembles that of mouse mutants that lack the activity of the AVE or the axial midline head organizers (Varlet et al., 1997; Shawlot et al., 1999; Bachiller et al., 2000). The phenotype most similar to that of nehe is seen in embryos homozygous for a partial loss of function allele of Ssdp1, Ssdp1hsk, a component of the Lhx1/Ldb1/Ssdp1 transcription factor complex (Nishioka et al., 2005). The Lhx1 complex has a variety of roles in early development, so that null alleles have complex phenotypes, whereas the Ssdp1hsk allele defines a specific role for the complex in forebrain specification. Both overall morphology and molecular marker analysis show that, as in Ssdp1hsk mutants, the nehe forebrain is absent from e8.5 onward, whereas other tissues are patterned normally. In Ssdp1hsk mutants, the loss of forebrain has been attributed to absence of the prechordal plate, whereas earlier signaling centers required for forebrain specification, the AVE and ADE, appear to be normal. Similarly, we observe that molecular markers of the prechordal plate (Foxa2, Gsc and Dkk1) are expressed at greatly reduced levels in nehe mutants. In addition, cells in the anterior axial midline of the mutants do not adopt the cellular organization characteristic of the prechordal plate.
The prechordal plate has a distinct origin and cellular organization from more posterior regions of the axial midline. The trunk notochord is derived from the node, whereas the anterior most cells of the axial midline in the prechordal plate and anterior head process derive from cells of anterior primitive streak (the early and mid-gastrula organizers) that migrate with the mesodermal wings and condense on the anterior midline at the stage when the node is forming (Kinder et al., 2001; Yamanaka et al., 2007). Prechordal plate cells express some genes that are characteristic of the entire axial midline, such as Foxa2, and also express prechordal-plate specific genes such as Gsc and Dkk1; expression of these markers fails in nehe embryos. Cells of the prechordal plate form a characteristic rosette-like structure, in contrast to the narrow stripe of cells characteristic of the more posterior parts of the midline. Some cells at the anterior end of the axial midline of nehe mutants have small apical surfaces, but they are interspersed with cells that resemble the endoderm that flanks the normal midline. Because of the specific disruption of gene expression and morphology of the prechordal plate in nehe mutants, we suggest that the cells of the prechordal plate depend on a higher level of mitochondrial activity than other cell types in the early embryo.
Defects in proliferation are not responsible for the anterior truncation and prechordal plate phenotypes of nehe embryos
Analysis of the nehe phenotype revealed that the requirement for the mitochondrial citric acid cycle changes during early development. Prior to e7.5 and after the early somite stage, Liasnehe embryos develop at the same rate and show the same mitotic index as their wild-type littermates. Only between the headfold and early somite stages did the reduction in activity of the citric acid cycle in Liasnehe homozygotes or Liasnehe/Liasnull embryos lead to a three-fold decrease in the mitotic index, a three-fold decrease in cyclin E and a three-fold increase in activated AMPK. This suggests that the balance between the glycolysis and mitochondrial metabolism as energy sources is under dynamic regulation during these stages of early embryogenesis.
Prior to the critical headfold stage, embryonic patterning is indistinguishable in wild-type and nehe embryos. During the headfold stage, nehe embryos show two obvious phenotypes: they grow more slowly than their littermates and they fail to specify the prechordal plate. Despite this temporal coincidence, we do not see regional differences in the reduced number of mitotic cells that would account for a defect in the prechordal plate. It is important to note that although most of the cells in the wild-type headfold embryo divide rapidly, the cells of the axial midline and prechordal plate are quiescent at this stage (Bellomo et al., 1996). Therefore, despite the temporal association of the two phenotypes, it is unlikely that the proliferation defect is responsible for the failure of prechordal plate specification. We therefore infer that specification of the prechordal plate in the late streak-headfold embryo depends on a different energy-dependent process.
AMPK is a central regulator of the response to energy: it is activated by phosphorylation at high AMP/ATP ratios and the active enzyme implements a set of homeostatic responses (Carling, 2004). The level of activated AMPK is three-fold higher in headfold stage nehe mutants than in stage-matched wild-type embryos, indicating that the mutants are responding to lowered levels of ATP. Studies in Drosophila have shown that AMPK can regulate apical-basal cell polarity in epithelia (Lee et al., 2007), as well as the cell cycle. Because determinants of apical-basal polarity can be crucial for coordinated cellular rearrangements (Zallen and Blankenship, 2008), activation of AMPK might affect the cell movements required for organization of the prechordal plate.
Given the striking similarity between the Ssdp1hsk and the nehe phenotypes, it is interesting to consider how the activity of the Lhx1/Ldp1/Ssdp1 complex and high ATP levels (or normal AMPK activity) might act together to promote specification of the prechordal plate. Although it is possible that the Lhx1 complex might regulate the expression of genes that control mitochondrial metabolism or that decreased ATP levels might decrease Lhx1 expression, it seems more likely that the Lhx1 complex and high ATP levels work in concert to control development of the prechordal plate. Although the precise mechanism of action of the Lhx1 complex in the promotion of forebrain development has not been defined, embryological experiments in both Xenopus and the mouse have suggested that Lhx1 is required for the morphogenetic movements of the axial mesoderm and/or endoderm that bring the inducing tissues into the correct apposition to promote anterior neural development (Hukriede et al., 2003; Tam et al., 2004). Given the requirement for AMPK activity in cell polarity, the lowered level of active AMPK in nehe embryos, and the similarity between the nehe and Lhx1 complex phenotypes, we suggest that one explanation for the nehe phenotype is that high ATP levels are required for the specialized morphogenetic movements that generate the prechordal plate.
The specificity of the nehe phenotype is remarkable. Prior to the critical headfold stage, embryonic patterning and growth are indistinguishable in wild-type and nehe embryos: migration of the AVE, the initiation of gastrulation, germ layer formation and germ layer migration are all normal in early nehe mutants. After the four somite stage, nehe embryos resume the wild-type rate of development, so that they remain ~12h delayed relative to littermates. Patterning and morphogenesis of tissues other than the prechordal plate are also normal in later nehe embryos, such that development of the somites, tail bud and neural tissues other than the forebrain all proceed on schedule.
There are two categories of explanation for the specific failure to specify the forebrain in nehe embryos. One possibility is that the signaling events and/or morphogenetic movements that generate the prechordal plate demand higher levels of ATP than the other morphogenetic movements that proceed normally in the mutant embryos. Alternatively, the decrease in energy levels in nehe embryos at the headfold stage may disrupt many signaling and/or morphogenetic processes that occur at that stage but the inductive interaction between the prechordal plate and the neighboring neural progenitors is constrained so that the embryo cannot compensate for the lack of that interaction when energy levels increase after the 4 somite stage.
nehe as a model for human microcephaly
In contrast to the focus of the mouse studies on signaling molecules and transcription factors important for forebrain specification, studies of congenital human syndromes have identified a variety of different genes required for normal forebrain size (Chae and Walsh, 2007). In addition to genes that promote neural proliferation, human genetic studies have shown that efficient activity of the mitochondrial citric acid cycle is important for normal brain size. The pyruvate dehydrogenase complex (PDH) converts pyruvate to acetyl-coenzyme A, the first substrate in the citric acid cycle, and pyruvate dehydrogenase deficiency causes complex symptoms including microcephaly (Matthews et al., 1994). Amish microcephaly is caused by mutations in SLC25A19 that deplete mitochondrial thiamine pyrophosphate, a cofactor of both pyruvate dehydrogenase and α-ketoglutarate dehydrogenase in the citric acid cycle (Lindhurst et al., 2006; Rosenberg et al., 2002). Amish microcephaly is associated with high levels of urinary α-ketoglutarate, a hallmark of defects in the α-ketoglutarate dehydrogenase complex, one of the complexes that depends on lipoic acid for its activity.
A targeted null mutation in the mouse Slc25a19 gene causes an open neural tube, and lethality at approximately e11 (Lindhurst et al., 2006). Although no analysis of forebrain development was carried out, the general morphology of these embryos is similar to that of the Liasnehe embryos. Similarly, mouse embryos that lack all activity of the E1 enzyme of the pyruvate dehydrogenase complex (Pdha1) die before e10.5 and have a morphology that could be consistent with a defect in forebrain development (Johnson et al., 2001). Although the microcephaly associated with defects in Slc25a19 or pyruvate dehydrogenase has been attributed to the requirement of the brain for high levels of oxidative metabolism (Brown et al., 1994; Lindhurst et al., 2006; Palmieri, 2008), our results demonstrate a crucial requirement for activity of the mitochondrial citric acid cycle early in development, during the initial specification of the forebrain. We therefore suggest that a requirement for high levels of ATP for early embryonic patterning may contribute to the defects seen in Amish microcephaly, pyruvate dehydrogenase deficiency and perhaps other human microcephalies. In addition, it seems likely that environmental factors such as maternal diabetes or maternal consumption of tobacco or alcohol could decrease energy production in the early embryo and thereby have a differential deleterious effect on development of the forebrain.
Supplementary Material
At early (A–C) and late (D–F) headfold stage, the prechordal plate marker Gsc is barely detectable in the anterior midline of nehe embryos, compared to stage-matched wild types. Scale bar in A–F: 0.25mm (G–I) Foxa2 is expressed normally in both the axial midline and the prechordal plate (G), and is expressed in the axial midline of nehe embryos but barely detectable in the anterior midline of the mutants. (J–L) Anterior mesendoderm marker Dkk1 is barely detectable at the anterior mesendoderm of the mutant embryos. Arrows point to position of prechordal plate. Scale bar in G–L: 0.25mm.
3D reconstructions of confocal images of whole mount embryos stained with DAPI (blue) and phalloidin (green); ventral views of a wild-type (A) and three stage-matched mutant embryos (B, C, D). The prechordal plate (brackets) lies at the anterior end of the axial midline. The regions within the brackets are shown at higher magnification in A′-D′. Cells of the wild-type prechordal plate, which can be recognized by their small apical surfaces, are arranged in a coherent cluster that is wider than the more posterior axial midline. The size and arrangement of these cells is irregular in the nehe embryos. Actin bundles are present in the anterior midline of mutant, but not wild-type, embryos (red arrows in B′, C′ and D′) Scale bars for A–D: 100 μm.
Acknowledgments
We thank Xianwen Yi and Nobuyo Maeda for the gift of the Lias null mice, and the MSKCC Molecular Cytology core facility for assistance with imaging. We thank Sarah Goetz and Isabelle Migeotte for comments on the manuscript. This work was supported by NIH grants NS044385 and HD035455 to KVA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acampora D, Mazan S, Lallemand Y, Avantaggiato V, Maury M, Simeone A, Brûlet P. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development. 1995;121:3279–3290. doi: 10.1242/dev.121.10.3279. [DOI] [PubMed] [Google Scholar]
- Ang SL, Conlon RA, Jin O, Rossant J. Positive and negative signals from mesoderm regulate the expression of mouse Otx2 in ectoderm explants. Development. 1994;120:2979–2989. doi: 10.1242/dev.120.10.2979. [DOI] [PubMed] [Google Scholar]
- Bachiller D, Klingensmith J, Kemp C, Belo JA, Anderson RM, May SR, McMahon JA, McMahon AP, Harland RM, Rossant J, DeRobertis EM. The organizer factors Chordin and Noggin are required for mouse forebrain development. Nature. 2000;403:658–661. doi: 10.1038/35001072. [DOI] [PubMed] [Google Scholar]
- Belo JA, Bouwmeester T, Leyns L, Kertesz N, Gallo M, Follettie M, De Robertis EM. Cerberus-like is a secreted factor with neutralizing activity expressed in the anterior primitive endoderm of the mouse gastrula. Mech, Dev. 1997;68:45–57. doi: 10.1016/s0925-4773(97)00125-1. [DOI] [PubMed] [Google Scholar]
- Bellomo D, Lander A, Harragan I, Brown NA. Cell proliferation in mammalian gastrulation: The ventral node and notochord are relatively quiescent. Dev Dyn. 1996;205:471–485. doi: 10.1002/(SICI)1097-0177(199604)205:4<471::AID-AJA10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Blum M, Gaunt SJ, Cho KWY, Steinbeisser H, Blumberg B, Bittner D, De Robertis EM. Gastrulation in the mouse: The role of the homeobox gene goosecoid. Cell. 1992;69:1097–1106. doi: 10.1016/0092-8674(92)90632-m. [DOI] [PubMed] [Google Scholar]
- Brown GK, Otero LJ, LeGris M, Brown RM. Pyruvate dehydrogenase deficiency. J Med Genet. 1994;31:875–879. doi: 10.1136/jmg.31.11.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus A, Davidson BP, Billiards S, Khoo P, Rivera-Perez JA, Wakamiya M, Behringer RR, Tam PPL. The morphogenetic role of midline mesendoderm and ectoderm in the development of the forebrain and the midbrain of the mouse embryo. Development. 2000;127:1799–1813. doi: 10.1242/dev.127.9.1799. [DOI] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade--a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Chae TH, Walsh CA. Genes that control the size of the cerebral cortex. Novartis Found Symp. 2007;288:79–90. doi: 10.1002/9780470994030.ch6. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–93. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC. Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524. doi: 10.1016/j.ntt.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118:1255–66. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Anderson KV. Dorsal and lateral fates in the mouse neural tube require the cell-autonomous activity of the open brain gene. Dev Biol. 2000;227:648–60. doi: 10.1006/dbio.2000.9918. [DOI] [PubMed] [Google Scholar]
- Frances F, Meyer G, Fallet-Bianco C, Moreno S, Kappeler C, Socorros AC, Tuy FP, Beldjord C, Chelly J. Human disorders of cortical development: from past to present. Eur J Neurosci. 2006;23:877–893. doi: 10.1111/j.1460-9568.2006.04649.x. [DOI] [PubMed] [Google Scholar]
- García-García M, Eggenschwiler J, Caspary T, Alcorn H, Wyler MR, Huangfu D, Rakeman AS, Lee JD, Feinberg EH, Timmer JR, Anderson KV. Analysis of mouse embryonic patterning and morphogenesis by forward genetics. Proc Natl Acad Sci U S A. 102:5913–5919. doi: 10.1073/pnas.0501071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallonet M, Kaestner KH, Martin-Parras L, Sasaki H, Betz UAK, Ang S-L. Maintenance of the Specification of the Anterior Definitive Endoderm and Forebrain Depends on the Axial Mesendoderm: A Study Using HNF3[beta]/Foxa2 Conditional Mutants. Dev Biol. 2002;243:20–33. doi: 10.1006/dbio.2001.0536. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–30. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukriede NA, Tsang TE, Habas R, Khoo PL, Steiner K, Weeks DL, Tam PPL, David IB. Conserved requirement of Lim1 function for cell movements during gastrulation. Dev Cell. 2003;4:83–94. doi: 10.1016/s1534-5807(02)00398-2. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Yang HS, Magnuson T, Patel MS. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci U S A. 1997;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MT, Mahmood S, Hyatt SL, Yang H-S, Soloway PD, Hanson RW, Patel MS. Inactivation of the murine pyruvate dehydrogenase (Pdha1) gene and its effect on early embryonic development. Mol Genet Metab. 2001;74:293–302. doi: 10.1006/mgme.2001.3249. [DOI] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kasarskis A, Manova K, Anderson KV. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci USA. 1998;95:7485–90. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinder SJ, Tsang TE, Wakamiya M, Sasaki H, Behringer RR, Nagy A, Tam PPL. The organizer of the mouse gastrula is composed of a dynamic population of progenitor cells for the axial mesoderm. Development. 2001;128:3623–3634. doi: 10.1242/dev.128.18.3623. [DOI] [PubMed] [Google Scholar]
- Krauss MJ, Morrissey AE, Winn HN, Amon E, Leet TL. Microcephaly: An epidemiologic analysis. Am J Obstet Gynecol. 2003;188:1484–1490. doi: 10.1067/mob.2003.452. [DOI] [PubMed] [Google Scholar]
- Layer G, Heinz DW, Jahn D, Schubert W-D. Structure and function of radical SAM enzymes. Curr Opin Chem Biol. 2004;8:468–476. doi: 10.1016/j.cbpa.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Lee JD, Anderson KV. Morphogenesis of the node and notochord: The cellular basis for the establishment and maintenance of left-right asymmetry in the mouse. Dev Dyn. 2008;237:3464–3476. doi: 10.1002/dvdy.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, Chung J. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–20. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- Lindhurst MJ, Fiermonte G, Song S, Struys E, De Leonardis F, Schwartzberg PL, Chen A, Castegna A, Verhoeven N, Mathews CK, Palmieri F, Biesecker LG. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci USA. 2006;103:15927–15932. doi: 10.1073/pnas.0607661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe LA, Yamada S, Kuehn MR. Genetic dissection of nodal function in patterning the mouse embryo. Development. 2001;128:1831–43. doi: 10.1242/dev.128.10.1831. [DOI] [PubMed] [Google Scholar]
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843–54. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Marquet A, Bui BT, Florentin D. Biosynthesis of biotin and lipoic acid. Vitam, Horm. 2001;61:51–101. doi: 10.1016/s0083-6729(01)61002-1. [DOI] [PubMed] [Google Scholar]
- Matthews PM, Brown RM, Otero LJ, Marchington DR, LeGris M, Howes R, Meadows LS, Shevell M, Scriver CR, Brown GK. Pyruvate dehydrogenase deficiency. Clinical presentation and molecular genetic characterization of five new patients. Brain. 1994;117:435–443. doi: 10.1093/brain/117.3.435. [DOI] [PubMed] [Google Scholar]
- Meno C, Shimono A, Saijoh Y, Yashiro K, Mochida K, Ohishi S, Noji S, Kondoh H, Hamada H. lefty-1 Is Required for Left-Right Determination as a Regulator of lefty-2 and nodal. Cell. 1998;94:287–297. doi: 10.1016/s0092-8674(00)81472-5. [DOI] [PubMed] [Google Scholar]
- Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Jr, Marletta MA. Escherichia coli LipA is a lipoyl synthase: in vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochem. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- Morikawa T, Yasuno R, Wada H. Do mammalian cells synthesize lipoic acid?: Identification of a mouse cDNA encoding a lipoic acid synthase located in mitochondria. FEBS Lett. 2001;498:16–21. doi: 10.1016/s0014-5793(01)02469-3. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, Chen L, Tsukui T, Gomer L, Dorward DW, Glinka A, Grinberg A, Huang S-P, Niehrs C, Ispizúa-Belmonte JC, Westphal H. Dickkopf1 Is Required for Embryonic Head Induction and Limb Morphogenesis in the Mouse. Dev Cell. 2001;1:423–434. doi: 10.1016/s1534-5807(01)00041-7. [DOI] [PubMed] [Google Scholar]
- Niehrs C. Regionally specific induction by the Spemann-Mangold organizer. Nat Rev Genet. 2004;5:425–34. doi: 10.1038/nrg1347. [DOI] [PubMed] [Google Scholar]
- Nishioka N, Nagano S, Nakayama R, Kiyonari H, Ijiri T, Taniguchi K, Shawlot W, Hayashizaki Y, Westphal H, Behringer RR, et al. Ssdp1 regulates head morphogenesis of mouse embryos by activating the Lim1-Ldb1 complex. Development. 2005;132:2535–2546. doi: 10.1242/dev.01844. [DOI] [PubMed] [Google Scholar]
- Palmieri F. Diseases caused by defects of mitochondrial carriers: a review. Biochim et Biophys Acta. 2008;1777:564–578. doi: 10.1016/j.bbabio.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Perham RN. Swinging arms and swinging domains in multifunctional enzymes: Catalytic Machines for Multistep Reactions. Ann Rev Biochem. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Hackert ML. Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem. 1990;265:8971–8974. [PubMed] [Google Scholar]
- Rhinn M, Dierich A, Shawlot W, Behringer RR, Le Meur M, Ang SL. Sequential roles for Otx2 in visceral endoderm and neuroectoderm for forebrain and midbrain induction and specification. Development. 1998;125:845–856. doi: 10.1242/dev.125.5.845. [DOI] [PubMed] [Google Scholar]
- Robb L, Tam PP. Gastrula organiser and embryonic patterning in the mouse. Semin Cell Dev Biol. 2004;15:543–554. doi: 10.1016/j.semcdb.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Rosenberg MJ, Agarwala R, Bouffard G, Davis J, Fiermonte G, Hilliard MS, Koch T, Kalikin LM, Makalowska I, Morton DH, et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat, Genet. 2002;32:175–179. doi: 10.1038/ng948. [DOI] [PubMed] [Google Scholar]
- Shawlot W, Behringer RR. Requirement for Lim1 in head-organizer function. Nature. 1995;374:425–430. doi: 10.1038/374425a0. [DOI] [PubMed] [Google Scholar]
- Shawlot W, Wakamiya M, Kwan KM, Kania A, Jessell TM, Behringer RR. Lim1 is required in both primitive streak-derived tissues and visceral endoderm for head formation in the mouse. Development. 1999;126:4925–4932. doi: 10.1242/dev.126.22.4925. [DOI] [PubMed] [Google Scholar]
- Shawlot W, Deng JM, Behringer RR. Expression of the mouse cerberus-related gene, Cerr1, suggests a role in anterior neural induction and somitogenesis. Proc Natl Acad Sci USA. 1998;95:6198–6203. doi: 10.1073/pnas.95.11.6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulik K, Dehart DB, Iangaki T, Carson JL, Vrablic T, Gesteland K, Schoenwolf GC. Morphogenesis of the murine node and notochordal plate. Dev Dyn. 1994;201:260–278. doi: 10.1002/aja.1002010309. [DOI] [PubMed] [Google Scholar]
- Tam PP, Steiner KA. Anterior patterning by synergistic activity of the early gastrula organizer and the anterior germ layer tissues of the mouse embryo. Development. 1999;126:5171–5179. doi: 10.1242/dev.126.22.5171. [DOI] [PubMed] [Google Scholar]
- Tam PPL, Khoo P-L, Wong N, Tsang TE, Behringer RR. Regionalization of cell fates and cell movement in the endoderm of the mouse gastrula and the impact of loss of Lhx1(Lim1) function. Dev Biol. 2004;274:171–187. doi: 10.1016/j.ydbio.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Thomas P, Beddington R. Anterior primitive endoderm may be responsible for patterning the anterior neural plate in the mouse embryo. Curr Biol. 1996;6:1487–1496. doi: 10.1016/s0960-9822(96)00753-1. [DOI] [PubMed] [Google Scholar]
- Thomas PQ, Brown A, Beddington RS. Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development. 1998;125:85–94. doi: 10.1242/dev.125.1.85. [DOI] [PubMed] [Google Scholar]
- Varlet I, Collignon J, Robertson EJ. nodal expression in the primitive endoderm is required for specification of the anterior axis during mouse gastrulation. Development. 1997;124:1033–1044. doi: 10.1242/dev.124.5.1033. [DOI] [PubMed] [Google Scholar]
- Yamanaka Y, Tamplin OJ, Beckers A, Gossler A, Rossant J. Live Imaging and Genetic Analysis of Mouse Notochord Formation Reveals Regional Morphogenetic Mechanisms. Dev Cell. 2007;13:884–896. doi: 10.1016/j.devcel.2007.10.016. [DOI] [PubMed] [Google Scholar]
- Yasuno R, Wada H. Biosynthesis of lipoic acid in Arabidopsis: cloning and characterization of the cDNA for lipoic acid synthase. Plant Physiol. 1998;118:935–43. doi: 10.1104/pp.118.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuno R, Wada H. The biosynthetic pathway for lipoic acid is present in plastids and mitochondria in Arabidopsis thaliana. FEBS Lett. 2002;517:110–114. doi: 10.1016/s0014-5793(02)02589-9. [DOI] [PubMed] [Google Scholar]
- Yi X, Maeda N. Endogenous production of lipoic acid is essential for mouse development. Mol Cell Biol. 2005;25:8387–8392. doi: 10.1128/MCB.25.18.8387-8392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakin L, Reversade B, Virlon B, Rusniak C, Glaser P, Elalouf JM, Brûlet P. Gene expression profiles in normal and Otx2−/− early gastrulating mouse embryos. Proc Natl Acad Sci USA. 2000;97:14388–14393. doi: 10.1073/pnas.011513398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen JA, Blankenship JT. Multicellular dynamics during epithelial elongation. Semin Cell Dev Biol. 2008;19:263–70. doi: 10.1016/j.semcdb.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamorano L, Chuaqui B. Teratogenetic periods for the principal malformations of the central nervous system. Virchows Arch A Pathol Anat Histol. 1979;384:1–18. doi: 10.1007/BF00427147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
At early (A–C) and late (D–F) headfold stage, the prechordal plate marker Gsc is barely detectable in the anterior midline of nehe embryos, compared to stage-matched wild types. Scale bar in A–F: 0.25mm (G–I) Foxa2 is expressed normally in both the axial midline and the prechordal plate (G), and is expressed in the axial midline of nehe embryos but barely detectable in the anterior midline of the mutants. (J–L) Anterior mesendoderm marker Dkk1 is barely detectable at the anterior mesendoderm of the mutant embryos. Arrows point to position of prechordal plate. Scale bar in G–L: 0.25mm.
3D reconstructions of confocal images of whole mount embryos stained with DAPI (blue) and phalloidin (green); ventral views of a wild-type (A) and three stage-matched mutant embryos (B, C, D). The prechordal plate (brackets) lies at the anterior end of the axial midline. The regions within the brackets are shown at higher magnification in A′-D′. Cells of the wild-type prechordal plate, which can be recognized by their small apical surfaces, are arranged in a coherent cluster that is wider than the more posterior axial midline. The size and arrangement of these cells is irregular in the nehe embryos. Actin bundles are present in the anterior midline of mutant, but not wild-type, embryos (red arrows in B′, C′ and D′) Scale bars for A–D: 100 μm.