

Abstract
Activation of NK cells is a hallmark of infections with intracellular pathogens. We previously showed that the protozoan parasite Leishmania infantum triggered a rapid NK-cell response in mice that required TLR9-positive myeloid DC and IL-12, but no IFN-α/β. Here, we investigated whether IL-15 or IL-18 mediate the activity of IL-12 or function as independent activators of NK cells. In contrast to earlier studies that described IL-15 as crucial for NK-cell priming in response to TLR ligands, the expression of IFN-γ, FasL, perforin and granzyme B by NK cells in L. infantum-infected mice was completely preserved in the absence of IL-15, whereas the proliferative capacity of NK cells was lower than in WT mice. IFN-γ secretion, cytotoxicity and FasL expression of NK cells from infected IL-18−/− mice were significantly reduced compared with controls, but, unlike IL-12, IL-18 was not essential for NK-cell effector functions. Part of the NK-cell-stimulatory effect of IL-12 was dependent on IL-18. We conclude that IL-15 is not functioning as a universal NK-cell priming signal and that IL-18 contributes to the NK-cell response in visceral leishmaniasis. The cytokine requirements for NK-cell activation appear to differ contingent upon the infectious pathogen.
Keywords: IL-12, IL-15, IL-18, Leishmaniasis, NK cell
Introduction
NK cells are important effector cells of the innate immune response to infectious pathogens and malignant cells. They contribute to the control of infections and tumors by producing cytokines such as IFN-γ, lysing transformed or virally infected target cells, and by supporting the development of a protective T-cell response 1–4. Later during infection NK cells might also acquire an immunoregulatory activity 5.
An NK-cell response is readily induced during infections with intracellular pathogens and requires cytokine and receptor signals that are delivered by myeloid cells 6–8. Recently, myeloid DC (mDC) and neutrophils have been shown to prime NK cells during viral, bacterial or protozoan infections in vivo 9–11. Notably, various cytokines of myeloid cell origin, such as IFN-α/β 9, IL-12p70 10, IL-15 9 and IL-18 11, were identified to account for the activation of mature NK cells. This suggests that different pathogens might trigger distinct NK-cell activation pathways, or, alternatively, that the production or function of the NK-cell-activating cytokines is interdependent so that the respective gene-deficient mice exhibit a similar defect of NK-cell activation. In the case of IL-12 and IL-18, not only direct and independent, but also cooperative effects on NK cells have been described 11–13. A particular convincing example for the sequential action of two cytokines was observed after activation of NK cells by ligands of TLR3, TLR4, TLR7 or TLR9, which was strictly dependent on IFN-α/β signaling as well as IL-15. mDC from WT, but not from IL-15−/− or IFN-α/β-receptor−/− mice were able to prime NK cells in vitro 14 and in vivo 9. As TLR-induced IFN-α/β triggered the production of IL-15 9,14,15, the IFN-α/β-dependent NK-cell activation is thought to occur via the release of endogenous IL-15, which binds to the IL-15Rα chain on the surface of the mDC producer cell and is then transpresented to IL-15-responsive NK cells expressing the IL-15Rβ chain and the IL-2R common γ chain 16,17. As IL-15 also cooperates with IL-18 18, induces IL-12 by myeloid cells 19,20, and is essential for NK-cell expansion and activation during viral, bacterial and fungal infections 9,16,21–26, it can be considered as a central cytokine not only for normal NK-cell homeostasis 17,27,28 but also for an active NK-cell response.
When mice are cutaneously or intravenously infected with protozoan parasites of the species Leishmania (L.) major or L. infantum, which cause cutaneous or visceral leishmaniasis, NK cells become rapidly activated in the draining lymph node or spleen, respectively, and contribute to parasite control (reviewed in 29). We previously showed that the activation of NK cells in these models required mDC, sensing of the parasites by TLR9 and the release of IL-12, but was independent of IFN-α/β despite eliciting a strong IFN-α/β response in plasmacytoid DC 10,30. These studies, however, did not exclude the contribution of additional cytokine signals acting independently or up- or downstream of IL-12. In the present report we therefore addressed three questions using the mouse model of experimental visceral leishmaniasis: (i) Does IL-15 function as NK-cell-stimulatory cytokine as predicted from the results obtained in other systems? (ii) Is IL-18 similar to IL-12 a prerequisite for NK-cell activation in visceral leishmaniasis? (iii) Do IL-15 and IL-18 operate independently of IL-12? Our data clearly illustrate that the full induction of NK-cell effector functions in visceral leishmaniasis requires IL-12 and IL-18, but – unexpectedly – is entirely independent of IL-15. Whereas IL-12 is essential for the expression of NK-cell effector functions in vivo, IL-18 only plays a contributory role by supporting the NK-cell response to IL-12.
Results
Induction of NK-cell effector functions by L. infantum is independent of IL-15
Considering the known mode of action of IL-15 via transpresentation 16,17, we first tested the expression of the IL-15Rα-chain on splenic mDC. At 24 h after infection with L. infantum, we observed a 2- to 2.5-fold increase of the percentage of IL-15Rα+ mDC in the spleen, indicating that L. infantum might modulate the IL-15/IL-15R system (Supporting Information Fig. 1).
Next, we analysed whether IL-15 is required for the induction of NK-cell effector functions in vivo. As IL-15 is critical for NK-cell development and maturation 21,31, the spleen of IL-15−/− mice contained a dramatically reduced percentage and absolute number of mature NK cells compared with WT mice (0.16±0.03% versus 2.8±0.9% NK1.1+CD3− cells; mean±SD of seven or ten experiments, respectively; see also PBS-treated groups of Fig. 2B). We first investigated the L. infantum-induced expression of IFN-γ 12 h post infection (p.i.) within the residual endogenous NK1.1+CD3− NK-cell population of IL-15−/− mice as compared with NK cells of WT controls after a 4 h culture period, either in medium alone or in the presence of YAC-1 target cells. As expected, the in vitro restimulation augmented the IFN-γ release of in vivo primed NK cells (Fig. 1A). Irrespective of the culture condition we did not observe a significant reduction of the percentage of IFN-γ+ NK cells in L. infantum-infected mice in the absence of IL-15; in fact, without YAC cell restimulation the IFN-γ production by NK cells from infected IL-15−/− mice was even significantly higher than by NK cells from WT controls (Fig. 1A, medium). In contrast, the TLR3-dependent activation of NK cells elicited by poly(I:C) was drastically reduced in IL-15−/− mice confirming previously reported results 9. Thus, IL-15 was clearly dispensable for the L. infantum-induced expression of IFN-γ by NK cells in vivo.
Figure 2.
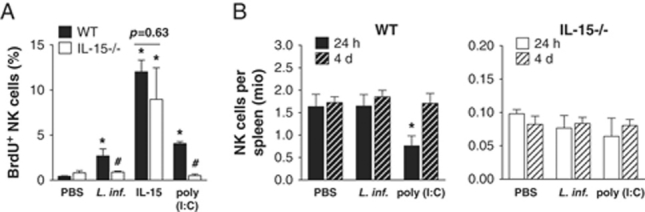
Infection with L. infantum causes an IL-15-dependent NK-cell proliferation, which does not augment the splenic NK-cell compartment. 1×107 L. infantum promastigote parasites or PBS were injected i.v. in C57BL/6 WT or IL-15−/− mice. (A) NK-cell proliferation in WT and IL-15−/− mice 24 h upon infection, poly(I:C) or IL-15/sIL-15Rα treatment measured by BrdU incorporation. Twenty-two hours p.i. 1 mg BrdU per mouse was injected i.p. and 2 h later the percentage of proliferated BrdU+ NK cells was analysed by FACS. Mean±SEM of two independent experiments with two mice per group. Significant differences by Mann–Whitney test between cells from infected, poly(I:C) or IL-15-treated and PBS treated mice (*p<0.05; **p<0.002) and between WT and IL-15−/− (♯p<0.05; ♯♯p<0.005) are indicated. (B) Absolute NK1.1+CD3− NK-cell numbers in the spleen were determined by FACS analysis at days 1 and 4 of infection. Mean±SEM of three independent experiments with two to three mice per group is given. Significant differences by Mann–Whitney test between cells from poly(I:C) and PBS treated mice (*p<0.05) are indicated.
Figure 1.
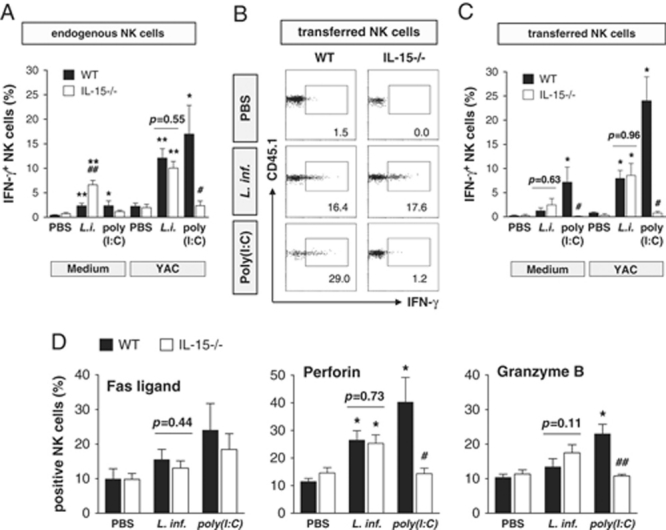
IL-15 is dispensable for NK-cell activation in L. infantum-infected mice. C57BL/6 WT or IL-15−/− mice were injected i.v. with 1×107 L. infantum promastigote parasites or PBS or i.p. with 50 μg poly(I:C). (A) Intracellular IFN-γ protein expression in endogenous NK cells taken from WT and IL-15−/− mice at 12 h after infection and cultured for 4 h in vitro in medium alone or in the presence of YAC-1 cells. Mean±SEM of four independent experiments with two to three mice per group. (B and C) Intracellular IFN-γ protein expression of donor NK cells after transfer into WT and IL-15−/− mice. Adoptive transfer of CD3−NK1.1+CD45.1+ NK cells from naïve WT donor mice was performed 2–4 h after L. infantum infection, PBS or poly(I:C) treatment of CD45.2+ WT or IL-15−/− recipients. Twelve hours later splenocytes were prepared, cultured in medium or in the presence of YAC-1 cells for 4 h, and stained for CD3−NK1.1+CD45.1+ NK cells and intracellular IFN-γ. (B) Dot plot analysis of one representative of three experiments with two to three mice per group after in vitro restimulation with YAC-1 cells is given. (C) Mean±SEM of three independent experiments with two to three mice per group. (D) Surface expression of FasL and intracellular expression of perforin or gzmB of endogenous CD3−NK1.1+ NK cells from WT and IL-15−/− directly ex vivo. Mean±SEM of three to four experiments with two to three mice per group. (A–D) Significant differences by Mann–Whitney test between cells from infected, poly(I:C) or IL-15-treated and PBS-treated mice (*p<0.05; **p<0.002) and between WT and IL-15−/− (♯p<0.05; ♯♯p<0.005) are indicated.
It was necessary to exclude the possibility that the residual NK-cell population in IL-15−/− mice had acquired IL-15-independent, alternative modes of activation and therefore did not exhibit the same stimulation requirements as NK cells in WT mice. To this end we analysed the L. infantum-induced expression of IFN-γ in highly purified NK cells from uninfected congenic CD45.1+ WT donor mice that had been transferred into L. infantum-infected WT or IL-15−/− recipient mice 2 to 4 h after infection (Fig. 1B and C). As the NK-cell activation was determined 12 h after cell transfer, the duration of the experiment was short enough to allow for the survival of transferred NK cells in the IL-15-negative microenvironment. Independent of the in vitro culture conditions the L. infantum-induced expression of IFN-γ in transferred CD45.1+ NK cells remained unaffected by the absence of IL-15 in the recipients. Neither the percentage (Fig. 1B and C) nor the MFI of IFN-γ+ donor NK cells (WT: 58.20±0.49, IL-15−/−: 53.89±5.78 (culture in medium); WT: 82.26±14.32, IL-15−/−: 74.59±16.46 (YAC-1 restimulation); mean±SEM of three experiments) or the absolute number of IFN-γ+ donor NK cells (WT: 651±99, IL-15−/−: 1641±324 (culture in medium); WT: 4733±744, IL-15−/−: 5444±1211 (YAC-1 restimulation); mean±SEM of three experiments) was significantly lower in infected IL-15−/− recipient mice compared with WT controls. In contrast, the TLR3-dependent activation of NK cells elicited by poly(I:C) was entirely dependent on IL-15 (Fig. 1B and C).
To test whether IL-15 is required for the induction of NK-cell cytotoxicity in vivo, we analysed the expression of critical components of the cytotoxic machinery of NK cells (Fas-FasL intercellular linkage-mediated pathway; granule-dependent exocytosis pathway), because the very low number of differentiated NK cells in IL-15−/− mice stymies the direct measurement of NK-cell cytotoxicity. L. infantum caused a modest upregulation of the percentages of FasL+, perforin+ and granzyme (gzm) B+ NK cells in the spleen, which was indistinguishable in WT and IL-15−/− mice, whereas the induction of perforin and gzmB in response to poly(I:C) was clearly impaired in NK cells from IL-15−/− mice (Fig. 1D). Thus, the induction of both NK-cell effector functions (IFN-γ release and cytotoxicity) by L. infantum is completely preserved in the absence of IL-15.
IL-15-dependent proliferation of NK cells in L. infantum-infected mice
Since signals through the IL-2Rγ/IL-15Rβ on NK cells are also known to stimulate NK-cell proliferation and accumulation 17,22,24,32,33, we tested the NK-cell expansion in L. infantum-infected WT versus IL-15−/− mice 24 h p.i. by BrdU incorporation. As a positive control both mouse groups were treated i.p. with pre-complexed IL-15 and sIL-15R-Fc, a “superagonist” of NK-cell proliferation 34. The injection of the parasites triggered a small, but significant NK-cell proliferation in the spleen of WT mice during the first day of infection, which was not observed in the absence of IL-15 (Fig. 2A). The L. infantum-induced and IL-15-dependent NK-cell proliferation, however, did not lead to an expansion of the splenic NK-cell compartment. During the early phase of infection (day 1 until day 4) the absolute number of NK cells per spleen remained unaltered in both WT and IL-15−/− mice compared with the respective uninfected control mice (Fig. 2B). In contrast, poly(I:C) treatment caused an IL-15-dependent decrease of the splenic NK-cell number, which presumably reflects the migration of primed and activated NK cells to the periphery 9.
The activation of NK cells in IL-15−/− mice requires IL-12
Having seen that IL-15 is dispensable for induction of NK-cell effector functions by L. infantum, we addressed the question, whether the L. infantum-induced NK-cell response in IL-15−/− mice is still strictly dependent on IL-12 as seen in WT mice 10 (see also below in text and Fig. 4). We first checked the secretion of IL-12 by splenic mDC of WT and IL-15−/− mice following infection (Fig. 3A). In both mouse groups IL-12 was induced upon infection, although mDC-derived IL-12p40/p70 was slightly reduced in IL-15-deficient mice at the 12 h time point. The fact that this difference was no longer detectable at 24 h p.i. (Fig. 3A) argues against a profound and biologically relevant difference in the production of IL-12 by mDC of both mouse strains. This view is strongly supported by the fact that in all our NK-cell transfer experiments (see Fig. 1C) the percentage of IL-12p40/p70+ splenic mDC (∼4–5%) 14–16 h p.i. was comparable in infected WT and IL-15−/− mice (data not shown). Furthermore, neutralization of IL-12 by Ab-treatment prior to infection completely prevented NK cell IFN-γ production in both WT and IL-15−/− mice (Fig. 3B). Anti-IL-12 had exactly the same effect, when the NK cell IFN-γ expression was determined without in vitro restimulation, except that the percentages of IFN-γ+ NK cells were lower in all groups (data not shown). Thus, even under IL-15-deficient conditions IL-12 is the key cytokine for L. infantum-induced NK-cell activation.
Figure 4.
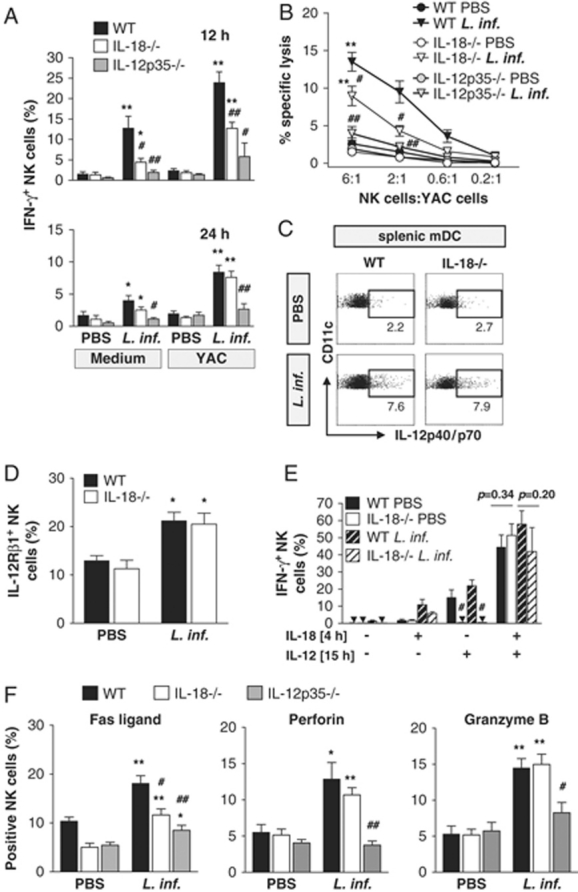
Mechanism of NK-cell activation by IL-18 in L. infantum-infected mice. At 12 and 24 h after i.v. injection of PBS or L. infantum promastigotes into C57BL/6 WT, IL-18−/− and IL-12p35−/− mice, splenocytes were prepared. (A) Frequency of IFN-γ+ NK cells (CD3−NK1.1+) as determined by intracellular cytokine staining and FACS analysis after a 4 h culture in medium or with YAC cells. Mean±SEM of six (WT versus IL-18−/−) and three (WT versus IL-18−/− and IL-12p35−/−) independent experiments with two to three mice per group. (B) NK-cell cytotoxicity at 24 h after infection. Mean±SEM of four (WT versus IL-18−/−) and three (WT versus IL-18−/− and IL-12p35−/−) independent experiments with two to three mice per group. (C) Intracellular IL-12p40/p70 protein expression of splenic mDC (restimulated in culture medium for 4 h; gated on CD11bintCD11chi cells) is shown in a dot plot analysis representative for three independent experiments with two to three mice per group. (D) Surface expression of IL-12Rβ1 on CD3-NK1.1+ NK cells directly ex vivo. Mean±SEM of three experiments with two to three mice per group. (E) Intracellular IFN-γ protein expression in NK cells from WT and IL-18−/− mice 12 h after infection or PBS treatment, which were cultured for 4 h (±IL-18), washed, and further incubated for 15 h (±IL-12). Mean±SEM of three independent experiments with two mice per group. (F) Surface expression of FasL and intracellular expression of perforin or gzmB of CD3-NK1.1+ NK cells directly ex vivo. FasL: mean±SEM of five (WT versus IL-18−/−) and three (WT versus IL-18−/− and IL-12p35−/−) experiments, two to three mice per group; perforin: mean±SEM of three experiments, two mice per group; gzmB: mean±SEM of four (WT versus IL-18−/−) and three (WT versus IL-18−/− and IL-12p35−/−) experiments, two to three mice per group. (A–F) Significant differences by Mann–Whitney test between cells from infected and PBS treated mice (*p<0.05; **p<0.002) and between WT and IL-18−/− or IL-12p35−/− (♯p<0.05; ♯♯p<0.005) are indicated.
Figure 3.
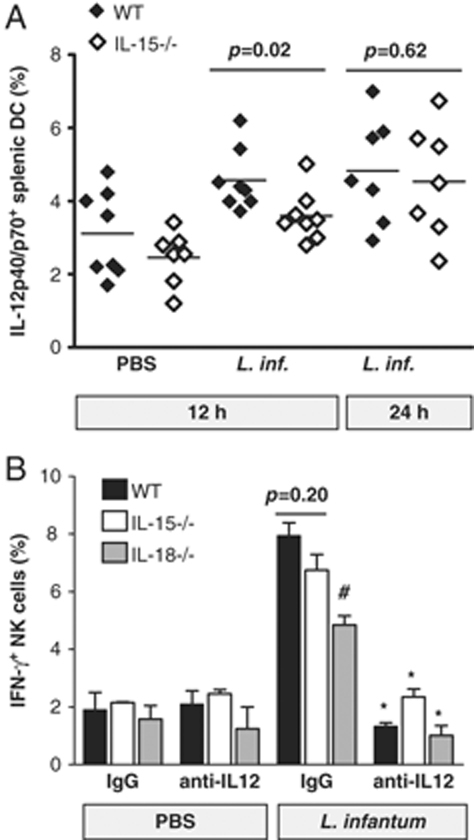
Activation of NK cells in IL-15−/− mice strictly depends on IL-12. C57BL/6 WT, IL-15−/− or IL-18−/− mice were injected i.v. with 1×107 L. infantum promastigote parasites or PBS. (A) Intracellular IL-12 p40/p70 expression in splenic mDC (CD11bintCD11chi cells) taken from WT and IL-15−/− mice at 12 or 24 h after infection and cultured in vitro for 4 h in medium. Three independent experiments with two to three mice per group. Each diamond represents a single mouse; the mean is given as horizontal line. (B) Intracellular IFN-γ protein expression in NK cells taken from WT, IL-15−/− or IL-18−/− mice 12 h after infection and co-cultured for 4 h in vitro with YAC-1 cells; 30 min before injection of parasites or PBS IL-12 p40-neutralizing mAb or rat IgG control Ab were applied i.p. Mean±SEM of two independent experiments with two mice per group. Significant differences by Mann–Whitney test between anti-IL-12p40 and control IgG treatment are indicated as *p<0.05; between WT and IL-15−/− or IL-18−/− NK cells as ♯p<0.05.
Endogenous IL-18 contributes to the activation of NK cells in visceral leishmaniasis
As IL-18 is known to activate mouse NK cells in vitro and in vivo 12,35, we tested whether IL-18 contributes to the NK-cell activation in visceral leishmaniasis. At the height of innate IFN-γ production in the spleen (12 h p.i.) the percentage of IFN-γ protein-positive NK cells from IL-18−/− mice was reduced by roughly 50% compared with WT mice (with or without in vitro restimulation) (Fig. 4A). Notably, infection with L. infantum did lead to an upregulation of IFN-γ mRNA in partially (70%) or highly purified splenic NK cells (>99.9%) and the levels of IFN-γ mRNA were comparable in WT and IL-18−/− mice (Supporting Information Fig. 2; data not shown). The cytotoxic activity of NK cells from IL-18−/− mice was significantly lower compared with WT NK cells (Fig. 4B). As seen before 10 the IFN-γ production as well as the cytotoxic activity of NK cells in L. infantum-infected IL-12−/− mice was nearly completely blocked (Fig. 4A and B). Thus, NK-cell activation is partially maintained in IL-18−/− mice, but abolished in IL-12−/− mice.
Functional relationship between IL-12 and IL-18 during activation of NK cells in visceral leishmaniasis
Earlier studies, which used naïve, non-infected mice, proposed that IL-18 either acts on NK cells in an IL-12-independent manner or that it increases the IL-12 responsiveness of NK cells 12,13. We therefore performed experiments to define how IL-18 might promote the IFN-γ production and the cytotoxicity of NK cells in visceral leishmaniasis. First, we searched for a possible impact of IL-18 on the expression of IL-12, which is essential for NK-cell activation in L. infantum-infected mice 10. We obtained no evidence that IL-18 regulates the induction of IL-12 by L. infantum in splenic DC in vivo (Fig. 4C) or in BM-derived DC or macrophages in vitro (Supporting Information Fig. 3), thus excluding the possibility that IL-18 functions via increasing the availability of IL-12. Second, we observed that the surface expression of the IL-12Rβ1-chain and the mRNA expression of IL-12Rβ2-chain of NK cells from WT and IL-18−/− mice were comparable (Fig. 4D and Supporting Information Fig. 4), which argues for an IL-12R-independent effect of IL-18. Third, we tested whether IL-18 might prime NK cells from L. infantum-infected mice for a response to IL-12 as previously reported for naïve mice 13. For this purpose spleen cells were harvested from PBS-treated or L. infantum-infected WT or IL-18−/− mice 12 h p.i. and cultured in the presence or absence of IL-18 for 4 h, followed by a 15 h incubation with or without IL-12. Thereafter, the IFN-γ production of NK cells was measured. Confirming earlier results 13, the in vitro induction of IFN-γ in naïve NK cells by IL-12 required prior in vivo priming by IL-18. In NK cells from IL-18−/− mice the IFN-γ expression could be restored by in vitro pre-stimulation with IL-18, which further augmented the IFN-γ release by NK cells of both mouse strains. Identical results were obtained with NK cells from infected mice (Fig. 4E). These ex vivo data suggest that priming by IL-18 is required to achieve full IL-12-mediated NK-cell activity during the early phase of L. infantum infection. However, it is important to emphasize that the induction of NK cell IFN-γ secretion and NK-cell cytotoxicity in vivo were only partially blocked in IL-18−/− mice, whereas both NK-cell functions were almost completely abolished in the absence of IL-12 (Fig. 4A and B). In addition, the production of IFN-γ by NK cells from IL-18−/− mice was eliminated when IL-12 was neutralized (Fig. 3B). Finally, we investigated whether IL-18 regulates FasL, perforin and/or gzmB expression. Although L. infantum infection caused an increase of the percentages of FasL+ NK cells in the spleen of both WT and IL-18−/− mice, the numbers of FasL+ NK cells were significantly lower in the case of IL-18−/− mice (Fig. 4F). In contrast, a deficiency of IL-18 did not affect the expression of gzmB or perforin mRNA and protein in NK cells (Fig. 4F and Supporting Information Fig. 5). In IL-12−/− mice none of these cytotoxic effector molecules was induced upon infection (Fig. 4F).
Together, these data support the notion that IL-18 contributes to the IL-12-mediated NK cell IFN-γ response by increasing the IL-12 responsiveness of NK cells. In addition, the results clearly illustrate that IL-12 also exerts IL-18-independent NK-cell-stimulatory effects.
Discussion
We previously reported an essential role of mDC-derived IL-12 for NK-cell activation in visceral leishmaniasis, whereas IFN-α/β was largely dispensable 10. In the present study we provide novel insights into the cytokine signals and mechanisms that underlie the process of NK-cell activation during L. infantum infection. First, our results demonstrate that the IL-12-dependent stimulation of NK cell IFN-γ production and cytotoxicity did not require the activity of IL-15 as shown by the analysis of residual NK cells in IL-15−/− mice or the transfer of WT NK cells into L. infantum-infected IL-15−/− mice. Only the modest enhancement of NK-cell proliferation in L. infantum-infected mice was mediated by IL-15. Second, even in the strict absence of IL-15 no compensatory IL-12-independent mechanism for the activation of NK-cell effector functions emerged. Third, IL-18 helped to trigger NK-cell effector functions in L. infantum infection, but comparing the NK-cell response in WT, IL-12−/− and IL-18−/− mice it became readily apparent that IL-18 played a contributory, but not an essential role. Finally, in vivo and ex vivo experiments demonstrated that IL-18 facilitated the response of NK cells to IL-12, but also proved that IL-12 is capable to stimulate NK cells in the absence of IL-18. There was clearly a differential regulation of FasL, perforin and gzmB in IL-18−/− versus IL-12−/− mice.
The IL-18-independent effect of IL-12 on NK-cell activation in visceral leishmaniasis might result from an increased nuclear translocation of STAT4 and/or NF-κB as previously seen in NK cells present in unseparated mouse macrophage populations 36,37. The enhancement of NK-cell cytotoxicity by IL-18 in L. infantum-infected mice is most likely due to the observed increased FasL-expression, which was shown to account for the antitumor effects of a treatment with IL-18 38,39.
Our results contrast with earlier findings obtained in other infection models using viral, bacterial or fungal pathogens, where IFN-α/β and/or IL-15 enhanced the activation of NK cells or were critical for NK cell IFN-γ production and/or for NK-cell cytotoxicity 1,9,22,25,26. With respect to the regulation of the expression of NK-cell cytotoxic granule proteins Fehniger et al. identified IL-2 and IL-15 as cytokines that were most potent in switching on the translation of perforin and gzmB in mouse NK cells in vitro, whereas IL-12 showed only a mild effect 40. These data differ markedly from our in vivo observations in visceral leishmaniasis, where we noted a strictly IL-12-dependent, but IL-15-independent increase of perforin and gzmB protein in NK cells (Figs. 1D and 4F). In accordance with earlier in vitro studies 40 our data provide evidence that perforin and gzmB are regulated by a post-transcriptional mechanism also in vivo, because up-regulation of both molecules was only visible on the protein level, but not on the mRNA level (Fig. 4F and Supporting Information Fig. 5).
There is evidence that an IL-15-independent activation of NK cells might also occur during other protozoan infections. Although the necessary in vivo experiments have not yet been reported, at least in vitro splenic DC from Plasmodium chabaudi-infected IL-15−/− mice were as potent as DC from WT mice to activate partially purified NK cells for the expression of IFN-γ 41. Even during certain viral infections IL-15 might be dispensable for NK-cell activation. During the preparation of our manuscript Sun et al. published that an infection with mouse CMV (MCMV) elicits a massive expansion of NK cells (72-fold within 7 days) in IL-15Rα-deficient mice and that these NK cells can be activated in vitro for the expression of IFN-γ and target cell cytotoxicity 42. As shown in the present study, L. infantum also caused a proliferation of NK cells (Fig. 2A), but unlike to the MCMV model it required IL-15 and did not lead to a measurable expansion and accumulation of NK cells in the spleen (Fig. 2B). This is most likely due to several circumstances. First, the NK-cell proliferation induced by L. infantum was quite weak. Only approximately 2.5% of all NK cells incorporated BrdU during the final 2 h of a 24 h infection period, whereas in the MCMV model a vigorous NK-cell proliferation with 21% BrdU+ NK cells was reported at day 1.5 of infection 43. Second, the modest NK-cell proliferation conveyed by IL-15 might reflect the less than twofold upregulation of the expression of IL-15Rα on mDC at day 1 of Leishmania infection (Supporting Information Fig. 1). Third, in the viral model the proliferation of the NK cells in the absence of IL-15 (or IFN-α/β) was maintained by the activating NK-cell receptor Ly49H, which is directly targeted by a single, highly stimulatory viral glycoprotein m157, and by IL-12 42,43. Although surface lipophosphoglycan purified from L. major was reported to activate human NK cells in vitro 44, we obtained no evidence for a direct activation of NK cells by L. infantum parasites 10. In fact, recent data suggest that Leishmania parasites might even express surface molecules that directly suppress NK-cell proliferation 45.
In conclusion, our analysis of the early NK-cell effector response in visceral leishmaniasis revealed differential functions for the cytokines IL-12, IL-15 and IL-18. In contrast to other models, IL-15 was required for NK-cell proliferation, but not for the IL-12-dependent NK cell IFN-γ production and cytotoxicity. We propose that the NK-cell effector response elicited by different pathogens in vivo requires distinct sets of cytokine signals and does not converge on a single or dominant NK-cell activation pathway.
Materials and methods
Mice, parasites and infection
Female C57BL/6 mice were obtained from Charles River (Sulzfeld, Germany). IL-12p35−/− 46, IL-18−/− 35 (both on a C57BL/6 background) and B6.SJL-PtprcaPepcb/BoyJ (CD45.1+) mice were from the Jackson Laboratories (Bar Harbor, ME, USA). IL-15−/− mice 31 (C57BL/6 background) were purchased from Taconic (Germantown, NY, USA). All mice were housed under specific pathogen-free conditions and used at the age of 6–16 wk.
The origin, propagation and preparation of L. infantum promastigotes (strain MHOM/00/98/LUB1) were described before 10.
Mice were injected into the retro-orbital plexus with 300 μL PBS or 1×107 stationary-phase L. infantum promastigotes in 300 μL PBS. The animal experiments were approved by the governmental animal welfare committee.
Generation, purification and stimulation of DC and macrophages
Preparation of splenic mDC, generation of BMMΦ and BM-mDC and cultivation of the cells was performed as described 10,37. For stimulation CpG ODN 1668 (1 μM, Thermo Electron, Ulm, Germany) and L. infantum promastigotes (parasite:cell ratio (MOI) indicated in the text) were used.
Flow cytometry analysis and intracellular cytokine staining
For surface phenotyping and cell sorting the following fluorochrome (FITC-, PE-, PerCP or APC)-labeled or biotinylated mAb were used (all from BD Biosciences, Heidelberg, Germany, unless otherwise stated): anti-CD11b (M1/70), anti-CD11c (HL3), anti-I-A/I-E (M5/114.15.2), anti-F4/80 (CI:A3-1, AbD Serotec, Düsseldorf, Germany), anti-IL-15Rα (BAF551, R&D Systems, Wiesbaden, Germany), anti-NK1.1 (PK136), anti-NKp46 (29A1.4; eBioscience, Hatfield, UK), anti-CD45.1 (A20), anti-CD3 (145–2C11), anti-IL12Rβ1 chain (114) and anti-FasL (MFL3). The specificity of the stainings was verified by the use of isotype control mAb. PI was included at 1 μg/mL in the final wash to detect dead cells.
The intracellular cytokine staining of IFN-γ in splenic NK cells or of IL-12p40/70 in splenic CD11bintCD11c+ cells was performed after a 4 h in vitro restimulation of spleen cells in medium alone or with YAC-1 cells in the presence of brefeldin A 10. In some experiments splenocytes were restimulated in vitro with rmIL-18 (10 ng/mL, R&D Systems) for 4 h, washed three times with PBS and stimulated with rmIL-12 (1 ng/mL, R&D Systems) for further 15 h before analysis of intracellular IFN-γ expression in NK cells 10. The intracellular expression of gzmB and perforin in splenic NK cells was measured by anti-gzmB (16G6, eBioscience) and anti-perforin (eBioOMAK, eBioscience) directly ex vivo. All FACS analyses were run on a FACS Calibur® (BD Biosciences) using the Cell Quest Pro® software.
NK-cell proliferation in vivo
To investigate cell proliferation in vivo the thymidine analog BrdU (1 mg/mouse) was injected i.p. into L. infantum-infected, IL-15/sIL-15R-Fc- or poly(I:C)-treated or control mice 22 h p.i., respectively. Two hours later the number of cells that incorporated BrdU into the DNA during proliferation was determined via the APC BrdU Flow Kit (BD Biosciences) according to the manufacturer's instructions.
NK-cell cytotoxicity assay
After determination of the percentage of NK1.1+CD3− or NKp46+CD3− NK cells within whole spleen cells, the NK-cell cytotoxicity against YAC-1 tumor cells was determined in a standard chromium-51 release assay 10.
NK-cell transfer and in vivo treatments
For cell transfer experiments splenic NK cells of CD45.1+ C57BL/6 mice were purified by MACS technology using anti-DX5 MicroBeads (Miltenyi, Bergisch-Gladbach, Germany) and by subsequent MoFlo® sorting gating on NK1.1+CD3− cells (purity ≥99%). Purified NK cells were injected i.v. For activation of NK cells in vivo, mice received 50 μg poly(I:C) (Invivogen, Toulouse, France) i.p. For neutralization of IL-12 mice were injected with 100 μg rat anti-IL-12p40 mAb (C17.8, eBioscience) or control rat IgG (The Jackson Laboratory) 30 min before i.v. infection with L. infantum. As positive control for the NK-cell proliferation assay mice were injected i.p. with pre-complexed 0.5 μg/mouse huIL-15 and 3 μg/mouse sIL-15Rα-Fc (both R&D Systems) as described 34.
mRNA expression analysis
Total RNA of homogenized tissue or of purified cells was extracted using the TRIZOL reagent (Invitrogen, Karlsruhe, Germany) or the RNeasy Micro Kit (Qiagen, Hilden, Germany) and was reverse transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems, Darmstadt, Germany). To assess the amount of mRNA expression levels we exactly followed our previously described protocol 30. The following assays (Applied Biosystems) were used: murine hypoxanthine guanine phosphoribosyl transferase 1 (mHPRT-1) (Mm00446968_m1), mIFN-γ (Mm00801778_m1), perforin (Mm00812512_m1), gzmB (Mm00442834_m1) and IL-12Rβ2 (Mm00434200_m1).
Statistical analysis
The non-parametric Mann–Whitney test was performed where indicated and p values are given.
Acknowledgments
We thank Uwe Appelt (Sorting Core Facility, Erlangen, Germany) for cell sorting and are grateful to the members of the lab for helpful discussions. This study was supported by grants from the Deutsche Forschungsgemeinschaft to C.B. and U.S. (DFG Bo996/3-3; SFB 643 project A6) and by a scholarship from the Schering Foundation to S.H.
Glossary
Abbreviations
- gzmB
granzyme B
- MCMV
mouse CMV
- mDC
myeloid DC
- p.i.
post infection
Conflict of interest:
The authors declare no financial or commercial conflict of interest.
Supplemental material
References
- 1.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 2.Diefenbach A, Raulet DH. The innate immune response to tumors and its role in the induction of T-cell immunity. Immunol. Rev. 2002;188:9–21. doi: 10.1034/j.1600-065x.2002.18802.x. [DOI] [PubMed] [Google Scholar]
- 3.Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-γ for Th1 priming. Nat. Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 4.Yokoyama WM. Mistaken notions about natural killer cells. Nat. Immunol. 2008;9:481–485. doi: 10.1038/ni1583. [DOI] [PubMed] [Google Scholar]
- 5.Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity. 2008;29:295–305. doi: 10.1016/j.immuni.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Della Chiesa M, Sivori S, Castriconi R, Marcenaro E, Moretta A. Pathogen-induced private conversations between natural killer and dendritic cells. Trends Microbiol. 2005;2005:128–136. doi: 10.1016/j.tim.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Newman KC, Riley EM. Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nat. Rev. Immunol. 2007;7:279–291. doi: 10.1038/nri2057. [DOI] [PubMed] [Google Scholar]
- 9.Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin-15. Immunity. 2007;26:1–15. doi: 10.1016/j.immuni.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, Fischer JA, et al. NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J. Exp. Med. 2007;204:893–906. doi: 10.1084/jem.20061293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sporri R, Joller N, Hilbi H, Oxenius A. A novel role for neutrophils as critical activators of NK cells. J. Immunol. 2008;181:7121–7130. doi: 10.4049/jimmunol.181.10.7121. [DOI] [PubMed] [Google Scholar]
- 12.Hyodo Y, Matsui K, Hayashi N, Tsutsui H, Kashiwamura S, Yamauchi H, Hiroishi K, et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999;162:1662–1668. [PubMed] [Google Scholar]
- 13.Chaix J, Tessmer MS, Hoebe K, Fuseri N, Ryffel B, Dalod M, Alexopoulou L, et al. Cutting edge: Priming of NK cells by IL-18. J. Immunol. 2008;181:1627–1631. doi: 10.4049/jimmunol.181.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koka R, Burkett P, Chien M, Chai S, Boone DL, Ma A. Cutting edge: murine dendritic cells require IL-15R alpha to prime NK cells. J. Immunol. 2004;173:3594–3598. doi: 10.4049/jimmunol.173.6.3594. [DOI] [PubMed] [Google Scholar]
- 15.Mattei F, Schiavoni G, Belardelli F, Tough DF. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 2001;167:1179–1187. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 16.Mortier E, Woo T, Advincula R, Gozalo S, Ma A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J. Exp. Med. 2008;205:1213–1225. doi: 10.1084/jem.20071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huntington ND, Legrand N, Alves NL, Jaron B, Weijer K, Plet A, Corcuff E, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009;206:25–34. doi: 10.1084/jem.20082013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.French AR, Holroyd EB, Yang L, Kim S, Yokoyama WM. IL-18 acts synergistically with IL-15 in stimulating natural killer cell proliferation. Cytokine. 2006;35:229–234. doi: 10.1016/j.cyto.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 19.D'Agostino P, Milano S, Arcoleo F, Di Bella G, La Rosa M, Ferlazzo V, Caruso R, et al. Interleukin-15, as interferon-gamma, induces the killing of Leishmania infantum in phorbol-myristate-acetate-activated macrophages increasing interleukin-12. Scand. J. Immunol. 2004;60:609–614. doi: 10.1111/j.0300-9475.2004.01522.x. [DOI] [PubMed] [Google Scholar]
- 20.Kuwajima S, Sato T, Ishida K, Tada H, Tezuka H, Ohteki T. Interleukin 15-dependent crosstalk between conventional and plasmacytoid dendritic cells is essential for CpG-induced immune activation. Nat. Immunol. 2006;7:740–746. doi: 10.1038/ni1348. [DOI] [PubMed] [Google Scholar]
- 21.Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu. Rev. Immunol. 1999;17:9–19. doi: 10.1146/annurev.immunol.17.1.19. [DOI] [PubMed] [Google Scholar]
- 22.Hirose K, Nishimura H, Matsuguchi T, Yoshikai Y. Endogenous IL-15 might be responsible for early protection by natural killer cells against infection with an avirulent strain of Salmonella choleraesuis in mice. J. Leukoc. Biol. 1999;66:382–390. doi: 10.1002/jlb.66.3.382. [DOI] [PubMed] [Google Scholar]
- 23.Ahmad A, Sharif-Askari E, Fawaz L, Menezes J. Innate immune response of the human host to exposure with herpes simplex virus type 1: in vitro control of the virus infection by enhanced natural killer activity via interleukin-15 induction. J. Virol. 2000;74:7196–7203. doi: 10.1128/jvi.74.16.7196-7203.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen KB, Salazar-Mather TP, Dalod MY, van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, et al. Coordinated and distinct roles for IFN-α/β, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002;169:4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- 25.Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, Suzuki T, et al. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J. Immunol. 2003;171:5423–5429. doi: 10.4049/jimmunol.171.10.5423. [DOI] [PubMed] [Google Scholar]
- 26.Tran P, Ahmad R, Xu J, Ahmad A, Menezes J. Host's innate immune response to fungal and bacterial agents in vitro: up-regulation of interleukin-15 gene expression resulting in enhanced natural killer cell activity. Immunology. 2003;109:263–270. doi: 10.1046/j.1365-2567.2003.01659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper MA, Bush JE, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, Aguila HL, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100:3633–3638. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- 28.Burkett PR, Koka R, Chien M, Chai S, Boone DL, Ma A. Coordinate expression and trans presentation of interleukin (IL)-15Ralpha and IL-15 supports natural killer cell and memory CD8+T cell homeostasis. J. Exp. Med. 2004;200:825–834. doi: 10.1084/jem.20041389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liese J, Schleicher U, Bogdan C. The innate immune response against Leishmania parasites. Immunobiology. 2008;213:377–387. doi: 10.1016/j.imbio.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Liese J, Schleicher U, Bogdan C. TLR9 signalling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur. J. Immunol. 2007;37:3424–3434. doi: 10.1002/eji.200737182. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 2000;191:771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, Münz C. Human dendritic cells activate resting natural killer cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med. 2002;195:343–351. doi: 10.1084/jem.20011149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Umemura M, Nishimura H, Hirose K, Matsuguchi T, Yoshikai Y. Overexpression of IL-15 in vivo enhances protection against Mycobacterium bovis bacillus Calmette-Guerin infection via augmentation of NK and T cytotoxic 1 responses. J. Immunol. 2001;167:946–956. doi: 10.4049/jimmunol.167.2.946. [DOI] [PubMed] [Google Scholar]
- 34.Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, Surh CD, Sprent J. Converting IL-15 to a superagonist by binding to soluble IL-15Rα. Proc. Natl. Acad. Sci. USA. 2006;103:9166–9171. doi: 10.1073/pnas.0600240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 36.Schindler H, Lutz MB, Röllinghoff M, Bogdan C. The production of IFN-γ by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J. Immunol. 2001;166:3075–3082. doi: 10.4049/jimmunol.166.5.3075. [DOI] [PubMed] [Google Scholar]
- 37.Schleicher U, Hesse A, Bogdan C. Minute numbers of contaminant CD8+ T cells or CD11b+CD11c+ NK cells are the source of IFN-g in IL-12/IL-18-stimulated mouse macrophage populations. Blood. 2005;105:1319–1328. doi: 10.1182/blood-2004-05-1749. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto W, Osaki T, Okamura H, Robbins PD, Kurimoto M, Nagata S, Lotze MT, et al. Differential antitumor effects of administration of recombinant IL-18 or recombinant IL-12 are mediated primarily by Fas-Fas ligand- and perforin-induced tumor apoptosis, respectively. J. Immunol. 1999;163:583–589. [PubMed] [Google Scholar]
- 39.Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, Kaneda K. IFN-gamma-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol. 1996;157:3967–3973. [PubMed] [Google Scholar]
- 40.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26:798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 41.Ing R, Stevenson MM. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun. 2009;77:770–782. doi: 10.1128/IAI.00994-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun JC, Ma A, Lanier LL. IL-15-Independent NK cell response to mouse cytomegalovirus infection. J. Immunol. 2009;183:2911–2914. doi: 10.4049/jimmunol.0901872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geurs TL, Zhao YM, Hill EB, French AR. Ly49H engagement compensates for the absence of type I interferon signaling in stimulating NK cell proliferation during murine cytomegalovirus infection. J. Immunol. 2009;183:5830–5836. doi: 10.4049/jimmunol.0901520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Gutierrez-Kobeh L, Ruiz A, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like-receptor-2. Mol. Biochem. Parasitol. 2003;130:65–74. doi: 10.1016/s0166-6851(03)00160-9. [DOI] [PubMed] [Google Scholar]
- 45.Lieke T, Nylen S, Eidsmo L, McMaster WR, Mohammadi AM, Khamesipour A, Berg L, et al. Leishmania surface protein gp63 binds directly to human natural killer cells and inhibits proliferation. Clin. Exp. Immunol. 2008;153:221–230. doi: 10.1111/j.1365-2249.2008.03687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, et al. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur. J. Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.