

Abstract
Ena/VASP proteins mediate the effects of guidance cues on the actin cytoskeleton. The single C. elegans homolog of the Ena/VASP family of proteins, UNC-34, is required for the migrations of cells and growth cones. Here we show that unc-34 mutant alleles also interact genetically with Wnt mutants to reveal a role for unc-34 in the establishment of neuronal polarity along the C. elegans anterior-posterior axis. Our mutant analysis shows that eliminating UNC-34 function results in neuronal migration and polarity phenotypes that are enhanced at higher temperatures, revealing a heat-sensitive process that is normally masked by the presence of UNC-34. Finally, we show that the UNC-34 protein is expressed broadly and accumulates in axons and at the apical junctions of epithelial cells. While most mutants lack detectable UNC-34, three unc-34 mutants that contained missense mutations in the EVH1 domain produced full-length UNC-34 that failed to localize to apical junctions and axons, supporting the role for the EVH1 domain in localizing Ena/VASP family members.
Keywords: C. elegans, UNC-34, neuronal polarity, cell migration, axon guidance
Introduction
The generation of neuronal polarity and the ability of neuronal growth cones to navigate to their synaptic targets contribute to the connectivity of the metazoan nervous system. When cultured, hippocampal and cortical neurons polarize with a single axon and multiple dendrites, indicating that these cells have an intrinsic polarity when they are cultured (Barnes and Polleux, 2009). In the developing nervous system, however, neuronal polarity is oriented by cues. The C. elegans HSN neuron, for example, extends a single axon ventrally and this extension is initiated by the guidance cue UNC-6/netrin (Adler et al., 2006). Cells and growth cones also migrate in culture in the absence of cues, but to reach their destinations, migrating cells and growth cones respond to both attractive and repulsive cues. In the past two decades, investigators have identified many conserved guidance cues and their receptors, and more recently, these molecules have also been shown to regulate neuronal polarity. How these receptors transduce extracellular cues to orient polarity and to guide migrating cells and growth cones, however, is not as well understood.
One important class of molecules thought to convey signals from guidance receptors to the actin cytoskeleton are members of the Ena/VASP family of proteins. Enabled (Ena) was first defined in Drosophila by mutations that dominantly suppress the lethality caused by mutations in the homolog of the Abelson oncogene (Gertler et al., 1990). Later, Ena mutants were found to have defects in axon morphology (Gertler et al., 1995). Vasodilator-Stimulated Phosphoprotein (VASP) was isolated as a protein in platelets that was phosphorylated in response to high levels of cAMP or cGMP (Haffner et al., 1995; Halbrugge and Walter, 1989; Waldmann et al., 1987). Mena, VASP and Ena-VASP like protein (Evl) are the three mammalian Ena/VASP family proteins (Gertler et al., 1996). These paralogs provide overlapping functions in the formation of filopodia and endothelial junctions that are only revealed in mouse mutants lacking function of all three genes (Furman et al., 2007; Kwiatkowski et al., 2007). C. elegans contains a single Enabled homolog known as UNC-34. Ena/VASP members all share three domains: an N-terminal Ena/VASP homology 1 (EVH1) domain that can interact with several proteins, a central proline rich region (PRR) that can interact with Profilin and proteins containing SH3 domains, and a C-terminal EVH2 domain that mediates the formation of tetramers and interactions with actin (Krause et al., 2003).
In order to study the role of the Ena/VASP family in neuronal development, we performed a genetic analysis of unc-34, the single C. elegans Ena/VASP homolog. All unc-34 mutants were heat sensitive for unc-34-dependent phenotypes, revealing a temperature-sensitive process that is normally masked by UNC-34. We discovered a role for UNC-34 in ALM neuronal polarity, a process that also requires Wnts, suggesting that Ena/VASP family members could function in Wnt signaling. All unc-34 mutants that express full-length UNC-34 protein contain amino acid substitutions in the EVH1 domain. UNC-34 is expressed broadly throughout the nervous system as well as in epithelial cells. In particular, we find UNC-34 localized to axons and apical junctions of epithelial cells. Our phenotypic analysis shows that the EVH1 domain is essential for UNC-34 localization.
Materials and Methods
Strains and genetics
Strains were grown at 20°C unless stated otherwise, and were maintained as described by Brenner (1974). In addition to the wild-type strain N2, strains with the following mutations or transgenes were used in this work:
LGI: lin-44(n1792 and n2111) (Herman and Horvitz, 1994), zdIs5 [Pmec-4∷gfp] (Prasad and Clark 2006)
LG II: cwn-1(ok546) (Zinovyeva and Forrester, 2005), juIs76 [Punc-25∷gfp] (Huang et al. 2002);
LG III: sup-5(e1464) (Waterston and Brenner 1978; Wills et al. 1983), mig-10(ct41) (Manser and Wood, 1990);
LG IV: egl-20(n585) (Trent et al., 1983), cwn-2(ok895) (Zinovyeva and Forrester, 2005), ced-10(n1993) (Ellis et al., 1991), ced-10(n3246) (Reddien and Horvitz, 2000), ced-10(tm597) (National Bioresource Project of Japan);
LG V: unc-34 alleles included e315, e951, e566 (Brenner 1974), gm104, gm114, gm115, gm134 (Forrester and Garriga 1997), ev561, ev562, ev553, ev564 (Colavita and Culotti 1998), zd117, zd132, zd133 (S. Clark), rh403 (E. M. Hedgecock), s138 (D. L. Baillie), n1877, n1889, and n1890 (L. Bloom and B. Horvitz), mom-2(or309) (Thorpe et al., 1997), mom-2(ne874ts) (Zinovyeva et al., 2008), kyIs5 [Pceh-23∷unc-76∷gfp] (Forrester and Garriga 1997);
Rearrangements and extrachromosomal arrays: nT1[qIs50] and dnT2[qIs50] (Belfiore et al., 2002), kyEx710 [Punc-86∷GFP∷unc-34, Podr-1∷dsRed] and kyEx926 [Punc-86∷mig-10∷YFP, Podr-1∷dsRed] (Adler et al. 2006).
unc-34 cDNA cloning and sequencing
A full-length unc-34 cDNA was generated. RNA was isolated from embryos and poly(A) RNA was selected using a Qiagen poly(A) selection kit using the manufacturer’s protocol. The RNA was reversed transcribed by incubating 5 μl of Poly(A) selected RNA at 37° for 60 minutes in 50 μl of a solution containing M-MLV buffer (50 mM Tris-HCl pH 8.3, 10 mM MgCl2, 7.5 mM KCl), 0.5 mM of each dNTP, 0.02 mM oligo dT, 25 units RNAsin and 200 units M-MLV enzyme (Promega, Madison, WI, USA). To amplify the unc-34 cDNA, we incubated 1 μl of the reverse transcription reaction in 50 μl PCR reaction. The oligonucleotides EN23 and EN24 were used to amplify by PCR an unc-34 cDNA. EN23 contained sequences (5’-CAGCCGCGGATGTTGTGTAATACGCATTCT-3’) from the cosmid Y50D4C with a SacII site at its 5’ end. EN24 contained sequences (5’-GCCGGTACCCTATCGACGGCCACCAATCGC-3’ and a KpnI site at its 5’ end. The PCR product was digested with SacII and KpnI and cloned into Bluescript KS- digested with SacII and KpnI to produce pMD1.
5’ RACE revealed that pMD1 contained the incorrect ATG. We corrected this error by amplifying by PCR from pMD1 using the oliginucleotide primers EN24 and EN37(5’ATGTTGTGTAATACGCATTCTCAGAACTTAGTTTCATTTTTTCCCACCA AAAAATTCAAATTTTCCATGAGTAGCGAAGCATGTAT 3’). The SacII site at the 5’ end of EN37 was used to clone the revised unc-34 PRC product into Bluescript KS- to produce pMD2. The sequence analysis of pMD2 revealed a silent point mutation at nucleotide 1026 of cDNA (GGT to GGC; Gly to Gly). Otherwise there are no other variations from the wild-type sequence.
Sequence analysis of unc-34
To determine the molecular lesions of unc-34 mutants, we generated from genomic DNA PCR products containing one or two unc-34 exons that were then sequenced. All primers are written 5’ to 3’. PCR using the primers TF34f (GCAAAAGGAGATTAGATTGC) and TF31r (GTCGTGTGAAAGCTATATTTG) generated a PCR product containing the first two exons of unc-34. Exon 3 was obtained by PCR with the primers TF35f (TTCACGAGGAATCTCAATTCG) and TF37r (GAATTTGCATGCTCATTTCAAAC). The PCR product generated with the primers, TF38f (TCTACTTAAACAATCGTTTCTG) and EN33r (CTTTCTATGCGTCTTCGGACTATC) included exon 4 and a portion of exon 5. The other portion of exon 5 along with exons 6 and 7 was obtained by PCR using the primers, EN32f (CAGCCGGCAATCTAATGTCCGAAT) and TF32r (GAAAATGGTTCTATGCAGCACCG).
RNA interference
A PCR product generated from the primers EN7 (5’-GCCGGTACCAGTAGCGAAGCATGT-3’) and EN15 (5’-CAGCCGCGGCGATGCATCTGTTGA-3’) and from an unc-34 cDNA as template was subcloned into Bluescript KS- using KpnI and SacII sites at the 5’ end of the primers, respectively. RNA was transcribed by T3 and T7 primers using Promega T3/T7 kits. The RNA was annealed by mixing equal molar concentrations of sense and antisense RNA in 3X injection buffer (20mM KPO4 pH7.5, 3mM KCitrate pH7.5, 2% PEG6000). Annealing took place at 68°C for 10 minutes and then 37°C for 30 minutes. The generation of double-stranded RNA was confirmed by reduced mobility of the RNA band on an agarose gel. The resulting double stranded RNA was injected into kyIs5 at 20°C at a concentration of 10ng/ μl. The progeny of the injected animals were scored for CAN migration defects.
unc-34 constructs and germline transformation
In order to assess if UNC-34 functions cell autonomously, we used a 7 kb ceh-23 promoter to drive unc-34 cDNA. The ceh-23 promoter was obtained by PCR of genomic DNA using the primers TF1 (5’-CGCGCATGCGTTGCACAATTTCTACGCCC-3’) and TF2 (5’CTTTATTTTCTAGATGGACACCC-3’). The PCR product was cut with SphI and XmaI and then cloned into the SphI and XmaI sites of pPD95.77 (Fire lab 1995 vector kit) to produce pTF1. An unc-34 translational fusion with GFP was generated by PCR of pMD2 with the primers, EN38 (5’-CAGCCCGGGATGAGTAGCGAAGCATG-TAT-3’) and EN39 (5’-TATCCCGGGATCGACGGCCACCAATCGC-3’), which both contain XmaI sites at the 5’ end. The PCR product was cut with XmaI and cloned into the XmaI site of pTF1. The resulting Pceh-23∷unc-34∷gfp transgene was injected at 5ng/μl with rol-6(su1006) DNA into wild-type animals. The resulting array gmEx249 was crossed into unc-34(e951) mutants. The CAN positions were then scored in this strain.
Anti-UNC-34 antisera
A His-tagged UNC-34 fusion gene was constructed by inserting the C-terminal two-thirds of the unc-34 cDNA into the pRSET-B vector beginning with the BamHI site in exon 2 (Invitrogen) A fusion protein of the predicted size of 31kd was purified on Ni agarose from E. coli and injected subcutaneously into mice and rabbits (Harlow and Lane, 1988). Antibody experiments used either polyclonal ascites from mice or polyclonal antisera from rabbits purified against fusion protein immobilized on nitrocellulose strips.
Immunoblot analysis
Animals were washed three times with M9 to remove bacteria. After the final wash, the animals were suspended in three volumes of sample buffer (0.125 M Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 10% ß-mercaptoethanol and 0.004% bromophenol blue). The suspended animals were heated to 100°C for 5 to 10 minutes. The protein from this extract was separated on 10% SDS-PAGE polyacrylamide gel. Proteins were transferred to 0.45 μm nitrocellulose filters (Schleicher and Schuell, Keene, NH, USA) by electroblotting at 100 mA overnight at room temperature in 20 mM Tris, pH 8.8, 150 mM glycine, 20% methanol. Blots were rinsed twice in water, wet briefly in TPBS (0.05% Tween-20 in PBS), then blocked for 1 h at room temperature in 5% dried, non-fat milk and 0.1% bovine serum albumin (BSA; Sigma Chemical Co., St. Louis, MO, USA) in TPBS. Blots were rinsed briefly in TPBS and incubated in primary antibody (diluted to 1:500 in 0.1% BSA, 0.5% NGS in TPBS) for 1 hour at room temperature. Excess primary antibody was removed from the blots by washing three times for 10 min each in TPBS. Secondary antibody was diluted to 1:10,000 in 0.1% BSA, 0.5% NGS in TPBS. Incubation in secondary antibody was carried out at room temperature for 1 to 2 hours. Blots were then washed three times for 10 min each in TPBS. Antibody detection was done using the SuperSignal Pico West Chemoluminescent substrate from Pierce (Rockford, IL, USA) as per manufacturer’s instructions. Molecular weights were determined using prestained protein molecular weight standards (Sigma Chemical Co., St. Louis, USA).
Immunocytochemistry
Embryos were prepared by treating gravid hermaphrodites and freshly laid eggs with 5 ml of hypochlorite solution (0.7M NaOH, and 4.4% NaOCl) for 10-15 minutes while vortexing intermittently. Embryos were fixed and permeabilized as described (Guenther and Garriga, 1996). To reduce the background, rabbit anti-EVH2 antibodies were diluted 1:500 in PBST-A (1X PBS, 1% BSA, 0.5% Triton-X 100, 0.05% NaN3, 1 mM EDTA), and the dilution pre-absorbed overnight 4x at room temperature with fixed unc-34(e951) embryos. After washing, the embryos were rocked overnight at room temperature in Cy3 goat anti-rabbit diluted 1:500 in PBST-A (Molecular Probes).
Larvae and adults were obtained as described by Guenther and Garriga (1996). Animals were washed, fixed, permeabilized and stained by the method of Finney and Ruvkun (Finney and Ruvkun, 1990). Pictures were obtained from Hamamatsu ORCA-ER digital camera attached to a Zeiss Axioskop2 microscope using Openlab software.
Scoring of cell migration defects
To determine the extent of CAN, HSN and ALM migration in wild-type and mutant animals, cell positions were scored relative to the positions of hypodermal nuclei using Normarski optics. Only newly hatched first larval stage (L1) hermaphrodites were scored. When CANs could not be detected they were scored in the most anterior position since CANs that fail to migrate out of the head were not distinguishable from other neurons. All p values were determined using a two-sample Z-test formula to compare the percentage of cells in a specific position from two genotypes.
Scoring of process outgrowth defects
The processes of the DD neurons of L1 animals were scored using the Zeiss Axioskop2 microscope to visualize the GFP fluorescence of juIs76 [Punc-25∷gfp] transgenic animals. Observed process morphologies were divided into five classes that are described in the legend to Fig. 5.
Fig. 5.

Process guidance defects of unc-34 mutants. (A) Images were taken at early L1 larval stage of a wild-type animal and an unc-34(e951) null mutant expressing the integrated array juIs76 used to visualize the DD processes. In juIs76 the unc-25 promoter drives GFP expression in the six DD neurons located in the ventral nerve cord (VNC) as well as in their processes. The DD processes extend along the VNC, send commissures to the dorsal nerve cord (DNC) and then extend in the DNC. The arrows indicate DD processes that reach the DNC but then fail to branch properly and would be scored as class III defects. The arrowheads point to DD processes that exit the VNC but fail to reach the DNC (class IV defects). The asterisk corresponds to a DD process that fails to exit the VNC and represents the most severe class V defect. A-anterior and P-posterior. (B) Diagrams representing the wild-type (classes I and II) process morphology and the three classes of defective process morphologies. Class II processes extended to the DNC and branched in both directions, but failed to fully extend along the DNC in one or both directions. Processes were scored as class III defects if they extended to the DNC but then either failed to branch completely or only sent out one branch. Class IV defects included processes that exited the VNC, but failed to reach the DNC and may or may not have extended branches. Processes in class V failed to exit the VNC. (C) Graph depicting the DD process defects of wild-type and unc-34 mutants. In a wild-type background all of the DD processes displayed normal morphology. The unc-34 mutants, e951, gm114, and gm104 all showed similar phenotypes with 40-50% of the DD processes failing to reach the DNC (classes IV and V). A slightly milder phenotype was observed in unc-34(ev561), in which 30% of DD axons failed to reach the DNC. In contrast, unc-34(ev562) displayed a hypomorphic phenotype in which all but 9% of DD processes reached the DNC. (D) Graph showing wild type and unc-34(e951) DD process defects at different temperatures. A mild temperature-sensitive effect was seen in wild-type animals while unc-34(e951) defects strongly depend on temperature. At 25°C 59% of DD processes were in class IV or V but at 15°C only 24% of processes were scored as class IV or V. 70 animals (350 processes) were scored for each genotype and temperature.
Scoring of the ALM polarity phenotype
Neuronal polarity of ALM was scored in L4 animals using the integrated array zdIs5 [Pmec-4∷gfp], which is expressed in the six mechanosensory neurons, ALMs, PLMs, AVM and PVM. For ALM, the bipolar phenotype was defined as a normal anterior process and a posterior process that is longer than five ALM cell diameters in length.
unc-34(e315) genetics
Backcrossing of the original unc-34(e315) mutation revealed that the severity of the cell migration defects could be variable in different strains. To determine if the original unc-34(e315) strain contained an unlinked genetic modifier, we crossed the original strain to wild-type males and generated multiple strains in which the unc-34(e315) mutant bred true for the Unc phenotype. The original unc-34(e315) strain displayed only weak cell migration defects, but after backcrossing, we isolated Unc strains that displayed cell migration defects comparable to unc-34 null mutants (data not shown). Out of seven backcrossed unc-34(e315) strains, two exhibited weak CAN migration defects, indicating the presence of the suppressor. The remaining five strains displayed a range of CAN phenotypes with 17% to 36% of the CANs in wild-type positions (data not shown). We isolated strains with CAN migration defects indistinguishable from strains that carried null alleles of unc-34. One of these strains, named NG4611, was used in this study. These results are consistent with the canonical strain containing a recessive mutation that is unlinked to unc-34(e315) and able to suppress its cell migration defects. We have not yet mapped this mutation. In screens for unc-34 suppressors, mutations in two genes, crml-1 and abl-1, have been identified (Hurwitz et al., 2009; Vanderzalm et al., 2009). The abl-1 and crml-1 mutations suppress the cell migration and Unc phenotypes of unc-34 mutations, whereas the e315 mutation primarily suppresses the cell migration defects of the unc-34 mutant and thus could define a new suppressor locus.
Results
Most unc-34 mutants produced no detectable UNC-34 protein
C. elegans contains a single Ena/VASP homolog known as UNC-34 (Withee et al., 2004; Yu et al., 2002). Ena/VASP family proteins have three domains: EVH1, PRR and EVH2 (Fig. 1A). Although UNC-34 has all three domains, it has weak homology to the EVH2 domain (16% identity overall with Mena EVH2). The EVH2 domain appears to lack conserved sequences necessary to bind globular actin but contains conserved residues involved in filamentous actin binding and tetramerization (data not shown). We raised antibodies to the EVH2 domain of UNC-34. Anti-EVH2 antibodies detected a protein of approximately 60kD present in wild-type animals (Fig. 1A). The size of UNC-34 is larger than the 50kD predicted from the sequence of its cDNA.
Fig. 1.

unc-34 mutations and UNC-34 mutant proteins. (A) Diagram of the protein domains of UNC-34 and of the changes caused by the mutations. C. elegans UNC-34 has an N-terminal Ena/VASP homology 1 (EVH1) domain, a central proline rich region (PRR) and a C-terminal EVH2 domain. Also shown are the positions of the altered amino acids of the sequenced unc-34 alleles. The asterisk indicates the presence of the additional mutation E433K in the EVH2 domain of unc-34(ev561) and unc-34(ev562) mutants. (B) Immunoblots of extracts from wild-type and unc-34 mutant embryos using antibodies raised against the UNC-34 C-terminus. Prominent background bands ran at 90kD (seen in top immunoblot) and 40kD. These 90 kD protein that reacted with the anti-UNC-34 was present in all of the unc-34 mutant strains and served as a control for normalizing the amount of UNC-34 in the different strains. Based on this criterion, there was less UNC-34 protein in the gm144, ev561 and ev562 mutant strains. The subtle shift of the UNC-34 protein from the ev561 and ev562 mutants is an artifact of removing several intervening lanes between the wild-type and mutant lanes. The wild-type and mutant proteins are of the same size.
To determine if existing unc-34 alleles produce detectable UNC-34 protein, we performed immunoblot analysis of embryonic extracts from 19 unc-34 mutants using the antibodies raised against the EVH2 domain. The 16 unc-34 mutants e315, e951, ev553, ev564, ev566, gm104, gm115, gm134, n1877, n1889, n1890, rh403, s138, zd117, zd132, and zd133 produced no detectable UNC-34 (Fig. 1B and data not shown). The lack of detectable protein suggests that most mutations eliminate unc-34 function. This hypothesis is supported by the similarity of the phenotypes of the mutants (see below and data not shown).
We attempted to identify the e315, e951 and gm104 mutations by sequencing the mutant unc-34 genes. We were unable to amplify by PCR unc-34 sequences from e951 genomic DNA, suggesting that most if not all of the gene is deleted in these mutants. Consistent with this finding, probing a Southern blot of e951 mutant DNA with a cDNA probe did not detect unc-34 sequences (data not shown). The allele gm104 is an early amber nonsense mutation at codon 24 (Fig. 1A), and both the locomotion and cell migration defects of gm104 hermaphrodites were suppressed by a sup-5 amber suppressor, confirming that the gm104 amber mutation is responsible for these phenotypes (data not shown). These observations indicate that the e951 and gm104 mutations eliminate unc-34 function. The unc-34(e315) mutation results in an amber stop at codon 258 and is predicted to result in a fragment that contains the EVH1 and PRR domains, but lacks the EVH2 domain (Fig. 1A). As expected, anti-EVH2 antibodies failed to detect UNC-34 protein from unc-34(e315) mutants (Fig. 2B).
Fig. 2.

UNC-34 localizes to apical junctions and neuronal processes. We stained embryos with anti-UNC-34 antibodies raised to the C-terminal EVH2 domain. (A-B) Pre-comma stage, wild-type embryo stained with an anti-UNC-34 antiserum (A) and DAPI (B). UNC-34 localized to the apical junctions (arrows indicate some of the junctions). (C) Two-fold and (D-H) late-stage embryos stained with the anti-UNC-34 antiserum. Late-stage embryos were dislodged from their eggshells during the staining protocol. Arrows in C, D, G and H indicate the nerve ring labeled by the antibody. Arrowheads in D indicate the apical junctions labeled by the antibody. (C, D) UNC-34 localized to axon bundles of late-stage, wild-type embryos. (E) unc-34(e951) mutants lacked all nervous system and epithelial cell staining. (F) The cell bodies but not the neuronal processes of unc-34(gm114) mutants contained UNC-34. (G-H) The nerve rings of (G) unc-34(ev561) and (H) unc-34(ev562) mutants contained reduced levels of UNC-34. scale bar is 5 μm.
Three unc-34 mutants produced full-length protein
Of the 19 unc-34 mutants tested, only three alleles, gm114, ev561 and ev562, expressed full-length UNC-34 (Fig. 1B). Sequence analysis of these alleles revealed that all three have missense mutations in the EVH1 domain (Fig. 1A). The gm114 mutation changes a highly conserved alanine to threonine at amino acid 99. In unc-34(ev561) mutants, the threonine at codon 83, which lies adjacent to a highly conserved region, is converted to an isoleucine. The unc-34(ev562) allele encodes a valine at codon 56 instead of a conserved aspartate. Both unc-34(ev561) and unc-34(ev562) also contain the same additional mutation in the EVH2 domain that changes a glutamate to a lysine at amino acid 433 in the tetramerization motif. Both alleles were generated in the same lab, and the existence of the same mutation in both strains suggests that it was present in the original strain.
UNC-34 localizes to neuronal processes and at apical junctions
The anti-EVH2 antibodies detected cytoplasmic protein in both wild-type and unc-34(e951) early embryos (data not shown). Because unc-34(e951) mutants contain no UNC-34 protein (Fig. 1B), this nonspecific staining prevented us from using these antibodies to determine the distribution of UNC-34 protein during the early stages of embryonic development that occur before morphogenesis. We first detected specific UNC-34 staining with anti-EVH2 antibodies in embryos at the beginning of ventral enclosure, a stage of embryogenesis where polarized epithelial cells originating on the dorsal side of the embryo migrate ventrally to meet at the ventral midline and attach via apical junctions (Chisholm and Hardin, 2005). UNC-34 specifically localized to apical junctions of these migrating epidermal cells as previously described using an UNC-34∷GFP reporter (Fig. 2A) (Sheffield et al., 2007). Sheffield et al. showed that the junctional protein AJM-1 and UNC-34 physically interact and that AJM-1 recruits UNC-34 to apical junctions (Sheffield et al., 2007). Later in embryogenesis, UNC-34 was also found in a pattern that suggested it localizes to junctions of the epithelial cells that line the inside of the pharynx. UNC-34 localization to these structures decreased after the first larval stage, but was observed strongly at apical junctions of the developing vulva (data not shown).
Anti-EVH2 antibodies revealed broad expression of UNC-34 in the nervous system that was missing from the mutants unc-34(e951) and unc-34(gm104) (Fig. 2C, D, E and data not shown). UNC-34 localized to axons with fainter cytoplasmic staining in neuronal cell bodies. The nerve ring, the major C. elegans neuropil that contains the highest concentration of axons, displayed the most intense staining, while the dorsal nerve cord (DNC) and ventral nerve cord (VNC) also expressed detectable UNC-34 (Fig. 2C, D and not shown). Nerve ring staining was detected as early as the two-fold stage of embryogenesis and the intensity of axonal staining increased until late embryogenesis (Fig. C, D). UNC-34 axon expression continued into adulthood although it became fainter in older larvae and adults (data not shown).
Staining of unc-34 mutants that produced full-length UNC-34 protein revealed a requirement for the EVH1 domain in UNC-34 localization. First, UNC-34 failed to localize to the apical junctions in unc-34(gm114) mutants (data not shown). This observation is consistent with the finding that a mutant form of AJM-1 that cannot bind to UNC-34 fails to recruit UNC-34 to apical junctions (Sheffield et al., 2007). Because of high background staining, we do not know whether mutant UNC-34 protein accumulated in the cytoplasm of these epithelial cells. Second, axonal staining was noticeably absent from unc-34(gm114) mutants even though high levels of the protein accumulated in the cell bodies of neurons in the head and along the VNC (Fig. 2F). The lack of UNC-34 in axons and its accumulation in the cytoplasm of neuronal cell bodies in unc-34(gm114) mutants indicated that the mutant UNC-34 localized abnormally and was not simply undetectable in axons due to lower expression levels. These observations suggest an important role for the EVH1 domain in UNC-34 localization.
The other mutants with EVH1 mutations, unc-34(ev561) and unc-34(ev562), also exhibited a decreased level of staining in the nerve ring, VNC and DNC staining (Fig. 2G and H). We observed UNC-34 in the nerve ring of unc-34(ev561) mutants only occasionally and at high levels in neuronal cell bodies. We detected faint but consistent UNC-34 levels in the nerve ring of unc-34(ev562) mutants. Both unc-34(ev561) and unc-34(ev562) mutants failed to show any staining of apical junctions (data not shown). Because unc-34(gm114) mutants also displayed the axonal localization defect, we favor the hypothesis that the deficiency in nerve ring staining and stronger cell body staining of unc-34(ev561) and unc-34(ev562) mutants resulted from changes in the EVH1 domain rather than the altered EVH2 domain that they share. This hypothesis is also supported by the different strengths of the localization defects between the two mutants, but we cannot eliminate the possibility that the EVH2 change shared by the two mutants contributes to the localization defect.
The EVH1 domain is important for cell migration and axon guidance
unc-34 mutants displayed defects in the positions of the CAN, HSN and ALM neurons (Forrester and Garriga, 1997). These bilaterally symmetric neurons all migrate during embryogenesis. The CANs migrate from the head to the middle of the embryo, stopping at positions near the anterior end of the gonad primordium. The HSNs migrate forward from the tail to positions that flank the embryonic gonad. The ALMs migrate toward the posterior within the mid-anterior region over a shorter distance than the CANs and HSNs (Sulston et al., 1983).
The unc-34 null mutants e951 and gm104 displayed similar cell migration defects. Only about 25% of CANs migrated to wild-type positions in these mutants, with the remaining neurons stopping short at various positions along the migratory route (Fig. 3). The HSN migration defects of these mutants were not as severe: 63% of unc-34(e951) HSNs and 48% of unc-34(gm104) HSNs migrated to wild-type positions with most of the remaining neurons clustered in slightly posterior positions (Fig. 4). Approximately 35% of the ALMs failed to reach wild-type positions in these mutants (data not shown). The unc-34 mutations affect multiple migrations in both the posterior and anterior directions, but affect CAN migration more severely.
Fig. 3.

CAN migration defects of unc-34 mutants. At the top is a merged fluorescence and Nomarski image of a newly hatched first larval stage (L1), Pceh-23∷gfp transgenic animal that expresses GFP in the CANs as well as sensory neurons in the head and tail. Anterior is to the left and dorsal is up. Only the left CAN is visible. While the ceh-23∷gfp transgenic animal shown is used to illustrate the normal CAN position, this transgene sensitizes the background to CAN defects. To avoid this, we scored CAN positions by Nomarski optics in a background lacking this transgene. The schematic diagram represents the anterior half of the L1 larval stage body and depicts the hypodermal nuclei used to score the terminal positions of migrated CANs. The arrow illustrates the posterior migration route of the CAN. Each box in the lower part of the Fig. contains the percentage of CANs in that position relative to the hypodermal nuclei. “n” is the number of CANs scored for each genotype.
Fig. 4.

HSN migration defects of unc-34 mutations. At the top is a merged fluorescence and Nomarski image of a newly hatched L1 animal containing the transgene Punc-86∷gfp. These transgenic animals express GFP in the HSNs as well as many other neurons, and this image is only used here to illustrate the positions of the HSNs. As in Fig. 3, the transgene was not used to score HSNs because it sensitizes the background and enhances the HSN migration defects of the mutants. Anterior is to the left and dorsal is up. The schematic diagram shows the posterior half of a first stage larvae. The arrow represents the migration path of the HSN. The hypodermal nuclei shown in the diagram provide reference positions for scoring the positions of the HSNs. Each box in the lower part of the Fig. contains the percentage of HSNs in that position along the anterior-posterior axis. “n” is the number of CANs scored for each genotype.
The CAN, HSN and ALM migration defects of unc-34(gm114) and unc-34(e315) mutants were similar to those of the null mutants (Fig.s 3, 4 and data not shown), indicating that unc-34(gm114) and unc-34(e315) eliminated UNC-34 function in cell migration. Thus, the EVH1 domain and in particular the conserved residue altered in unc-34(gm114) are essential for UNC-34’s function in cell migration.
The defects in unc-34(e315) mutants could reflect an essential requirement for the EVH2 domain in UNC-34 function. However, we could not confirm the presence of a truncated UNC-34 in unc-34(e315) mutants because our antibodies recognize the C-terminal EVH2 domain (data not shown). Because unc-34(e315) mutants behave genetically like nulls, if a mutant protein is made in unc-34(e315) mutants, it would lack UNC-34 function. The hypomorphic unc-34(lq17) mutation is predicted to produce a protein that lacks the last 43 amino acids of UNC-34 and hence should fail to form tetramers (Shakir et al., 2006). Because the mutant phenotype of unc-34(e315) animals is more severe than that of unc-34(lq17) animals and because unc-34(lq17) mutants presumably lack tetramerization functions, EVH2 sequences other than those involved in tetramerization would be important for UNC-34 function if a truncated protein is made in unc-34(e315) mutants. An alternative possibility is that instability of unc-34 mRNA or protein in unc-34(e315) mutants leads to the severe migration defects observed.
It is noteworthy that the original unc-34(e315) strain CB315 contains a second mutation that partially suppressed the cell migration defects in this strain (see Materials and methods). Once this suppressor was removed, the e315 allele behaved like all of the other strong unc-34 mutant alleles. Based on its effects on the unc-34(e315) mutant phenotype, this suppressor is different from other characterized unc-34 suppressors (Hurwitz et al., 2009; Vanderzalm et al., 2009) (see Materials and methods).
The unc-34(ev561) and unc-34(ev562) alleles are hypomorphic, resulting in weaker locomotion and migration defects compared to other unc-34 alleles (Figs. 3, 4 and data not shown). In unc-34(ev561) mutants, 57% of the CANs were found in wild-type positions, which is significantly higher than the 25% in null mutants (p<0.0001). The remaining CANs were found at various positions along the migratory path (Fig. 3). The CANs of unc-34(ev562) mutants, by contrast, all migrated to near wild-type positions. Thus, the EVH1 missense mutation of unc-34(ev561) partially disrupts UNC-34 activity in CAN migration, while the EVH1 missense mutation of unc-34(ev562) had only a minor effect on UNC-34’s ability to function in CAN migration. In contrast to their different effects on CAN migration, both unc-34(ev561) and unc-34(ev562) mutants displayed similar HSN migration defects, which were not easily distinguished from null mutants (Fig. 4).
Because UNC-34 is expressed throughout the nervous system and is known to function in multiple cell migrations, it could promote proper migration from within a migrating cell or through its activity in neighboring cells. To test for a cell autonomous role of UNC-34 in CAN cell migration, we expressed an unc-34 cDNA tagged with GFP from a ceh-23 promoter, which drove expression in the CAN and in a small subset of sensory neurons (Wang et al., 1993). In the null mutant unc-34(e951), only 24% of the CAN cells migrate to the wild-type location. Expression of UNC-34∷GFP within the CAN increases the number of cells that reach the wild-type location to 75% (Fig. 3). Thus, UNC-34 expression within the CAN is capable of rescuing a significant portion of the unc-34(e951) CAN cell migration defects. These results indicate that UNC-34 acts in the CAN cell to promote its migration.
The locomotion deficits of unc-34 mutants presumably result from widespread defects in axon extension, guidance and fasciculation (Forrester and Garriga, 1997; Hedgecock et al., 1985; McIntire et al., 1992). As with the CAN migration defects, unc-34 mutants carrying EVH1 missense mutations displayed varying degrees of uncoordinated locomotion. The Unc phenotype of unc-34(gm114) mutants was similar to that of unc-34 null mutants. unc-34(ev561) mutants were less severely uncoordinated, and unc-34(ev562) displayed only a very mild Unc phenotype.
To test whether the Unc phenotypes observed correlate with axonal defects, we scored the process morphology of the DD GABAergic motor neurons in first larval stage animals using the Punc-25∷gfp transgene juIs76 (Huang et al., 2002) (Fig. 5A). The cell bodies of the six DD motor neurons (DD1-DD6) are located in the ventral nerve cord and extend their processes during embryonic development (Durbin, 1987; White et al., 1976, 1986). We use the term “process” instead of axon or dendrite because DDs are not polarized with an axon emerging from one part of the cell and dendrites from the other. Rather, the branched process of each DD has both presynaptic and postsynaptic domains. Each DD neuron extends a long anterior process that branches near its terminus, extending a commissure that leaves the ventral nerve cord and continues dorsally to the dorsal nerve cord. Once it reaches the dorsal nerve cord, the commissure branches to form a short anterior process and a longer posterior process (Fig. 5A and B).
The observed process morphologies were divided into five classes ranging from wild type (class I and class II) to the most severe defect where processes failed to exit the ventral cord (class V) (Fig. 5B). Because juIs76 animals express GFP in some head neurons whose projections overlap with those of DD1, we present the results for DD2-DD6. The defects of unc-34(gm114), unc-34(gm104) and unc-34(e951) mutants were similar with about 40% of the processes failing to reach the DNC (class IV and class V) in all three mutants. The unc-34(ev561) mutants, by contrast, showed only 30% of processes in class IV or V categories and unc-34(ev562) mutants, which are the least Unc, displayed no class V defects and only about 9% of the processes were classified as class IV defects (Fig. 5C). These process phenotypes parallel the observed Unc phenotypes.
UNC-34 functions in Wnt-regulated neuronal polarity
The three Wnt genes cwn-1, cwn-2 and egl-20 regulate the polarity of the ALM neuron (Hilliard and Bargmann, 2006; Prasad and Clark, 2006). While ALM development is normal in cwn-1, cwn-2 and egl-20 single mutants, the polarity of the ALMs is often disrupted in cwn-1; cwn-2 and cwn-1; egl-20 double mutants. The ALMs normally extend a long single process to the head (Fig. 6A, D). The process forms a branch that innervates the nerve ring (White et al., 1986). In Wnt double mutants, the ALMs can be bipolar or their polarity can be reversed, extending a single process toward the tail (Fig. 6B, C, E, F).
Fig. 6.
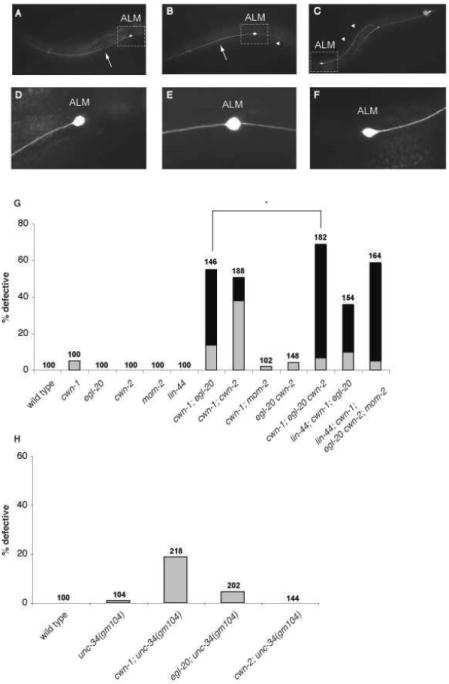
unc-34and cwn-1 mutations synergize to produce defects in ALM neuronal polarity. (A–F) Photomicrographs of L4 zdIs5 (pmec-4∷gfp) animals showing ALM neuronal morphology. The ALM cell bodies are labeled. Arrows indicate anterior processes, and arrowheads indicate posterior processes. Anterior is to the left, dorsal is up. unc-34 mutants exhibit defects in ALM neuronal polarity. The boxed regions of A-C are shown in D-F. (A, D) A wild-type ALM extends a single, anterior process. (B, E) A cwn-1; egl-20 ALM is bipolar, extending both a normal anterior process and an ectopic posterior process. (C, F) A cwn-1; egl-20 ALM extends a single posterior process, indicating a reversal of polarity. (G and H): Graph shows the percentage of ALM neurons with defective polarity for each genotype. Gray bars indicate the bipolar phenotype and black bars indicate reversed polarity. In G, the lin-44 mutation was n2111 in the single wnt mutant strain and n1972 in the strains with multiple wnt mutations. The mom-2 mutation was or309 in the single wnt mutant and ne874 in the multiple wnt mutant strain. Numbers for each genotype are provided. N.S., not significant; *p < 0.01 (Fisher’s exact test).
We found similar ALM polarity defects in cwn-1; cwn-2 and cwn-1; egl-20 mutants to those reported previously (Fig. 6G). Because the cwn-2 and egl-20 genes are linked, the egl-20 cwn-2 double mutant was not constructed and analyzed previously. We find that an egl-20 cwn-2 double mutant has a weak bipolar phenotype but that a cwn-1; egl-20 cwn-2 mutant shows a significantly stronger defect than any of the double mutants (Fig. 6G). These findings indicate that each of these three Wnts provide overlapping functions in ALM polarity.
C. elegans has two additional Wnt genes: lin-44 and mom-2. Because a mom-2 mutation does not enhance the cwn-1 ALM defect, this Wnt gene may not have a role in ALM polarity (Fig. 6G). One caveat of this interpretation is that we could only analyze homozygous mom-2 mutants coming from heterozygous mothers since mom-2 loss leads to a completely penetrant maternal effect embryonic lethality. Thus, a maternally provided gene product could mask a role for mom-2 in this process. A lin-44 mutation suppresses the ALM polarity defects of the cwn-1; egl-20 double mutant (Hilliard and Bargmann, 2006; Prasad and Clark, 2006). We confirmed this result and also showed that a strain containing mutations in all five Wnt genes is less severely affected than the cwn-1; egl-20 cwn-2 triple mutant (Fig. 6G), supporting the hypothesis that LIN-44 antagonizes the effects of one or more of the other Wnts in this process. These quintuple mutants, however, were derived from mothers heterozygous for the egl-20, cwn-2 and mom-2 mutations and hence maternal contribution of wild-type activity could also ameliorate the effects of the egl-20 and cwn-2 mutations.
By screening existing mutations for an effect on ALM polarity, we found that mutations in unc-34 produced a synthetic bipolar phenotype in a cwn-1 but not an egl-20 or a cwn-2 mutant background (Fig. 6H). Similar to their effects on cell migration and DD process outgrowth, the strong alleles gm104 and gm114 resulted in a higher penetrance of ALM defects than the weaker ev561 and ev562 alleles (Fig. 7A). We were also able to rescue partially the synthetic ALM defect of cwn-1; unc-34(gm104) mutants by expressing unc-34 in the ALM from the unc-86 promoter, which is expressed in many neurons including the ALM, but not in non-neurnonal cells (Baumeister et al., 1996) (Fig. 7A). This finding suggests that UNC-34 functions in the ALM to establish its polarity.
Fig. 7.

Genetic interactions among genes involved in ALM polarity. (A-C) Graph shows the percentage of ALM neurons with defective polarity for each genotype. Gray bars indicate the bipolar phenotype and black bars indicate reversed polarity. (A) Multiple alleles of unc-34 interact with cwn-1 and unc-34 functions cell- autonomously in ALM polarity. (B) A mutation in cwn-1 significantly enhanced polarity defect of putative null alleles of unc-34 (gm104 and gm114) but not putative hypomorphic alleles of unc-34 (ev561 and ev562). Driving unc-34 expression from the neural promoter unc-86 significantly rescued polarity defect in cwn-1; unc-34(gm104). (B) The roles of ced-10 and mig-10 in ALM polarity. (C) unc-34(gm104) is temperature-sensitive for ALM polarity. Numbers for each genotype are provided. N.S., not significant; *p < 0.01 (Fisher’s exact test).
UNC-34 and the Rac CED-10 were found to act in parallel in UNC-6-mediated ventral guidance of the AVM axon, and these two pathways were shown to act downstream of the UNC-6 receptor UNC-40 (Gitai et al., 2003). To address whether CED-10 also acts in ALM polarity, we scored ced-10, cwn-1; ced-10 and ced-10; unc-34 mutants (Fig. 7B). None of the three ced-10 alleles tested had much of an effect on ALM polarity, but the cwn-1; ced-10 double mutants displayed significant ALM polarity defects. The strongest ced-10 allele, tm597, is a maternal effect lethal, and unlike the other ced-10 mutants that were analyzed, animals homozygous for tm597 came from heterozygous mothers. The weaker effect of the tm597 allele presumably reflects rescue by maternally supplied gene product.
Because mutations in both unc-34 and ced-10 generate a synthetic polarity defect in combination with the cwn-1 mutation, we attempted to construct a ced-10; unc-34 double mutant, but were unable to maintain the strains as homozygous stocks because of lethality. Instead, we balanced both mutations over the nT1 balancers. Because the balancers are marked by a transgene that expresses GFP in the pharynx, we scored animals that lacked the GFP marker. Very few double homozygous larvae are generated from the balanced strain and those that were produced arrested development as larvae. The ced-10; unc-34 double mutants lacked significant ALM polarity defects. The lack of a phenotype could result from the presence of maternally supplied ced-10, unc-34 or both.
The lamellopodin/RIAM homolog MIG-10 physically interacts with both UNC-34 and CED-10 (Quinn et al., 2006; Quinn et al., 2008). We found that like mutations in unc-34 and ced-10, a mig-10 mutation did not cause an ALM polarity defect but did generate a synthetic phenotype with cwn-1 (Fig. 7B). The ability of a Punc-86∷mig-10∷gfp transgene to partially rescue the synthetic phenotype suggests that mig-10 also acts in the ALM to regulate its polarity.
unc-34 mutants are heat sensitive for neuronal migrations and polarity
The locomotion phenotype of unc-34 mutants has been reported to be heat sensitive (Bloom, 1993). We confirmed that the Unc phenotype of all unc-34 mutants, including the null mutants e951 and gm104, increased in severity at elevated temperatures, leading to the prediction that the mutants would display more pronounced defects in process morphology at higher temperatures. To test this hypothesis, we analyzed the morphology of the DD processes. We scored DD2 process morphology in unc-34 mutants raised at 15°, 20° and 25°C and found that the severity of the defects increased at higher temperatures (Fig. 5D and data not shown). For instance, the null allele e951 showed less than 24% of processes with the most severe class IV and V morphologies at 15°C, but 59% of the processes were in these classes at 25°C (Fig. 5D). The DD and VD processes form a single bundle in the ventral and dorsal nerve cords that can be scored using the unc-25∷gfp reporter. We also found that unc-34 mutants raised at higher temperatures exhibited increased defasciculation of the DD and VD processes (data not shown).
We also found that the cell migration defects of strains carrying either null or hypomorphic alleles of unc-34 were more severe at higher temperatures (Fig. 8 and data not shown). The cell migration defects of hermaphrodites carrying the null allele e951, for example, has only 11% of its CAN cells failing to migrate out of the head region at 20°C, but 34% of the CANs remain in these positions at 25°C (Fig. 8). The CAN defects of gm104 and gm114 mutants were also stronger when the animals were raised at 25°C and weaker when raised at 15°C (Fig. 8 and data not shown). Similar temperature effects were seen for ALM and HSN migrations in all three mutants (data not shown).
Fig. 8.

Temperature-sensitive CAN migration defects of unc-34(e951) mutants. Diagram of CAN migration is the same as in Fig. 3. CAN migration in wild-type animals was not strongly affected by temperature, whereas the unc-34(e951) null allele showed a significant increase in CAN migration defects with increasing temperature. Each box in the lower part of the Fig. contains the percentage of CANs in that position relative to the hypodermal nuclei. “n” is the number of CANs scored for each genotype.
Finally, the ALM polarity defects of unc-34 mutants were also affected by temperature. Whereas the penetrance of ALM polarity defects in cwn-1; egl-20 double mutants was not elevated significantly at higher temperatures, the penetrance of the cwn-1; unc-34(gm104) double mutant increased significantly with increasing temperature (Fig. 7C).
Because the neuronal migration and polarity in wild-type animals were largely unaffected in this same temperature range, these phenotypic differences reveal a temperature-sensitive process that is normally masked by wild-type unc-34 function.
Discussion
UNC-34 and neuronal polarity
Ena/VASP family members are essential for polarization of cortical neurons grown in culture (Kwiatkowski et al., 2007). These neurons usually go through several stages to polarize, generating a single axon and multiple dendrites. In the first stage, neurons extend filopodia that develop into neurites, and these filopodia fail to form in cortical neurons derived from mice lacking all three Ena/VASP family members (Kwiatkowski et al., 2007). The finding that these triple mutant mice are almost devoid of cortical axon tracts is consistent with Ena/VASP family members playing an essential role in the early stages of neuronal polarization. Unlike the cortical neurons of the mouse triple mutants, C. elegans unc-34 neurons lacking all Eva/VASP function polarize normally. One explanation for this difference is that cortical neurons lack redundant pathways that can generate polarity in the absence of Ena/VASP family members. The finding that either expression of the actin nucleating protein mDia in the mutant cortical neurons or growing the neurons on laminin can rescue the defect in neuritogenesis is consistent with this hypothesis (Dent et al., 2007).
UNC-34 was implicated in the initial ventral polarization of the HSN motor neuron and the AVM mechanosensory neuron (Adler et al., 2006; Quinn et al., 2006). While unc-34 mutants rarely have defects in the ventral growth of the HSN and AVM neurons, genetic interaction studies show unc-34 mutations can interact synergistically with other mutations during ventral growth. For example, loss of UNC-34 and the Rac CED-10 leads to defects in AVM ventral guidance, and these molecules are thought to mediate the polarizing effects of the UNC-6-receptor UNC-40 (Gitai et al., 2003). We also tested whether CED-10 plays a role in ALM polarity and found that mutations in ced-10 produced a synthetic phenotype in a cwn-1 mutant background. The ced-10; unc-34 double mutants, however, did not have an ALM polarity defect. We balanced both the ced-10 and unc-34 mutations and scored the double homozygous animals coming from the heterozygous mothers, so the lack of a phenotype could reflect rescue by maternal products. Alternatively, additional pathways might function in ALM polarity. We do not believe that the presence of other Racs are responsible for the lack of a phenotype because reducing either mig-2 or rac-2 in a cwn-1 mutant background did not produce a significant ALM phenotype (data not shown). A recent report showed that CED-10 mediates the effects of Wnts in the engulfment of apoptotic cell corpses, the orientation of mitotic spindles during asymmetric cell divisions and the migration of the distal tip cells, somatic gonadal cells that shape the structure of the gonad (Cabello et al., 2010). Our results suggest that Wnts also act through CED-10 to control neuronal polarity.
We also tested the role of MIG-10, a lamellopodin/RIAM homolog that physically interacts with both UNC-34 and CED-10 (Quinn et al., 2006; Quinn et al., 2008), and found that it plays a cell autonomous role in ALM polarity. Our findings indicate that the same signal transduction molecules involved in polarizing the AVM along the dorsoventral axis also function in polarizing the ALM along the anterioposterior axis.
The lack of interactions between unc-34 and either egl-20 or cwn-2 suggests that unc-34 could mediate the effects of these Wnts and act in parallel to CWN-1. Alternatively, the cwn-1 mutant background might provide a more sensitized background than that of the other Wnt mutants. In either case, UNC-34 might mediate the effects of these Wnts on ALM polarity. It is noteworthy that unc-34 and the three Wnts involved in ALM polarity also control HSN migration, consistent with a general role for UNC-34 in Wnt mediated events (Forrester and Garriga, 1997; Pan et al., 2006). How Wnts regulate cell motility and neuronal polarity is poorly understood. Recent papers have implicated an atypical protein kinase C, PI3 kinase (Wolf et al., 2008) and a Rac in Wnt regulated motility (Cabello et al., 2010). Our results support the role of Racs in this process and implicate Ena for the first time in the Wnt regulated neuronal polarity. While our results are consistent with UNC-34, CED-10 and MIG-10 mediating the effects of the Wnts, they are also consistent with these molecules acting in parallel to the Wnts. Further experiments will be necessary to distinguish between these possibilities.
The EVH1 domain is essential for UNC-34 function
The gm114 mutation disrupts the EVH1 domain of UNC-34 by changing an alanine to threonine at amino acid 99. This allele results in behavioral defects as well as cell and growth cone defects that are similar to those produced by the e951 and gm104 mutations, indicating that this residue is essential for UNC-34 function. We propose that the defect in UNC-34 function in the gm114 mutant results from its failure to localize properly. unc-34(gm114) is a lesion that is similar to a mutation identified in Drosophila ena210 mutants. The ena210 mutation changes the same conserved alanine altered in unc-34(gm114) to valine. ena210 mutants are embryonic lethal and dominantly suppress the lethality caused by mutations in Abl (Ahern-Djamali et al., 1998; Gertler et al., 1995). Modeling of the EVH1 domain predicts that the A97V change will disrupt EVH1/Zyxin interactions (Renfranz and Beckerle, 2002). These findings support a requirement for the EVH1 domain and in particular the conserved alanine in UNC-34 function.
The complete loss of UNC-34 localization to axons and apical junctions in unc-34(gm114) mutants demonstrates that the EVH1 domain is required for proper localization of UNC-34. This idea is further supported by the absence of UNC-34 staining at apical junctions in unc-34(ev562) and unc-34(ev561) mutants as well as the decreased axonal staining in these mutants. The localization of Ena/VASP proteins to the lamellipodia is essential for Ena/VASP function in fibroblast motility (Bear et al., 2000). In addition, the recruitment of Ena/VASP to the surface of Listeria correlates with Ena/VASP function to promote Listeria motility (Chakraborty et al., 1995). Therefore, we propose that the loss of proper UNC-34 localization in unc-34(gm114) mutants accounts for their severe cell and axon migration defects and that the partial defects in UNC-34 localization in unc-34(ev562) and unc-34(ev561) results in their weaker phenotypes. The accumulation of UNC-34 to the cell bodies of the neurons in these mutants is surprising and suggests that the EVH1 mediates interactions that transport UNC-34 out of the cell body into axons. Proof of this hypothesis will require the identification of the EVH1 binding partner and the demonstration that it is necessary for UNC-34 transport.
Acknowledgments
We thank David Baillie, Cori Bargmann, Scott Clark, Joe Culotti, Ed Hedgecock and Bob Horvitz for providing C. elegans strains. Some of the C. elegans strains used in the work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH National Center. This work was supported by National Institutes of Health (NIH) grant NS32057 to G. Garriga and HD37815 to W. Forrester. T. C. Fleming was supported by genetics training grant GM077232 from NIH. P.J.V. was supported by an NSF predoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler CE, Fetter RD, Bargmann CI. UNC-6/Netrin induces neuronal asymmetry and defines the site of axon formation. Nat Neurosci. 2006;9:511–518. doi: 10.1038/nn1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern-Djamali SM, Comer AR, Bachmann C, Kastenmeier AS, Reddy SK, Beckerle MC, Walter U, Hoffmann FM. Mutations in Drosophila enabled and rescue by human vasodilator-stimulated phosphoprotein (VASP) indicate important functional roles for Ena/VASP homology domain 1 (EVH1) and EVH2 domains. Molecular Biology of the Cell. 1998;9:2157–2171. doi: 10.1091/mbc.9.8.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes AP, Polleux F. Establishment of Axon-Dendrite Polarity in Developing Neurons. Annu Rev Neurosci. 2009 doi: 10.1146/annurev.neuro.31.060407.125536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister R, Liu Y, Ruvkun G. Lineage-specific regulators couple cell lineage asymmetry to the transcription of the Caenorhabditis elegans POU gene unc-86 during neurogenesis. Genes Dev. 1996;10:1395–1410. doi: 10.1101/gad.10.11.1395. [DOI] [PubMed] [Google Scholar]
- Bear JE, Loureiro JJ, Libova I, Fassler R, Wehland J, Gertler FB. Negative regulation of fibroblast motility by Ena/VASP proteins. Cell. 2000;101:717–728. doi: 10.1016/s0092-8674(00)80884-3. [DOI] [PubMed] [Google Scholar]
- Belfiore M, Mathies LD, Pugnale P, Moulder G, Barstead R, Kimble J, Puoti A. The MEP-1 zinc-finger protein acts with MOG DEAH box proteins to control gene expression via the fem-3 3’ untranslated region in Caenorhabditis elegans. RNA. 2002;8:725–739. doi: 10.1017/s1355838202028595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom L. Biology. Massachusetts Institute of Technology; Cambridge: 1993. Genetic and molecular analysis of genes required for axon outgrowth in Caenorhabditis elegans; p. 410. [Google Scholar]
- Cabello J, Neukomm LJ, Gunesdogan U, Burkart K, Charette SJ, Lochnit G, Hengartner MO, Schnabel R. The Wnt pathway controls cell death engulfment, spindle orientation, and migration through CED-10/Rac. PLoS Biol. 8:e1000297. doi: 10.1371/journal.pbio.1000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T, Ebel F, Domann E, Niebuhr K, Gerstel B, Pistor S, Temm-Grove CJ, Jockusch BM, Reinhard M, Walter U, et al. A focal adhesion factor directly linking intracellularly motile Listeria monocytogenes and Listeria ivanovii to the actin-based cytoskeleton of mammalian cells. Embo J. 1995;14:1314–1321. doi: 10.1002/j.1460-2075.1995.tb07117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm AD, Hardin J. Epidermal morphogenesis. WormBook. 2005:1–22. doi: 10.1895/wormbook.1.35.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, Alberts AS, Mori S, Gertler FB. Filopodia are required for cortical neurite initiation. Nat Cell Biol. 2007;9:1347–1359. doi: 10.1038/ncb1654. [DOI] [PubMed] [Google Scholar]
- Durbin R. Studies on the development and organisation of the nevrous system of Caenorhabditis elegans. University of Cambridge; 1987. [Google Scholar]
- Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- Forrester WC, Garriga G. Genes necessary for C. elegans cell and growth cone migrations. Development (Cambridge) 1997;124:1831–1843. doi: 10.1242/dev.124.9.1831. [DOI] [PubMed] [Google Scholar]
- Furman C, Sieminski AL, Kwiatkowski AV, Rubinson DA, Vasile E, Bronson RT, Fassler R, Gertler FB. Ena/VASP is required for endothelial barrier function in vivo. J Cell Biol. 2007;179:761–775. doi: 10.1083/jcb.200705002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Comer AR, Juang J-L, Ahern SM, Clark MJ, Liebl EC, Hoffmann FM. Enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3 domain-binding properties. Genes & Development. 1995;9:521–533. doi: 10.1101/gad.9.5.521. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Doctor JS, Hoffmann FM. Genetic suppression of mutations in the Drosophila abl proto-oncogene homolog. Science. 1990;248:857–860. doi: 10.1126/science.2188361. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- Gitai Z, Yu TW, Lundquist EA, Tessier-Lavigne M, Bargmann CI. The netrin receptor UNC-40/DCC stimulates axon attraction and outgrowth through enabled and, in parallel, Rac and UNC-115/AbLIM. Neuron. 2003;37:53–65. doi: 10.1016/s0896-6273(02)01149-2. [DOI] [PubMed] [Google Scholar]
- Guenther C, Garriga G. Asymmetric distribution of the C. elegans HAM-1 protein in neuroblasts enables daughter cells to adopt distinct fates. Development. 1996;122:3509–3518. doi: 10.1242/dev.122.11.3509. [DOI] [PubMed] [Google Scholar]
- Haffner C, Jarchau T, Reinhard M, Hoppe J, Lohmann SM, Walter U. Molecular cloning, structural analysis and functional expression of the proline-rich focal adhesion and microfilament-associated protein VASP. EMBO J. 1995;14:19–27. doi: 10.1002/j.1460-2075.1995.tb06971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbrugge M, Walter U. Purification of a vasodilator-regulated phosphoprotein from human platelets. Eur J Biochem. 1989;185:41–50. doi: 10.1111/j.1432-1033.1989.tb15079.x. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Thomson JN, Perkins LA. Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev Biol. 1985;111:158–170. doi: 10.1016/0012-1606(85)90443-9. [DOI] [PubMed] [Google Scholar]
- Herman MA, Horvitz HR. The Caenorhabditis elegans gene lin-44 controls the polarity of asymmetric cell divisions. Development. 1994;120:1035–1047. doi: 10.1242/dev.120.5.1035. [DOI] [PubMed] [Google Scholar]
- Hilliard MA, Bargmann CI. Wnt signals and frizzled activity orient anterior-posterior axon outgrowth in C. elegans. Dev Cell. 2006;10:379–390. doi: 10.1016/j.devcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Huang X, Cheng HJ, Tessier-Lavigne M, Jin Y. MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron. 2002;34:563–576. doi: 10.1016/s0896-6273(02)00672-4. [DOI] [PubMed] [Google Scholar]
- Hurwitz ME, Vanderzalm PJ, Bloom L, Goldman J, Garriga G, Horvitz HR. Abl kinase inhibits the engulfment of apoptotic cells and cell migration in Caenorhabditis elegans. PLOS Biology. 2009 doi: 10.1371/journal.pbio.1000099. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, Bronson RT, Mori S, Fassler R, Gertler FB. Ena/VASP Is Required for neuritogenesis in the developing cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Manser J, Wood WB. Mutations affecting embryonic cell migrations in Caenorhabditis elegans. Dev Genet. 1990;11:49–64. doi: 10.1002/dvg.1020110107. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Garriga G, White J, Jacobson D, Horvitz HR. Genes necessary for directed axonal elongation or fasciculation in C. elegans. Neuron. 1992;8:307–322. doi: 10.1016/0896-6273(92)90297-q. [DOI] [PubMed] [Google Scholar]
- Pan CL, Howell JE, Clark SG, Hilliard M, Cordes S, Bargmann CI, Garriga G. Multiple Wnts and frizzled receptors regulate anteriorly directed cell and growth cone migrations in Caenorhabditis elegans. Dev Cell. 2006;10:367–377. doi: 10.1016/j.devcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Prasad BC, Clark SG. Wnt signaling establishes anteroposterior neuronal polarity and requires retromer in C. elegans. Development. 2006;133:1757–1766. doi: 10.1242/dev.02357. [DOI] [PubMed] [Google Scholar]
- Quinn CC, Pfeil DS, Chen E, Stovall EL, Harden MV, Gavin MK, Forrester WC, Ryder EF, Soto MC, Wadsworth WG. UNC-6/netrin and SLT-1/slit guidance cues orient axon outgrowth mediated by MIG-10/RIAM/lamellipodin. Curr Biol. 2006;16:845–853. doi: 10.1016/j.cub.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Quinn CC, Pfeil DS, Wadsworth WG. CED-10/Rac1 mediates axon guidance by regulating the asymmetric distribution of MIG-10/lamellipodin. Curr Biol. 2008;18:808–813. doi: 10.1016/j.cub.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2:131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- Renfranz PJ, Beckerle MC. Doing (F/L)PPPPs: EVH1 domains and their proline-rich partners in cell polarity and migration. Curr Opin Cell Biol. 2002;14:88–103. doi: 10.1016/s0955-0674(01)00299-x. [DOI] [PubMed] [Google Scholar]
- Shakir MA, Gill JS, Lundquist EA. Interactions of UNC-34 Enabled with Rac GTPases and the NIK kinase MIG-15 in Caenorhabditis elegans axon pathfinding and neuronal migration. Genetics. 2006;172:893–913. doi: 10.1534/genetics.105.046359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield M, Loveless T, Hardin J, Pettitt J. C. elegans Enabled exhibits novel interactions with N-WASP, Abl, and cell-cell junctions. Curr Biol. 2007;17:1791–1796. doi: 10.1016/j.cub.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Thorpe CJ, Schlesinger A, Carter JC, Bowerman B. Wnt signaling polarizes an early C. elegans blastomere to distinguish endoderm from mesoderm. Cell. 1997;90:695–705. doi: 10.1016/s0092-8674(00)80530-9. see comments. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsuing N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderzalm PJ, Pandey A, Hurwitz ME, Bloom L, Horvitz HR, Garriga G. C. elegans CARMIL negatively regulates UNC-73/Trio function during neuronal development. Development. 2009;136:1201–1210. doi: 10.1242/dev.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Nieberding M, Walter U. Vasodilator-stimulated protein phosphorylation in platelets is mediated by cAMP- and cGMP-dependent protein kinases. Eur J Biochem. 1987;167:441–448. doi: 10.1111/j.1432-1033.1987.tb13357.x. [DOI] [PubMed] [Google Scholar]
- Wang BB, Muller-Immergluck MM, Austin J, Robinson NT, Chisholm A, Kenyon C. A homeotic gene cluster patterns the anteroposterior body axis of C. elegans. Cell. 1993;74:29–42. doi: 10.1016/0092-8674(93)90292-x. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the ventral nerve cord of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1976;275:327–348. doi: 10.1098/rstb.1976.0086. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Withee J, Galligan B, Hawkins N, Garriga G. Caenorhabditis elegans WASP and Ena/VASP proteins play compensatory roles in morphogenesis and neuronal cell migration. Genetics. 2004;167:1165–1176. doi: 10.1534/genetics.103.025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AM, Lyuksyutova AI, Fenstermaker AG, Shafer B, Lo CG, Zou Y. Phosphatidylinositol-3-kinase-atypical protein kinase C signaling is required for Wnt attraction and anterior-posterior axon guidance. J Neurosci. 2008;28:3456–3467. doi: 10.1523/JNEUROSCI.0029-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TW, Hao JC, Lim W, Tessier-Lavigne M, Bargmann CI. Shared receptors in axon guidance: SAX-3/Robo signals via UNC-34/Enabled and a Netrin-independent UNC-40/DCC function. Nat Neurosci. 2002;5:1147–1154. doi: 10.1038/nn956. [DOI] [PubMed] [Google Scholar]
- Zinovyeva AY, Forrester WC. The C. elegans Frizzled CFZ-2 is required for cell migration and interacts with multiple Wnt signaling pathways. Dev Biol. 2005;285:447–461. doi: 10.1016/j.ydbio.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Zinovyeva AY, Yamamoto Y, Sawa H, Forrester WC. Complex network of Wnt signaling regulates neuronal migrations during Caenorhabditis elegans development. Genetics. 2008;179:1357–1371. doi: 10.1534/genetics.108.090290. [DOI] [PMC free article] [PubMed] [Google Scholar]