

Abstract
Double-stranded (ds) DNA, DNA- or RNA-associated nucleoproteins are the primary autoimmune targets in SLE, yet their relative inability to trigger similar autoimmune responses in experimental animals has fascinated scientists for decades. While many cellular proteins bind non-specifically negatively charged nucleic acids, it was discovered only recently that several intracellular proteins are involved directly in innate recognition of exogenous DNA or RNA, or cytosol-residing DNA or RNA viruses. Thus, endosomal Toll-like receptors (TLR) mediate responses to double-stranded RNA (TLR-3), single-stranded RNA (TLR-7/8) or unmethylated bacterial cytosine (phosphodiester) guanine (CpG)-DNA (TLR-9), while DNA-dependent activator of IRFs/Z-DNA binding protein 1 (DAI/ZBP1), haematopoietic IFN-inducible nuclear protein-200 (p202), absent in melanoma 2 (AIM2), RNA polymerase III, retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) mediate responses to cytosolic dsDNA or dsRNA, respectively. TLR-induced responses are more robust than those induced by cytosolic DNA- or RNA- sensors, the later usually being limited to interferon regulatory factor 3 (IRF3)-dependent type I interferon (IFN) induction and nuclear factor (NF)-κB activation. Interestingly, AIM2 is not capable of inducing type I IFN, but rather plays a role in caspase I activation. DNA- or RNA-like synthetic inhibitory oligonucleotides (INH-ODN) have been developed that antagonize TLR-7- and/or TLR-9-induced activation in autoimmune B cells and in type I IFN-producing dendritic cells at low nanomolar concentrations. It is not known whether these INH-ODNs have any agonistic or antagonistic effects on cytosolic DNA or RNA sensors. While this remains to be determined in the future, in vivo studies have already shown their potential for preventing spontaneous lupus in various animal models of lupus. Several groups are exploring the possibility of translating these INH-ODNs into human therapeutics for treating SLE and bacterial DNA-induced sepsis.
Keywords: HIN-200, HMGB1, systemic lupus erythematosus, Toll-like receptors 7 and 9
Overview
Genetic, epigenetic, gender-related and environmental factors are believed to contribute to the pathogenesis of autoimmunity in systemic lupus erythematosus (SLE). Defective clearance of apoptotic cell debris [1] with release of nucleosomes and ‘alarmins’[e.g. high mobility group box 1 (HMGB1)], increased bioavailability of type I interferon (IFN), evidence of up-regulated IFN-inducible genes along with multiple autoantibodies targeting DNA or RNA-associated nucleoproteins are immunological hallmarks of SLE.
SLE genetics
It is broadly accepted that SLE is a complex genetic disease with multiple genes contributing to the disease phenotype [2]. The concordance rate among monozygotic twins is ∼30%, which is 10 times higher than the rate among dizygotic twins (∼3%) [3]. Recently established risk factors for human SLE include allelic variants of genes with known immune function, such as human leucocyte antigen (HLA), interferon regulatory factor 5 (IRF5), signal transducer and activators of transcription 4 (STAT4), Fc gamma receptor 2a gene (FCGR2A)/3A, protein tyrosine phosphatase, non-receptor type 22 (PTPN22), integrin alpha M (ITGAM), CTD-binding SR-like protein rA9 (KIAA1542), PX domain containing serine/threonine kinase (PXK), B cell scaffold protein with ankyrin repeats (BANK1), programmed cell death 1 (PDCD1), methyl cytosine (phosphodiester) guanine (CpG) binding protein 2 (MECP2) and tumour necrosis factor (ligand) superfamily, member 4 (TNFSF4) [4–6].
Epigenetic changes
The two most important epigenetic changes that can affect the gene functioning are histone modifications and DNA methylation. Environmentally driven epigenetic changes may explain partial discordance for SLE in monozygotic twins [7].
The role for abnormal DNA methylation in lupus has long been suspected. For example, lymphocytes from SLE patients exhibit globally hypomethylated DNA pattern [8,9]. Interestingly, many lupus-promoting drugs work through DNA demethylation (e.g. procainamide and hydralazine [10]).
A high-throughput approach was used to study monozygotic twins discordant for SLE, rheumatoid arthritis (RA) or dermatomyositis [7]. Only those discordant for SLE showed widespread changes in the DNA methylation status of a number of genes, particularly in those associated with immune function [e.g. interferon (IFN)-γR2, matrix metalloprotease 14 (MMP), lipocalin 2 (LCN2), colony-stimulating factor 3 receptor (CSF3R), platelet/endothelial cell adhesion molecule 1 (PECAM1), CD9 and absent in melanoma 2 (AIM-2)].
Infections and lupus: beyond molecular mimicry
It is well documented that infections coincide frequently with the occurrence of autoimmune diseases. The concept of molecular mimicry is one way of explaining this phenomenon because of the structural similarity between invading microbes and self-antigens [11]. A classical example of molecular mimicry is Guillain–Barré syndrome following infection with Campylobacter jejuni (reviewed in [11]. In relation to the lupus, Judith James found that autoantibodies against an epitope in 60 kD Ro antigen may cross-react with the peptide from the latent viral Epstein–Barr virus nuclear antigen-1 [12]. When animals are immunized with these cross-reactive epitopes they progressively develop autoantibodies which bind multiple epitopes of Ro autoantigens, a phenomenon known as epitope spreading.
Alternatively, viral infections (similar to ultraviolet lights) may increase the supply of lupus autoantigens by inducing cellular injury and promoting apoptotic cell death. Slow disposal of apoptotic cells by mononuclear phagocytes may result in spontaneous release of lupus autoantigens from apoptotic blebs, initiating autoantibody production in genetically susceptible hosts.
However, the role of infectious agents may extend beyond promoting cell death. Certain microbial products may act as immune adjuvants inducing bystander activation of the innate immune system via Toll-like receptors (TLRs). Interestingly, the most common autoimmune targets in SLE, DNA and/or RNA-associated nucleoproteins, either alone or combined with autoantibodies, may trigger B cell activation through endosomal TLRs. Thus, common autoantigens in systemic autoimmune diseases such as chromatin, nucleosomes, Ku, topoisomerases, Sm/snRNP, ribosomes and Ro/La are not only passive targets of autoimmunity, but active triggers of innate immunity. In concordance with this hypothesis, injection of synthetic ligands for TLR-3, -7 and -9 could worsen spontaneous animal lupus [13–17].
Role for nucleic acid-sensing endosomal TLRs in lupus
Several lines of evidence support a role for TLRs in SLE. Of 13 known TLRs, endosomal single-stranded RNA- and hypomethylated DNA-sensing innate receptors TLR-7 and -9, respectively [18–21], but not double-stranded RNA-sensing TLR-3 [22,23], have been implicated in the pathogenesis of SLE. These intracellular TLRs are responsible for activation of type I IFN-producing dendritic cells and autoreactive B cells upon recognition of immune complexes containing endogenous nucleic acids (RNA or DNA).
TLR-7 and -9 in lupus: in vitro studies
Transgenic AM14 B cells express a rheumatoid factor-like B cell receptor for antigen (BCR) which recognizes autologous IgG2a of the particular allotype. Because of the low affinity on a non-autoimmune background, AM14 B cells are not tolerized and can be found within the conventional mature B cell pool [24]. In vitro, AM14 B cells can be induced to proliferate upon exposure to immune complexes containing IgG2a antibodies directed against DNA or DNA-associated nuclear proteins [25], but not against unrelated haptens or proteins [26,27]. This proliferation depends heavily upon the presence of CpG motifs within the DNA [28]. RNA-containing immune complexes can also activate AM14 B cells, especially following priming with type I IFNs [29]. In these examples, BCR-mediated capture and endosomal delivery of nucleic-acid containing immune complexes [30] induces B cell proliferation dependent upon TLR-9 or TLR-7, respectively [26,29]. In anergic B cells, however, effective BCR/TLR co-localization fails to happen, blunting responses to self-DNA [31].
In dendritic cells (DCs), stimulation with nucleic acid-containing immune complexes induces IFN-α and proinflammatory cytokines. This requires a FcγR-mediated delivery of TLR ligands to TLR-7- or -9-expressing intracellular compartments [32,33].
TLR-7 and -9: in vivo studies
In Murphy–Roths large (MRL)-Faslpr/lpr mice, TLR-7 deficiency causes a significant drop in secretion of autoantibodies reactive with RNA-associated autoantigens, but not against chromatin. These animals also have a milder kidney disease [34]. A similar effect of TLR-7-deficiency was observed in pristane-induced SLE [35,36], and in glomerular injury induced with syngeneic late apoptotic cells [37].
Male BXSB mice develop a lupus-like disease because of the Y-linked autoimmune accelerator (Yaa). These mice represent a unique example how gene copy number variation may result in disease induction [38]. Noticeably, the Yaa phenotype is due to the translocation of X-chromosomal region that contains the TLR-7 gene (and 16 additional genes) to the Y chromosome resulting in TLR-7 gene duplication in males [39,40]. Deletion of the endogenous TLR-7 decreases autoantibody levels significantly and improves kidney disease in the Yaa strain [38,41].
B6 FcγRIIB−/− mice develop an SLE-like disease where secretion of IgG2a and IgG2b antibodies requires a TLR-9 [42]. Lupus-like disease in these mice is accelerated upon acquiring the Yaa accelerator, through a mechanism that absolutely requires IRF5 but is independent of the type I IFN secretion [43]. Interestingly, the autoantibody profile in these mice changes from predominantly DNA-biased to RNA-related [44].
Contrasting with the lack of TLR-7, TLR-9-deficiency in MRL-Faslpr/lpr strain results in fewer anti-chromatin/anti-dsDNA antibodies, but causes exacerbation of the clinical disease [34,45]. A similar ‘protective’ role of TLR-9 was observed in other models of animal lupus [46]. While the logical explanation for this is lacking, one of the possibilities is that TLR-9-dependent anti-chromatin antibodies may have a role in apoptotic cell clearance. Alternatively, TLR-9 (but not TLR-7) signalling in autoreactive B cells and DCs may induce cytokines with regulatory properties [e.g. interleukin (IL)-10][47,48] or may promote generation of forkhead box P3 (FoxP3)+ T cells. Interestingly, a TLR-7 by itself may play a role in acceleration of lupus in TLR-9-deficient mice [49]. While the deficiency of double-stranded RNA receptor TLR-3 does not affect the progression of spontaneous MRL-Faslpr/lpr disease, injection of viral dsRNA ligands aggravates lupus nephritis, presumably via TLR-3-expressing mesangial cells [50].
UNC93b1 shuttles TLR-3, -7 and -9 from endoplasmic reticulum (ER) to endosomes
Another layer of evidence supporting intracellular TLRs in the pathogenesis of SLE comes from autoimmune mice lacking the UNC93b1 protein [51]. Unc93b1 is an ER protein that is involved in ligand-induced trafficking of TLR-3, -7 and -9 from the ER to the endosomes [52,53]. Under normal circumstances, Unc93b1 biases TLR responses towards DNA, not RNA sensing [54]. Amino acid D34 in UNC93b1 suppresses TLR-7-mediated responses and favours responsiveness to TLR-9 ligands. Conversely, D34A mutation renders DCs hyperresponsive to TLR-7 ligand, but hyporesponsive to TLR-9 ligand. This may help to explain heightened TLR-7 responsiveness in TLR-9−/− lupus mice. Lupus-prone mice lacking the UNC93b1 protein develop a less severe lupus than their UNC93b1-sufficient counterparts [51].
TLR-7 and -9 expression and polymorphism in SLE
Several studies have suggested that TLR-9 may play an important role in human SLE. Papadimitraki found increased renal expression of TLR-9 in 50% of patients with SLE nephritis, particularly in those with higher activity index [55]. Moreover, antigen-presenting cells from SLE patients showed higher percentage of TLR-9+ cells as well as higher expression of TLR-9 compared to controls. However, response to CpG-ODNs correlated with disease activity, independently of the TLR-9 expression [56]. Another study by Wu [57] in untreated SLE patients showed increased percentages of both T and B cells expressing TLR-9. In addition to TLR-9, increased expression of TLR-7 was also documented, and this correlated with IFN-α transcription [58]. An interesting clinical observation linked acquired TLR-7/9 signalling defect and resulting antibody deficiency with long-lasting clinical remission in a patient with known history of SLE [59].
Several studies have investigated polymorphisms in TLR-7 and TLR-9 genes. A study in Chinese patients with SLE [60] explored the role of SNP (rs352140) in exon 2 of the TLR-9. A mild association between the T allele and susceptibility to SLE was found in a dominant model. A study in a Spanish SLE cohort revealed no significant difference in allele and genotype distribution of TLR-5 (rs5744168) and TLR-7 (rs179008) [61].
Is there a role for non-TLR DNA- and RNA-sensors in the pathogenesis of human SLE?
Over recent years several non-TLR proteins capable of binding and initiating responses to intracellular DNA or RNA molecules have been discovered. Indeed, increasing evidence suggests that TLR-9 engagement by DNA vaccines is not vital for the induction of immune responses in vivo[62–65]. Self-DNA may be delivered to cytosol under certain experimental conditions leading to increased IFN-β production. For example, DNAse II-deficient macrophages digest self-DNA poorly from engulfed apoptotic cells, causing IFN-β production through the IRF3-dependent pathway [66,67].
A search for additional dsDNA-sensors has identified several candidate proteins, e.g. haematopoietic IFN-inducible nuclear protein (HIN)-p202, AIM-2, DNA-dependent activator of IRFs (DAI)/ZBP1, RNA polymerase III (Pol-III) and HMGB1. A similar search has identified RNA-helicases retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) as TLR-7-independent cytoplasmic RNA sensors. Herein, we provide a brief summary about these novel nucleic acid sensors, particularly in relation to their putative role in lupus pathogenesis (Table 1).
Table 1.
Nucleic acid sensors.
| Sensor | Ligand | Reference |
|---|---|---|
| TLR-9 | Hypomethylated CpG-DNA | [18] |
| DAI/ZBP1 | dsDNA | [82] |
| RNA-Pol III | dsDNA (AT-rich) | [101,102] |
| HIN-200 (p202) | dsDNA | [72] |
| AIM-2 | dsDNA | [73] |
| TLR-3 | dsRNA, poly I : C | [22] |
| TLR-7 | ssRNA, imiquimod, Loxoribine, R848 | [20] |
| TLR-8 | ssRNA, R848 | [20] |
| RIG-I | dsRNA, 3P-ssRNA, short poly I : C | [104] |
| MDA-5 | dsRNA, long poly I : C | [105] |
| LGP2 | dsRNA, 3P-ssRNA | [104] |
AIM-2, absent in melanoma 2; DAI, DNA-dependent activator of IRFs/Z-DNA binding protein 1; HIN, haematopoietic IFN-inducible nuclear protein; LGP2, RIG-I-like RNA-helicase; MDA, melanoma differentiation-associated gene 5; RIG, retinoic acid-inducible gene-I; TLR, Toll-like receptor.
IFI200 (HIN-200) family
At least two newly identified cytoplasmic DNA-binding receptors belong to the subset of the IFN-stimulated genes–IFI200 family. This family includes several structurally related proteins in mice and in humans (reviewed in [68,69]). IFI200 family members function by negatively regulating cell growth [70] and inhibiting lymphocyte apoptosis [71].
The HIN-200 family is a human homologue of murine Ifi200. It was shown recently that the HIN-200 family member, p202, binds stably transfected cytoplasmic dsDNA and works by inhibiting DNA-induced caspase (1 and 3) activation in macrophages [72]. Conversely, another HIN-200 protein, AIM2 (absent in melanoma 2, p210) was necessary for cytoplasmic dsDNA-induced caspase-1 and nuclear factor (NF)-κB activation and inflammasome formation (but not type I induction) [72,73]. Therefore, recognition of cytoplasmic dsDNA ligands by different members of the HIN-200 family may result in different biological outcomes.
In BL6 mice congenic for the Nba2 locus, Ifi202 gene has been identified as a candidate gene for SLE susceptibility [68]. These mice develop splenomegaly and produce high-titre immunoglobulin G (IgG) anti-nuclear antibodies. Polymorphism in the coding region of the Ifi202 genes in the NZB mice accounts for the increased level of this protein in B6.Nba2 mice. Interestingly, Ifi202a−/− mice do not have any phenotype [74], probably because of the redundancy between Ifi202a and b proteins. Their B cells respond normally to various stimuli in vitro and to immunization with T-dependent antigens in vivo[75]. As well as B6.Nba2 mice, P202 protein levels are also elevated in the MRL-Faslpr/lpr strain [76], and correlate well with the development of lupus in these mice [77]. The way in which p202 protein in MRL-Faslpr/lpr contributes to the lupus pathogenesis is difficult to understand, as IFN-α appears to play a protective role in this strain. In relation to the known gender bias in SLE, steady state levels of Ifi202 mRNA were found to be higher in splenic cells from female mice than in males.
Human MNDA, IFIX, IFI16 and AIM2 proteins are increased in leucocytes from SLE patients [78], and increased MNDA expression was found in glomeruli from SLE patients [79].
A possible interplay between the TLRs and Ifi200 has not been studied in sufficient detail. Circumstantial evidence suggests that Ifi202 may cooperate with TLRs in cell activation. For example, overexpression of Ifi202 enhances lipopolysaccharide (LPS)-induced activation in Abelson murine leukaemia virus-transformed macrophage cell line 264·7 macrophages, presumably via TLR-4-dependent activation [80]. It remains to be determined whether Ifi202 and TLR-9 synergize or have antagonistic effects upon each other.
Similar to other nucleic acid associated proteins, IFI200 members may also serve as lupus autoantigens [81]. Antibodies against the IFI16 were found in both SLE and in Sjögren's syndrome [69,81]. These antibodies could differentiate further between patients with limited cutaneous systemic sclerosis and those with the systemic form [69].
DAI/ZBP1 (DNA-dependent activator of IRFs/Z-DNA binding protein 1)
DAI is another cytoplasmic DNA sensor [82] capable of activating IRF3 and NF-κB, resulting in type I IFN-production. DAI interacts directly with dsDNA in vitro[83] and this interaction in turn enhances DAI association with IRF3 [82]. DAI-induced IRF3 phosphorylation is dependent on TANK-binding kinase 1 (TBK1) [84], while the NF-kB signalling depends on recruitment of RIP1 and either RIP2 or 3 (receptor interacting proteins) [85,86]. Once IRF is phosphorylated it undergoes dimerization and nuclear translocation promoting IFN-β secretion. In a positive feedback loop manner, IFN-β signals through JAK/STAT molecules inducing IRF7 and amplifying type I IFN secretion. However, DAI may not be the only cytosolic DNA sensor in human cells [87] and DAI-deficient mice still can make type I IFN in response to B form DNA [65]. The possible role of DAI in lupus has not been studied.
HMGB1 (high mobility group box 1)
HMGB1 is a non-histone, chromatin-associated nuclear protein that binds DNA, regulates transcription and functions as a proinflammatory cytokine when released outside the cell. By itself HMGB1 is only mildly proinflammatory, but it can synergize with other cellular activators including TLR ligands [88,89]. For example, TLR-9-dependent IL-6 secretion in response to type B(K) CpG-ODNs is enhanced by HMGB1 [90]. While HMGB1 binds preferentially complex forms of DNA, such as cruciform DNA [91] and class A(D) CpG-ODNs [92], it appears that HMGB1 binds DNA without sequence specificity [88,92]. HMGB1 is an excellent example of an alarmin: once released from necrotic (and possibly late apoptotic) cells it ‘alarms’ the immune system about the presence of tissue damage [93]. Extracellular HMGB1 may signal through TLR-2, TLR-4 or receptor for advanced glycation end-products (RAGE). HMGB1/DNA complexes bind RAGE (or some other receptor on B cells) [94] which in turn causes recruitment of TLR-9 and enhances type I IFN secretion from pDCs and proliferation of autoreactive B cells [92]. The cross-talk between HMGB1 and TLR-9 appears to be a unique example of a synergistic interaction between two DNA receptors, resulting in cellular activation. HMGB1 does not affect the uptake of CpG-DNA into TLR-9 expressing early endosomes, but accelerates the recruitment of TLR-9 from the ER to endosomes [92]. While HMGB1 binds tightly to chromatin and is retained within apoptotic cells [88], novel data suggest that late apoptotic cells may release some HMGB1 [95]. This happens only after late apoptotic cells undergo secondary necrosis, when the permeability barrier breaks down [96]. The complex of HMGB1 with nucleosomes is more immunogenic and proinflammatory than naked nucleosomes and triggers more vigorous cell activation [97]. Highly relevant to the lupus, HMGB1 is expressed in skin lesions from patients with cutaneous lupus [98], and high levels of both HMGB1 and anti-HMGB1 antibodies can be found in patients with SLE [97,99,100]. HMGB1-containing nucleosomes from apoptotic cells, but not those from living cells, could induce anti-dsDNA and anti-histone antibody responses [97]. They were also capable of activating dendritic cells and macrophages to express co-stimulatory molecules and to secrete IL-1, IL-6, IL-10 and tumour necrosis factor (TNF)-α[97].
RNA polymerase III [101,102]
The RIG-I dependent pathway plays a role in recognition of viral RNA (see below), and also mediates responses to cytosolic dsDNA. In a recent study, AT-rich dsDNA served as a template for RNA polymerase III and was transcribed into dsRNA with a 5′-triphosphate moiety. dsRNA in turn engaged RIG-I resulting in type I IFN production and NF-κB activation [101,102]. Pol-III functions normally to transcribe 5S rRNA, tRNA and other small non-coding RNA. This Pol-III-mediated conversion of transfected cytosolic poly (dA-dT) (but not other forms of dsDNA) into 5′-pppRNA was also seen by Chiu [102]. However, because of the apparent redundancy between cytosolic DNA sensors, dsDNA could induce IFN production in mouse cells lacking RIG-I [103].
RNA-sensors: RIG-I, MDA5 and LGP2
Viral RNA induced IFN-α production in cell types other than pDCs is not dependent on TLR expression, but rather requires RIG-I family cytoplasmic RNA-helicases. Two family members, RIG-I [104] and MDA5 [105], are recently identified RNA sensors that recognize distinct viral and synthetic RNA patterns leading to the production of type I IFN and proinflammatory cytokines via IRF3, IRF7 and NF-κB. Single-stranded 5′-triphosphate RNA is a well-characterized RIG-I and RIG-I-like RNA-helicase (LGP2) ligand [106], while dsRNA synthetic analogue poly I : C can activate either MDA5 or RIG-I depending on its length [107]. LGP2 binds dsRNA, but functions as a negative regulator of the RIG-I/MDA5 signalling pathway [104]. Transient exposure of prediseased MRL-Fas lpr/lpr mice to 3P-RNA aggravates lupus nephritis via IFN-signalling [108].
Evidence for the role of type I IFN system in SLE
One common feature of all nucleic acid sensing receptors (with the possible exception of AIM-2) is their ability to induce type I IFN secretion upon recognition of their cognate ligands. We will therefore summarize recent evidence that links type I IFNs with the pathogenesis of SLE (reviewed in [109,110]).
Treatment of patients with chronic viral infections and certain types of malignancies with IFN-α may result in SLE-like clinical manifestations in patients without prior history of SLE [111]. Increased serum concentrations of IFN-α can be found in human SLE [112] and IFN-α levels correlate with disease activity and severity in human SLE [112,113]. Higher IFN-α levels are found in earlier stages of SLE. Nevertheless, some SLE patients never develop high IFN-α levels, not even during disease flares.
Type I IFN-inducible gene expression signature was found in peripheral blood mononuclear cells in approximately 50% of adult SLE patients, particularly in those with more severe disease [2,114], and in almost all paediatric SLE patients with recent onset [115]. Gene expression profile of peripheral blood mononuclear cells from SLE patients was suggestive of an active type I IFN signalling [2,116]. However, an extended longitudinal study in 11 SLE patients failed to show any association between IFN response scores and changes in disease severity or in risk for SLE flares [117]. This suggests a very limited usefulness of IFN-inducible gene profiling as a biomarker for lupus.
IFN-α inducers
TLR-7 and -9 ligands are very strong inducers of type I IFNs both in vitro and in vivo. TLR-7- [118] and TLR-9-mediated [119] induction of early IFN-inducible genes in pDCs is dependent on p38-mitogen-activated protein kinase (MAPK) signalling, STAT1 phosphorylation and IRF7 translocation but is, interestingly, IFN-α/β-independent. DNA-containing immune complexes are capable of inducing dendritic cell activation and type I IFN secretion both in humans and in rodents. This activation is dependent on the presence of unmethylated CpG dinucleotides within the dsDNA [120].
In vivo treatment of NZB autoimmune mice with complex TLR-9 agonists induces abnormally high levels of serum IFN-α[17]. Treatment with IFN-α accelerates disease and induces early lethal lupus in New Zealand black/white (NZB/W)-F1 mice [121]. Several investigators have shown that deficiency of the type I IFN receptor may protect lupus-prone mice from developing a lupus-like disease [122,123].
One way in which IFN-α may contribute to the SLE pathogenesis could be via up-regulation of TLR7 [124] and TLR-9 [125]. Type I IFN may also promote B cell activation and plasma cell differentiation with isotype switching to complement-fixing IgG2a, IgG2b, IgG3 antibodies in mice. IFN-α from SLE patients could induce differentiation of monocytes into potent DC-like antigen-presenting cells which may then present antigens derived from apoptotic cells to T cells [126]. In T helper cells, type I IFN promotes TH1 differentiation. They can also increase cytotoxic T cell and NK cell activity along with increased IFN-γ production. Therefore, there are multiple ways how type I IFN may contribute to the lupus pathogenesis. Attempts to block this activity, either by using neutralizing monoclonal anti-IFN-α/β antibodies, soluble IFNAR-Fc constructs, or by blocking the activity of nucleic acid inducers at the level of TLRs may provide a better control of disease activity in human SLE. Indeed, a recent phase Ia clinical trial in patients with mild to moderate SLE with skin involvement showed that an anti-IFN-α monoclonal antibody (MEDI-545) could (partially) reverse the overexpression of type I IFN-inducible genes in peripheral blood and lesional skin [127]. However, specific data on safety were not provided. As adequate type I IFN response is critical for fighting viral infections, it could be speculated as to whether blocking type I IFN activity may result in serious immunodeficiency. This could be a problem in patients with SLE whom suffer from acute or chronic viral infections or were vaccinated recently with live attenuated viruses [128].
Selective TLR-7 and TLR-9 antagonists as potential lupus therapeutics
There is a growing interest for developing TLR-7- and/or TLR-9-specific antagonists as therapeutics for human SLE. Anti-malarial agents such as hydroxychloroquine work, in part, by blocking endosomal acidification and TLR-7/9-dependent signalling. Hydroxychloroquine reduces frequency and severity of lupus flares and has a favourable therapeutic effect on lupus arthritis, serositis and skin disease [129]. However, its use is limited by suboptimal efficacy and toxicity. Therefore, selective TLR-7- and/or TLR-9- specific antagonists may be more beneficial, assuming a broadened therapeutic safety window.
Pisetsky's group was the first to discover that synthetic inhibitory oligonucleotides (INH-ODN) containing poly-G sequences could block bacterial DNA-induced activation [130,131]. However, this effect required relatively high micromolar concentrations of INH-ODNs and was not specific for TLR-9, as these INH-ODNs could also block other signalling pathways [132].
Krieg's group noticed that certain CpG sequences, including methylated CpG-ODNs [133,134], have not only lost the ability to stimulate TLR-9-responsive cells, but acted as antagonists when added to bacterial DNA-stimulated cultures [134].
Our group discovered that at the signalling level, the earliest steps in NF-κB [135] and MAPK-activation were potently inhibited [136] suggesting a very proximal place of INH-ODN action (Fig. 1), possibly at the level of TLR-9 itself [137]. Detailed mapping studies in mouse B and non-B cells [138,139] determined the following rules for TLR-9-inhibition: (i) the CpG motif is not required for inhibition; (ii) a stretch of three consecutive G nucleotides is necessary for inhibition; (iii) the 5′ end of an INH-ODN is important for inhibition: CCT triplet is optimal. An INH-ODN containing multiple CCT repeats could inhibit TLR-7/9 activation [140]; (iv) the optimal distance between the 5′ CCT and the downstream GGG triplet is 3–5 nucleotides long; (v) the order of 5′CCT → GGG-3′ is critical, as ODNs with the reverse order are non-inhibitory; (vi) the spacing between the CCT and GGG triplets has a minor impact on INH-ODN activity and can accept multiple substitutions; (vii) the overall length of an INH-ODN has an impact on activity; and (viii) inhibition of the TLR-9-pathway does not require G-rich segments to form intrachain or interchain Hoogsten bonds between the adjacent Gs [141].
Fig. 1.
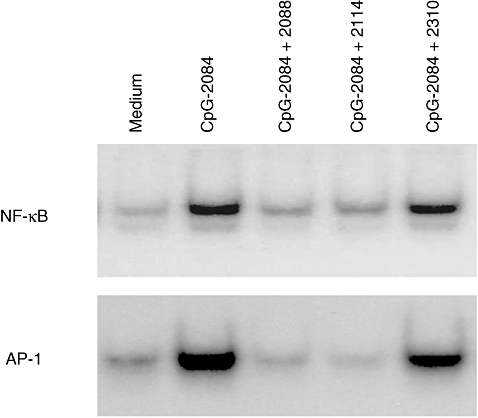
Toll-like receptor (TLR)-9 ligand cytosine (phosphodiester) guanine oligonucleotide (CpG-ODN 2084) (100 nM) induces rapid nuclear factor kappa-B (NF-κB) and activator protein-1 (AP-1) nuclear translocation as detected by electrophoretic mobility shift assay which can be inhibited by equimolar concentrations of inhibitory oligonucleotides (INH-ODNs) 2088 and 2114, but not with the control ODN 2310.
INH-ODNs are active in human peripheral blood B cells, B cell lines, pDCs [142,143] and in TLR-9-transfected HEK cells [144]. Our recent mapping studies showed that extending INH-ODNs for 4–5 bases at their 5′ end significantly enhanced their inhibitory activity in human cells. Similar to mouse studies, activity was not dependent on the ability of INH-ODN to self-aggregate [144]. TLR-9 bound (PO) INH-ODNs similarly to CpG-ODNs and the affinity for TLR-9 did not correlate with the biological activity (Ashman et al. submitted for publication). These results concur with the recent observation that sugar backbone 2-deoxyribose determines DNA recognition by TLR-9 [145]. Phosphorothioate-modified deoxyribose has much higher affinity for both TLR-7 and -9 compared to PO-deoxyribose, transforming these molecules into TLR-7 and TLR-9 antagonists [145]. Therefore, some other molecule, not TLR-9, must be responsible for sequence-specific recognition of INH-ODNs (unpublished data and [144]).
Concept of class R and class B INH-ODN
For our mapping studies we used INH-ODN-2114 ([141]; Table 2). This ODN is a very potent TLR-9-inhibitor in both human and mouse settings in vitro[138,139,141]. It also works in the MRL-Fas lpr/lpr strain of lupus in vivo[146]. INH-ODN-2114, and very similar ODNs developed by two other groups have no inhibitory activity on TLR-2, -3, -4 and -5 and BCR-induced stimulation when used at concentrations up to 1 µM [141,147,148].
Table 2.
Inhibitory oligonucleotides.
| No. | Sequence | Class | Effect in lupus | Reference |
|---|---|---|---|---|
| 2088 | TCCTGGCGGGGAAGT | B/G | Not tested | [141] |
| 2114 | TCCTGGAGGGGAAGT | B/G | ++ | [141] |
| 4024 | TCCTGGATGGGAAGT | B | Not tested | [138] |
| 4084F | CCTGGATGGGAA | B | Not tested | [150] |
| INH-1 | CCTGGATGGGAATTCCCATCCAGG | R | ++ | [150] |
| INH-4 | TTCCCATCCAGGCCTGGATGGGAA | R | Not tested | [150] |
| INH-13 | CTTACCGCTGCACCTGGATGGGAA | B | Not tested | [150] |
| INH-18 | CCTGGATGGGAACTTACCGCTGCA | B | −/+ | [150] |
| Poly-G | GGGGGGGGGGGGGGGGGGGG | G | Not tested | [130] |
| A151 | TTAGGGTTAGGGTTAGGGTTAGGG | G | ++ | [137] |
| GpG | TGACTGTGAAGGTTAGAGATGA | B | + | [170] |
| G-ODN | CTCCTATTGGGGGTTTCCTAT | B/G | Not tested | [148] |
| IRS-869 | TCCTGGAGGGGTTGT | B/G | Not tested | [143] |
| IRS-661 | TGCTTGCAAGCTTGCAAGCA | R/TLR7 | + | [147,180] |
| IRS-954 | TGCTCCTGGAGGGGTTGT | B/TLR7/9 | ++ | [147] |
| SAT05f | CCTCCTCCTCCTCCTCCTCCTCCT | B/TLR7/9 | Not tested | [140] |
| 6 | CTATCTG2-o-mrA2-o-mrCGTTCTCTGT | B/TLR7/9 | Not tested | [174] |
Because INH-ODN-2114, similar to poly-G ODNs [149], has four consecutive Gs and at higher concentrations can make G4-stacks, this ODN may have some non-specific effects on immune activation. In order to avoid any contribution from G4 aggregates we created INH-ODN 4024 (Table 2) [142]. This ODN contains canonical CCT and GGG triplets, but does not make G4-stacks and is as potent as INH-ODN 2114 for human and mouse TLR-9-expressing cells [139,142]. We further truncated INH-ODN-4024 to create a shortest active 12-mer INH-ODN 4084-F (Table 2) [150].
In order to understand the contribution from defined secondary structures other than G4 stacks, e.g. ability to make DNA duplexes or hairpins, we created 24 mer-ODNs in which the INH-ODN 4084F sequence was positioned either at the 5′ or the 3′ end, and was followed (or preceded) by 12 nucleotides complementary to the INH-ODN 4084F sequence, making a complete palindrome (INH-1, INH-4, Table 2) [150]. Based on their biological activity we named these new TLR-9-antagonists class R INH-ODNs (where ‘R’ stands for restricted activity [151]), as they showed similar inhibitory potency for TLR-9-activated IFN-α producing dendritic cells as their linear analogues (class B, broadly active, INH-18, INH-13, Table 2), but were between 10- and 30-fold less inhibitory in human and mouse B cells [150]. This difference in activity between classes R and B INH-ODNs in B cells could depend on the ability of these ODNs to reach different TLR-9-expressing compartments, e.g. early versus late endosomes [150,152–154]. In B cells, class R INH-ODNs, similar to mammalian DNA, have a restricted access to late endo/lysosomes. Because the BCR engagement allows B cells to respond to a wider range of TLR-9 ligands, including complex TLR-9-agonists [125,154–158], we hypothesized that the same principal may apply to class R TLR-9-antagonists. In order to test this hypothesis, we used autoreactive AM14 B cells as a model for BCR/TLR-9 cross-talk [24]. When AM14 B cells were stimulated with linear CpG-ODN ligands, similar to non-autoreactive B cells, class R INH-ODNs were still 10-fold less potent inhibitors compared to class B INH-ODNs [150]. However, when DNA-containing immune complexes were used for stimulation, the potency of class R INH-ODN increased and equalized that of class B INH-ODNs [150]. We wondered whether this selectivity could be advantageous for treating lupus. Indeed, studies in the MRL-Fas lpr/lpr strain showed that linear class B INH-18 was surprisingly less effective, while treatment with palindromic class R INH-1 improved survival, diminished renal pathology and restored the B cell phenotype (Figs 2 and 3) [150]. Furthermore, levels of anti-dsDNA and anti-Sm/RNP antibodies were reduced significantly in mice treated with INH-1. These results re-emphasized the fact that TLR-9 may have some protective effects in the MRL-Faslpr/lpr strain of lupus mice.
Fig. 2.
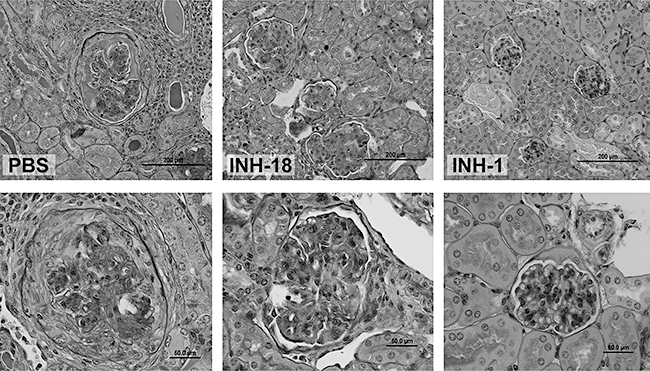
Superior efficacy of class R INH-1, compared to class B inhibitor-18 (INH-18) in preventing renal damage in MRL-Faslpr/lpr mice. Periodic acid-Schiff (PAS) staining. Magnification: ×50 (upper panels); ×200 (lower panels).
Fig. 3.
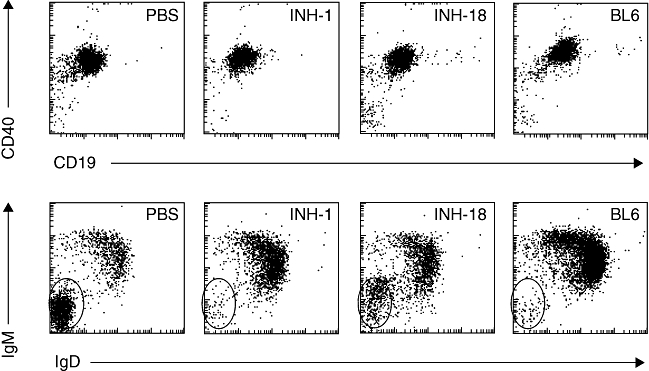
Class R inhibitor-1 (INH-1) completely, and class B INH-18 partially, restore resting B cell immunoglobulin (Ig)M+IgD+ phenotype in Murphy Roths large (MRL)-Faslpr/lpr mice. CD40+CD19+ gated splenic B cells were analysed for IgM and IgD expression in phosphate-buffered saline (PBS) or inhibitory oligonucleotide (INH-ODN)-treated MRL-Faslpr/lpr mice.
Telomeric TTAGGG repeats
Klinman's group created oligonucleotides containing telomeric TTAGGG repeats (A151 in Table 2). These INH-ODNs had multiple effects on immune activation [129]. For example, A151 blocked cytokine production induced by a variety of polyclonal activators and antigens, including TLR-9-ligands [129,159,160]. In vivo, these ODNs showed a remarkable potential for preventing inflammatory arthritis induced by intra-articular injection of CpG-ODNs [161], SLE in the NZB/W strain [162], uveitis [159], silicosis [163] and LPS-induced toxic shock [164]. Their activity depended on the ability to make G4-stacks. There is evidence that TTAGGG repeats may selectively bind STAT1 and STAT4 and block their phosphorylation [160,164]. Similar G-rich ODNs can also target STAT3 [165,166], scavenger receptors [149], nucleolin [167] and lupus autoantigen – Ku [168].
Combined TLR-7/TLR-9 antagonists
Barrat-Coffman's group created a novel TLR-9 inhibitor, IRS 869 [143]. Noticeably, this ODN differs from INH-ODN 2114 by having two A → T substitutions at the 3′ end where the ODN length but not the primary sequence counts [138,139]. This ODN was capable of preventing d-galactosamine plus CpG-ODN-induced systemic inflammation and cytokine storm [143].
The same group subsequently developed INH-ODNs suppressive for the TLR7-pathway. Their prototype IRS 661 contained five GC motifs buried within the complete palindrome [147]. This ODN was capable of blocking TLR-7/8 agonist (R848)-induced IL-6 secretion, but was ineffective against TLR-9-ligands. The same group combined TLR-7 and TLR-9 sequences and developed IRS 954 which blocked stimulation through TLR-7 and TLR-9. In vivo, IRS 954 proved efficacious in the NZB/W-F1 strain of lupus mice [169]. Interestingly, several groups have shown that (PS) ODNs including TLR-9-antagonists, control ODNs and CCT repeats [140], may have backbone-dependent and sequence-independent effects on TLR-7-induced activation [29,150,170–173].
Finally, Idera pharmaceuticals has developed INH-ODNs containing 2′-O-methylribonucleotide modifications but lacking the poly-G motif. These INH-ODNs were capable of blocking TLR-7 and TLR-9-induced activation, both in vitro and in vivo[174].
What is the mechanism of INH-ODN action?
There is a possibility that various types of INH-ODNs may work through different mechanisms. For example, INH-ODNs may compete for receptor-mediated endocytosis in a sequence-independent manner. This effect may depend on the length of an INH-ODN, as well as on its ability to make G4 stacks [149,175]. Longer and G-rich ODNs are taken up more effectively by macrophages than shorter ODNs. The opposite is true for primary B cells [175]. The next possibility is that INH-ODNs block TLR-9-trafficking from the ER to the endosomes or prevent TLR-9-processing into a functionally active product [176,177]. INH-ODNs may bind TLR-9 and prevent it from undergoing a conformational change critical for recruiting MyD88 [178]. INH-ODNs may promote Rab7-dependent degradation of TLR-9 in LAMP1+ late endosomes [179]. Certain INH-ODNs may work downstream of the TLR-7/9, e.g. at the level of STAT signalling.
Revised classification of INH-ODNs
We classified INH-ODNs into two major categories: class B and class R [151]. Class B are linear ODNs that potently block CpG-induced activation in all TLR-9-expressing cells. They also block TLR-7 activation in a sequence-independent manner. Interestingly, they are less protective in the MRL-Fas lpr/lpr strain. They may find applications for prevention/treatment of TLR-9-dependent microbial sepsis and chronic inflammation. On the other hand, class R INH-ODNs contain complete palindromes or short 5′ or 3′ single-stranded overhangs. Class R INH-ODNs are typically longer (20–28 mer) ODNs capable of either dimerizing or making hairpins. BCR cross-linking increases their potency for TLR-9-activated B cells for at least 10-fold, making them good candidates for targeting dsDNA-, nucleosome- or RF-specific autoreactive B cells. We initially included all complex INH-ODNs, including those capable of making G4 stacks, into the class R category [151]. However, it is now clear that G4-stacking ODNs and palindromic ODNs have different signalling targets. Therefore, we now propose a new category of INH-ODNs: class G (Table 3). We include into this class all INH-ODNs which are capable of making larger G-aggregates. These ODNs are much less specific for the TLR-7/9 pathway, and may have direct pro-apoptotic effects in tumour cells. They can block phosphorylation and nuclear translocation of multiple members of the STAT family. Besides TLRs, they can also interact with other cellular targets such as scavenger receptors Ku and nucleolin. Because of their promiscuity they can be more immunosuppressive than other classes of INH-ODNs.
Table 3.
Classification of inhibitory oligonucleotides.
| Class | Distinguishing feature | Example | TLR-9 inhibition in B cells | TLR-9 inhibition in DC | TLR-7 inhibition (backbone effect) | Effect in lupus | Reference |
|---|---|---|---|---|---|---|---|
| G | G4-stacking | A151 | + | +++ | ++ | ++ | [137] |
| R | Palindromic | INH-1 | + | +++ | ++ | ++ | [150] |
| B | Linear | INH-18 | +++ | +++ | ++ | −/+ | [150] |
TLR, Toll-like receptor.
All three classes of TLR-9-antagonists have sequence-independent phosphorothioate backbone-dependent effects on TLR-7 activation. TGC triplets may additionally increase their potency for the TLR-7 pathway [147]. Classes B and G INH-ODNs and combined TLR-7/9 inhibitors are effective in animal models of lupus [146,150,162,169,180]. These ODNs are very potent upstream inhibitors of TLR-7- or TLR-9-induced type I IFN secretion. However, it remains to be determined whether any of these INH-ODNs can interfere with the type I IFN-production induced following engagement of cytosolic DNA and RNA receptors and what the exact role of these nucleic acid sensors in the lupus pathogenesis might be. We envision application of INH-ODNs as therapeutic agents for human lupus and for DNA-dependent bacterial sepsis.
Acknowledgments
This study was supported by NIH grants AI047374 and AI064736 to PL. I am thankful for helpful advices from Dr Robert F. Ashman.
Disclosure
None.
References
- 1.Sheriff A, Gaipl US, Voll RE, Kalden JR, Herrmann M. Apoptosis and systemic lupus erythematosus. Rheum Dis Clin North Am. 2004;30:505–27. doi: 10.1016/j.rdc.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–15. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005;52:1138–47. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- 4.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci USA. 2007;104:6758–63. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN) Harley JB, Alarcón-Riquelme ME, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009;10:373–9. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Javierre BM, Fernandez AF, Richter J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 2010;20:170–9. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33:1665–73. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 9.Corvetta A, Della Bitta R, Luchetti MM, Pomponio G. 5-Methylcytosine content of DNA in blood, synovial mononuclear cells and synovial tissue from patients affected by autoimmune rheumatic diseases. J Chromatogr. 1991;566:481–91. doi: 10.1016/0378-4347(91)80265-e. [DOI] [PubMed] [Google Scholar]
- 10.Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol. 1988;140:2197–200. [PubMed] [Google Scholar]
- 11.Oldstone MB. Molecular mimicry and immune-mediated diseases. FASEB J. 1998;12:1255–65. doi: 10.1096/fasebj.12.13.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med. 2005;11:85–9. doi: 10.1038/nm1167. [DOI] [PubMed] [Google Scholar]
- 13.Carpenter DF, Steinberg AD, Schur PH, Talal N. The pathogenesis of autoimmunity in New Zealand mice. II. Acceleration of glomerulonephritis by polyinosinic-polycytidylic acid. Lab Invest. 1970;23:628–34. [PubMed] [Google Scholar]
- 14.Pawar RD, Patole PS, Zecher D, et al. Toll-like receptor-7 modulates immune complex glomerulonephritis. J Am Soc Nephrol. 2006;17:141–9. doi: 10.1681/ASN.2005070714. [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa K, Hayashi T. Synthetic CpG oligodeoxynucleotides accelerate the development of lupus nephritis during preactive phase in NZB × NZWF1 mice. Lupus. 2003;12:838–45. doi: 10.1191/0961203303lu483oa. [DOI] [PubMed] [Google Scholar]
- 16.Anders HJ, Vielhauer V, Eis V, et al. Activation of Toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004;18:534–6. doi: 10.1096/fj.03-0646fje. [DOI] [PubMed] [Google Scholar]
- 17.Lian ZX, Kikuchi K, Yang GX, Ansari AA, Ikehara S, Gerswin ME. Expansion of bone marrow IFN-alpha-producing dendritic cells in New Zealand Black (NZB) mice: high level expression of TLR9 and secretion of IFN-alpha in NZB bone marrow. J Immunol. 2004;173:5283–9. doi: 10.4049/jimmunol.173.8.5283. [DOI] [PubMed] [Google Scholar]
- 18.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 19.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 20.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science. 2004;303:1526–9. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 21.Lund JM, Alexopoulou L, Sato A, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598–603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 23.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–31. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hannum LG, Ni D, Haberman AM, Weigert MG, Shlomchik MJ. A disease-related rheumatoid factor autoantibody is not tolerized in a normal mouse: implications for the origins of autoantibodies in autoimmune disease. J Exp Med. 1996;184:1269–78. doi: 10.1084/jem.184.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monestier M, Kotzin BL. Antibodies to histones in systemic lupus erythematosus and drug-induced lupus. Rheum Dis Clin North Am. 1992;18:415–36. [PubMed] [Google Scholar]
- 26.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Schlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–7. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 27.Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–47. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 28.Uccellini MB, Busconi L, Green NM, et al. Autoreactive B cells discriminate CpG-rich and CpG-poor DNA and this response is modulated by IFN-alpha. J Immunol. 2008;181:5875–84. doi: 10.4049/jimmunol.181.9.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau CM, Broughton C, Tabor AS, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–7. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses toDNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Neill SK, Veselits ML, Zhang M, et al. Endocytic sequestration of the B cell antigen receptor and Toll-like receptor 9 in anergic cells. Proc Natl Acad Sci USA. 2009;106:6262–7. doi: 10.1073/pnas.0812922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulé MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation bychromatin-immunoglobulin G complexes. J Exp Med. 2004;199:1631–40. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody–DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–17. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Lee PY, Kumagai Y, Li Y, Takeuchi O, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205:2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savarese E, Steinberg C, Pawar RD, et al. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107–15. doi: 10.1002/art.23407. [DOI] [PubMed] [Google Scholar]
- 37.Pan ZJ, Maier S, Schwarz K, et al. TLR7 modulates anti-nucleosomal autoantibody isotype and renal complement deposition in mice exposed to syngeneic late apoptotic cells. Ann Rheum Dis. 2009 doi: 10.1136/ard.2009.108282. doi:10.1136/ard.2009.108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deane JA, Pisitkun P, Barrett RS, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–10. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 40.Subramanian S, Tus K, Li QZ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci USA. 2006;103:9970–5. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fairhurst AM, Hwang SH, Wang A, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. 2008;38:1971–8. doi: 10.1002/eji.200838138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006;203:553–61. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richez C, Yasuda K, Bonegio RG, et al. IFN regulatory factor 5 is required for disease development in the FcgammaRIIB-/-Yaa and FcgammaRIIB-/- mouse models of systemic lupus erythematosus. J Immunol. 2010;184:796–806. doi: 10.4049/jimmunol.0901748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(-/-) mice. J Exp Med. 2002;195:1167–74. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu X, Peng SL. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. 2006;54:336–42. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 46.Yu P, Wellmann U, Kunder S, et al. Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int Immunol. 2006;18:1211–19. doi: 10.1093/intimm/dxl067. [DOI] [PubMed] [Google Scholar]
- 47.Brummel R, Lenert P. Activation of marginal zone B cells from lupus mice with type A(D) CpG-oligodeoxynucleotides. J Immunol. 2005;174:2429–34. doi: 10.4049/jimmunol.174.4.2429. [DOI] [PubMed] [Google Scholar]
- 48.Lenert P, Brummel R, Field EH, Ashman RF. TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005;25:29–40. doi: 10.1007/s10875-005-0355-6. [DOI] [PubMed] [Google Scholar]
- 49.Santiago-Raber ML, Dunand-Sauthier I, Wu T, et al. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J Autoimmun. 2009 doi: 10.1016/j.jaut.2009.11.001. doi:10.1016/j.jaut.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 50.Patole PS, Gröne HJ, Segerer S, et al. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol. 2005;16:1326–38. doi: 10.1681/ASN.2004100820. [DOI] [PubMed] [Google Scholar]
- 51.Kono DH, Haraldsson MK, Lawson BR, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci USA. 2009;106:12061–6. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–8. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 53.Tabeta K, Hoebe K, Janssen EM, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–64. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 54.Fukui R, Saitoh S, Matsumoto F, et al. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206:1339–50. doi: 10.1084/jem.20082316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Papadimitraki ED, Tzardi M, Bertsias G, Sotsiou E, Boumpas DT. Glomerular expression of Toll-like receptor-9 in lupus nephritis but not in normal kidneys: implications for the amplification of the inflammatory response. Lupus. 2009;18:831–5. doi: 10.1177/0961203309103054. [DOI] [PubMed] [Google Scholar]
- 56.Zorro S, Arias M, Riaño F, et al. Response to ODN-CpG by B Cells from patients with systemic lupus erythematosus correlates with disease activity. Lupus. 2009;18:718–26. doi: 10.1177/0961203309103098. [DOI] [PubMed] [Google Scholar]
- 57.Wu O, Chen GP, Chen H, et al. The expressions of Toll-like receptor 9 and T-bet in circulating B and T cells in newly diagnosed, untreated systemic lupus erythematosus and correlations with disease activity and laboratory data in a Chinese population. Immunobiology. 2009;214:392–402. doi: 10.1016/j.imbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Komatsuda A, Wakui H, Iwamoto K, et al. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol. 2008;152:482–7. doi: 10.1111/j.1365-2249.2008.03646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Visentini M, Conti V, Cagliuso M, et al. Regression of systemic lupus erythematosus after development of an acquired Toll-like receptor signaling defect and antibody deficiency. Arthritis Rheum. 2009;60:2767–71. doi: 10.1002/art.24760. [DOI] [PubMed] [Google Scholar]
- 60.Xu CJ, Zhang WH, Pan HF, Li XP, Xu JH, Ye DQ. Association study of a single nucleotide polymorphism in the exon 2 region of Toll-like receptor 9 (TLR9) gene with susceptibility to systemic lupus erythematosus among Chinese. Mol Biol Rep. 2009;36:2245–8. doi: 10.1007/s11033-008-9440-z. [DOI] [PubMed] [Google Scholar]
- 61.Sánchez E, Callejas-Rubio JL, Sabio JM, et al. Investigation of TLR5 and TLR7 as candidate genes for susceptibility to systemic lupus erythematosus. Clin Exp Rheumatol. 2009;27:267–71. [PubMed] [Google Scholar]
- 62.Spies B, Hochrein H, Vabulas M, et al. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J Immunol. 2003;171:5908–12. doi: 10.4049/jimmunol.171.11.5908. [DOI] [PubMed] [Google Scholar]
- 63.Babiuk S, Mookherjee N, Pontarollo R, et al. TLR9-/- and TLR9+/+ mice display similar immune responses to a DNA vaccine. Immunology. 2004;113:114–20. doi: 10.1111/j.1365-2567.2004.01938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cortez-Gonzalez X, Pellicciotta I, Gerloni M, et al. TLR9-independent activation of B lymphocytes by bacterial DNA. DNA Cell Biol. 2006;25:253–61. doi: 10.1089/dna.2006.25.253. [DOI] [PubMed] [Google Scholar]
- 65.Ishii KJ, Kawagoe T, Koyama S, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–9. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- 66.Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S. Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med. 2005;202:1333–9. doi: 10.1084/jem.20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 68.Choubey D, Panchanathan R. Interferon-inducible Ifi200-family genes in systemic lupus erythematosus. Immunol Lett. 2008;119:32–41. doi: 10.1016/j.imlet.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mondini M, Vidali M, Airò P, et al. Role of the interferon-inducible gene IFI16 in the etiopathogenesis of systemic autoimmune disorders. Ann NY Acad Sci. 2007;1110:47–56. doi: 10.1196/annals.1423.006. [DOI] [PubMed] [Google Scholar]
- 70.Song LL, Ponomareva L, Shen H, Duan X, Alimirah F, Choubey D. Interferon-inducible IFI16, a negative regulator of cell growth, down-regulates expression of human telomerase reverse transcriptase (hTERT) gene. PLoS One. 2010;5:e8569. doi: 10.1371/journal.pone.0008569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jørgensen TN, Gubbels MR, Kotzin BL. Links between type I interferons and the genetic basis of disease in mouse lupus. Autoimmunity. 2003;36:491–502. doi: 10.1080/08916930310001605864. [DOI] [PubMed] [Google Scholar]
- 72.Roberts TL, Idris A, Dunn JA, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–60. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 73.Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–18. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang H, Chatterjee G, Meyer JJ, et al. Characteristics of three homologous 202 genes (Ifi202a, Ifi202b, and Ifi202c) from the murine interferon-activatable gene 200 cluster. Genomics. 1999;60:281–94. doi: 10.1006/geno.1999.5923. [DOI] [PubMed] [Google Scholar]
- 75.Gubbels Bupp MR, Li M, Pashine A, Aud D, Peng SL. The candidate lupus susceptibility gene Ifi202a is largely dispensable for B-cell function. Rheumatology (Oxf) 2008;47:103–4. doi: 10.1093/rheumatology/kem259. [DOI] [PubMed] [Google Scholar]
- 76.Santiago-Raber ML, Laporte C, Reininger L, Izui S. Genetic basis of murine lupus. Autoimmun Rev. 2004;3:33–9. doi: 10.1016/S1568-9972(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 77.Teramoto K, Negoro N, Kitamoto K, et al. Microarray analysis of glomerular gene expression in murine lupus nephritis. J Pharmacol Sci. 2008;106:56–67. doi: 10.1254/jphs.fp0071337. [DOI] [PubMed] [Google Scholar]
- 78.Kimkong I, Avihingsanon Y, Hirankarn N. Expression profile of HIN200 in leukocytes and renal biopsy of SLE patients by real-time RT–PCR. Lupus. 2009;18:1066–72. doi: 10.1177/0961203309106699. [DOI] [PubMed] [Google Scholar]
- 79.Peterson KS, Huang JF, Zhu J, et al. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J Clin Invest. 2004;113:1722–33. doi: 10.1172/JCI19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamauchi M, Hashimoto M, Ichiyama K, et al. Ifi202, an IFN-inducible candidate gene for lupus susceptibility in NZB/W F1 mice, is a positive regulator for NF-kappaB activation in dendritic cells. Int Immunol. 2007;19:935–42. doi: 10.1093/intimm/dxm054. [DOI] [PubMed] [Google Scholar]
- 81.Hueber W, Zeng D, Strober S, Utz PJ. Interferon-alpha-inducible proteins are novel autoantigens in murine lupus. Arthritis Rheum. 2004;50:3239–49. doi: 10.1002/art.20508. [DOI] [PubMed] [Google Scholar]
- 82.Takaoka A, Wang Z, Choi MK, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–5. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 83.Wang Z, Choi MK, Ban T, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA. 2008;105:5477–82. doi: 10.1073/pnas.0801295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA. 2004;101:233–8. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rebsamen M, Heinz LX, Meylan E, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10:916–22. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008;181:6427–34. doi: 10.4049/jimmunol.181.9.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lippmann J, Rothenburg S, Deigendesch N, et al. IFN beta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI) Cell Microbiol. 2008;10:2579–88. doi: 10.1111/j.1462-5822.2008.01232.x. [DOI] [PubMed] [Google Scholar]
- 88.Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev. 2007;220:35–46. doi: 10.1111/j.1600-065X.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- 89.Pisetsky DS, Erlandsson-Harris H, Andersson U. High-mobility group box protein 1 (HMGB1): an alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res Ther. 2008;10:209. doi: 10.1186/ar2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ivanov S, Dragoi AM, Wang X, et al. A novel role for HMGB1 in TLR9 mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–81. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bianchi ME, Beltrame M, Paonessa G. Specific recognition of cruciform DNA by nuclear protein HMG1. Science. 1989;243:1056–9. doi: 10.1126/science.2922595. [DOI] [PubMed] [Google Scholar]
- 92.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 93.Harris HE, Raucci A. Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Rep. 2006;7:774–8. doi: 10.1038/sj.embor.7400759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Avalos AM, Kiefer K, Tian J, et al. RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmunity. 2010;43:103–10. doi: 10.3109/08916930903384591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bell CW, Jiang W, Reich CF, III, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:C1318–25. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 96.Gauley J, Pisetsky DS. The translocation of HMGB1 during cell activation and cell death. Autoimmunity. 2009;42:299–301. doi: 10.1080/08916930902831522. [DOI] [PubMed] [Google Scholar]
- 97.Urbonaviciute V, Fürnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–18. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Popovic K, Ek M, Espinosa A, et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum. 2005;52:3639–45. doi: 10.1002/art.21398. [DOI] [PubMed] [Google Scholar]
- 99.Ayer LM, Rubin RL, Dixon GH, Fritzler MJ. Antibodies to HMG proteins in patients with drug-induced autoimmunity. Arthritis Rheum. 1994;37:98–103. doi: 10.1002/art.1780370115. [DOI] [PubMed] [Google Scholar]
- 100.Jiang W, Pisetsky DS. Expression of high mobility group protein 1 in the sera of patients and mice with systemic lupus erythematosus. Ann Rheum Dis. 2008;67:727–8. doi: 10.1136/ard.2007.074484. [DOI] [PubMed] [Google Scholar]
- 101.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA : dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–72. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–91. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ishii KJ, Coban C, Kato H, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–8. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 104.Yoneyama M, Kikuchi M, Natsukawa T, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 105.Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA. 2002;99:637–42. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kato H, Takeuchi O, Sato S, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 107.Kato H, Takeuchi O, Mikamo-Satoh E, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–10. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Allam R, Pawar RD, Kulkarni OP, et al. Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses. Eur J Immunol. 2008;38:3487–98. doi: 10.1002/eji.200838604. [DOI] [PubMed] [Google Scholar]
- 109.Ronnblom L, Alm GV. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol. 2001;22:427–31. doi: 10.1016/s1471-4906(01)01955-x. [DOI] [PubMed] [Google Scholar]
- 110.Crow MK, Kirou KA. Interferon-alpha in systemic lupus erythematosus. Curr Opin Rheum. 2004;16:541–7. doi: 10.1097/01.bor.0000135453.70424.1b. [DOI] [PubMed] [Google Scholar]
- 111.Gota C, Calabrese L. Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity. 2003;36:511–18. doi: 10.1080/08916930310001605873. [DOI] [PubMed] [Google Scholar]
- 112.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 113.Bengtsson AA, Sturfelt G, Truedsson L, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus. 2000;9:664–71. doi: 10.1191/096120300674499064. [DOI] [PubMed] [Google Scholar]
- 114.Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5:279–87. doi: 10.1186/ar1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;97:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feng X, Wu H, Grossman JM, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54:2951–62. doi: 10.1002/art.22044. [DOI] [PubMed] [Google Scholar]
- 117.Petri M, Singh S, Tesfasyone H, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. 2009;18:980–9. doi: 10.1177/0961203309105529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Di Domizio J, Blum A, Gallagher-Gambarelli M, Molens JP, Chaperot L, Plumas J. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood. 2009;114:1794–802. doi: 10.1182/blood-2009-04-216770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Takauji R, Iho S, Takatsuka H, et al. CpG-DNA-induced IFN-alpha production involves p38 MAPK-dependent STAT1 phosphorylation in human plasmacytoid dendritic cell precursors. J Leukoc Biol. 2002;72:1011–19. [PubMed] [Google Scholar]
- 120.Yasuda K, Richez C, Uccellini MB, et al. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J Immunol. 2009;183:3109–17. doi: 10.4049/jimmunol.0900399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mathian A, Weinberg A, Gallegos M, Banchereau J, Koutouzov S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black × New Zealand White) F1 but not in BALB/c mice. J Immunol. 2005;174:2499–506. doi: 10.4049/jimmunol.174.5.2499. [DOI] [PubMed] [Google Scholar]
- 122.Santiago-Raber ML, Baccala R, Haraldsson KM, et al. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003;197:777–88. doi: 10.1084/jem.20021996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Agrawal H, Jacob N, Carreras E, et al. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol. 2009;183:6021–9. doi: 10.4049/jimmunol.0803872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Green NM, Laws A, Kiefer K, et al. Murine B cell response to TLR7 ligands depends on an IFN-beta feedback loop. J Immunol. 2009;183:1569–76. doi: 10.4049/jimmunol.0803899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brummel R, Roberts TL, Stacey KJ, Lenert P. Higher-order CpG-DNA stimulation reveals distinct activation requirements for marginal zone and follicular B cells in lupus mice. Eur J Immunol. 2006;36:1951–62. doi: 10.1002/eji.200535734. [DOI] [PubMed] [Google Scholar]
- 126.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 127.Yao Y, Richman L, Higgs BW, et al. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60:1785–96. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]
- 128.Rönnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–9. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tsakonas E, Joseph L, Esdaile JM, et al. A long-term study of hydroxychloroquine withdrawal on exacerbations in systemic lupus erythematosus. The Canadian Hydroxychloroquine Study Group. Lupus. 1998;7:80–5. doi: 10.1191/096120398678919778. [DOI] [PubMed] [Google Scholar]
- 130.Halpern MD, Pisetsky DS. In vitro inhibition of murine IFN gamma production by phosphorothioate deoxyguanosine oligomers. Immunopharmacology. 1995;29:47–52. doi: 10.1016/0162-3109(95)00043-s. [DOI] [PubMed] [Google Scholar]
- 131.Pisetsky DS, Reich CF. Inhibition of murine macrophage IL-12 production by natural and synthetic DNA. Clin Immunol. 2000;96:198–204. doi: 10.1006/clim.2000.4897. [DOI] [PubMed] [Google Scholar]
- 132.Zhu FG, Reich CF, Pisetsky DS. Inhibition of murine dendritic cell activation by synthetic phosphorothioate oligodeoxynucleotides. J Leukoc Biol. 2002;72:1154–63. [PubMed] [Google Scholar]
- 133.Krieg AM, Wu T, Weeratna R, et al. Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc Natl Acad Sci USA. 1998;95:12631–6. doi: 10.1073/pnas.95.21.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen Y, Lenert P, Weeratna R, et al. Identification of methylated CpG motifs as inhibitors of the immune stimulatory CpG motifs. Gene Ther. 2001;8:1024–32. doi: 10.1038/sj.gt.3301482. [DOI] [PubMed] [Google Scholar]
- 135.Lenert P, Stunz L, Yi AK, Krieg AM, Ashman RF. CpG stimulation of primary mouse B cells is blocked by inhibitory oligodeoxyribonucleotides at a site proximal to NF-kB activation. Antisense Nucleic Acid Drug Dev. 2001;11:247–56. doi: 10.1089/108729001317022241. [DOI] [PubMed] [Google Scholar]
- 136.Lenert P, Yi AK, Krieg AM, Stunz LL, Ashman RF. Inhibitory oligonucleotides block the induction of AP-1 transcription factor by stimulatory CpG oligonucleotides in B cells. Antisense Nucleic Acid Drug Dev. 2003;13:143–50. doi: 10.1089/108729003768247600. [DOI] [PubMed] [Google Scholar]
- 137.Gursel I, Gursel M, Yamada H, Ishii KJ, Takeshita F, Klinman DM. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J Immunol. 2003;171:1393–400. doi: 10.4049/jimmunol.171.3.1393. [DOI] [PubMed] [Google Scholar]
- 138.Lenert P, Rasmussen W, Ashman RF, Ballas ZK. Structural characterization of the inhibitory DNA motif for the type A[D]-CpG-induced cytokine secretion and NK-cell lytic activity in mouse spleen cells. DNA Cell Biol. 2003;22:621–31. doi: 10.1089/104454903770238094. [DOI] [PubMed] [Google Scholar]
- 139.Ashman RF, Goeken JA, Drahos J, Lenert P. Sequence requirements for oligodeoxyribonucleotide inhibitory activity. Int Immunol. 2005;17:411–20. doi: 10.1093/intimm/dxh222. [DOI] [PubMed] [Google Scholar]
- 140.Sun R, Sun L, Bao M, et al. A human microsatellite DNA-mimicking oligodeoxynucleotide with CCT repeats negatively regulates TLR7/9-mediated innate immune responses via selected TLR pathways. Clin Immunol. 2010;134:262–76. doi: 10.1016/j.clim.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stunz LL, Lenert P, Peckham D, et al. Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol. 2002;32:1212–22. doi: 10.1002/1521-4141(200205)32:5<1212::AID-IMMU1212>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 142.Lenert P. Inhibitory oligodeoxynucleotides – therapeutic promise for systemic autoimmune diseases? Clin Exp Immunol. 2005;140:1–10. doi: 10.1111/j.1365-2249.2004.02728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Duramad O, Fearon KL, Chang B, et al. Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J Immunol. 2005;174:5193–200. doi: 10.4049/jimmunol.174.9.5193. [DOI] [PubMed] [Google Scholar]
- 144.Ashman RF, Lenert P. Structural requirements and applications of inhibitory oligodeoxyribonucleotides. Immunol Res. 2007;39:4–14. doi: 10.1007/s12026-007-0065-4. [DOI] [PubMed] [Google Scholar]
- 145.Haas T, Metzger J, Schmitz F, et al. The DNA sugar backbone 2′ deoxyribose determines Toll-like receptor 9 activation. Immunity. 2008;28:315–23. doi: 10.1016/j.immuni.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 146.Patole PS, Zecher D, Pawar RD, Grone H-J, Schlondorff D, Anders H-J. G-rich DNA suppresses systemic lupus. J Am Soc Nephrol. 2005;16:3273–80. doi: 10.1681/ASN.2005060658. [DOI] [PubMed] [Google Scholar]
- 147.Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Peter M, Bode K, Lipford GB, Eberle F, Heeg K, Dalpke AH. Characterization of suppressive oligodeoxynucleotides that inhibit Toll-like receptor-9-mediated activation of innate immunity. Immunology. 2008;123:118–28. doi: 10.1111/j.1365-2567.2007.02718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pearson AM, Rich A, Krieger M. Polynucleotide binding to macrophage scavenger receptors depends on the formation of base-quartet-stabilized four-stranded helices. J Biol Chem. 1993;268:3546–54. [PubMed] [Google Scholar]
- 150.Lenert P, Yasuda K, Busconi L, et al. DNA-like Class R inhibitory oligonucleotides (INH-ODNs) preferentially block autoantigen-induced B-cell and dendritic cell activation in vitro and autoantibody production in lupus-prone MRL-Faslpr/lpr mice in vivo. Arthritis Res Ther. 2009;11:R79. doi: 10.1186/ar2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lenert P. Targeting Toll-like receptor signaling in plasmacytoid dendritic cells and autoreactive B cells as a therapy for lupus. Arthritis Res Ther. 2006;8:R203. doi: 10.1186/ar1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Honda K, Ohba Y, Yannai H, et al. Spatiotemporal regulation of MyD88-IRF-7 signaling for robust type-I interferon induction. Nature. 2005;434:1035–40. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 153.Guiducci C, Ott G, Chan JH, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. 2006;203:1999–2008. doi: 10.1084/jem.20060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Avalos AM, Latz E, Mousseau B, et al. Differential cytokine production and bystander activation of autoreactive B cells in response to CpG-A and CpG-B oligonucleotides. J Immunol. 2009;183:6262–8. doi: 10.4049/jimmunol.0901941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bernasconi NL, Onai N, Lanzavecchia A. A role for Toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naïve B cells and constitutive expression in memory B cells. Blood. 2003;102:956–63. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- 156.Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol. 2006;36:810–16. doi: 10.1002/eji.200535744. [DOI] [PubMed] [Google Scholar]
- 157.Poeck H, Wagner M, Battiany J, et al. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood. 2004;103:3058–64. doi: 10.1182/blood-2003-08-2972. [DOI] [PubMed] [Google Scholar]
- 158.Busconi L, Bauer JW, Tumang JR, et al. Functional outcome of B cell activation by chromatin immune complex engagement of the B cell receptor and TLR9. J Immunol. 2007;179:7397–405. doi: 10.4049/jimmunol.179.11.7397. [DOI] [PubMed] [Google Scholar]
- 159.Klinman DM, Tross D, Klaschik S, Shirota H, Sato T. Therapeutic applications and mechanisms underlying the activity of immunosuppressive oligonucleotides. Ann NY Acad Sci. 2009;1175:80–8. doi: 10.1111/j.1749-6632.2009.04970.x. [DOI] [PubMed] [Google Scholar]
- 160.Shirota H, Gursel M, Klinman DM. Suppressive oligodeoxynucleotides inhibit Th1 differentiation by blocking IFN-gamma and IL-12-mediated signaling. J Immunol. 2004;173:5002–7. doi: 10.4049/jimmunol.173.8.5002. [DOI] [PubMed] [Google Scholar]
- 161.Zeuner RA, Verthelyi D, Gursel M, Ishii KJ, Klinman DM. Influence of stimulatory and suppressive DNA motifs on host susceptibility to inflammatory arthritis. Arthritis Rheum. 2003;48:1701–7. doi: 10.1002/art.11035. [DOI] [PubMed] [Google Scholar]
- 162.Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB × NZW mice. Arthritis Rheum. 2005;52:651–8. doi: 10.1002/art.20810. [DOI] [PubMed] [Google Scholar]
- 163.Sato T, Shimosato T, Alvord WG, Klinman DM. Suppressive oligodeoxynucleotides inhibit silica-induced pulmonary inflammation. J Immunol. 2008;180:7648–54. doi: 10.4049/jimmunol.180.11.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shirota H, Gursel I, Gursel M, Klinman DM. Suppressive oligodeoxynucleotides protect mice from lethal endotoxic shock. J Immunol. 2005;174:4579–83. doi: 10.4049/jimmunol.174.8.4579. [DOI] [PubMed] [Google Scholar]
- 165.Jing N, Li Y, Xu X, et al. Targeting Stat3 with G-quartet oligodeoxynucleotides in human cancer cells. DNA Cell Biol. 2003;22:685–96. doi: 10.1089/104454903770946665. [DOI] [PubMed] [Google Scholar]
- 166.Jing N, Li Y, Xiong W, Sha W, Jing L, Tweardy DJ. G-quartet oligonucleotides: a new class of signal transducers and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004;64:6603–9. doi: 10.1158/0008-5472.CAN-03-4041. [DOI] [PubMed] [Google Scholar]
- 167.Hanakahi LA, Sun H, Maizels N. High affinity interactions of nucleolin with G-G-aspired rDNA. J Biol Chem. 1999;274:15908–12. doi: 10.1074/jbc.274.22.15908. [DOI] [PubMed] [Google Scholar]
- 168.Bianchi A, de Lange T. Ku binds telomeric DNA in vitro. J Biol Chem. 1999;274:21223–7. doi: 10.1074/jbc.274.30.21223. [DOI] [PubMed] [Google Scholar]
- 169.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–6. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 170.Graham KL, Lee LY, Higgins JP, Steinman L, Utz PJ, Ho PP. Treatment with a Toll-like receptor inhibitory GpG oligonucleotide delays and attenuates lupus nephritis in NZB/W mice. Autoimmunity. 2010;43:140–55. doi: 10.3109/08916930903229239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vollmer J, Tluk S, Schmitz C, et al. Immune stimulation medicated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. 2005;202:1575–85. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jurk M, Kritzler A, Schulte B, et al. Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich phosphorothiate oligodeoxynucleotides. Eur J Immunol. 2006;36:1815–26. doi: 10.1002/eji.200535806. [DOI] [PubMed] [Google Scholar]
- 173.Gorden KK, Qiu X, Batiste JJ, Wightman PP, Vasilakos JP, Alkan SS. Oligodeoxynucleotides differentially modulate activation of TLR7 and TLR8 by imidazoquinolines. J Immunol. 2006;177:8164–70. doi: 10.4049/jimmunol.177.11.8164. [DOI] [PubMed] [Google Scholar]
- 174.Wang D, Bhagat L, Yu D, et al. Oligodeoxyribonucleotide-based antagonists for Toll-like receptors 7 and 9. J Med Chem. 2009;52:551–8. doi: 10.1021/jm8014316. [DOI] [PubMed] [Google Scholar]
- 175.Trieu A, Roberts TL, Dunn JA, Sweet MJ, Stacey KJ. DNA motifs suppressing TLR9 responses. Crit Rev Immunol. 2006;26:527–44. doi: 10.1615/critrevimmunol.v26.i6.50. [DOI] [PubMed] [Google Scholar]
- 176.Ewald SE, Lee BL, Lau L, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–62. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sepulveda FE, Maschalidi S, Colisson R, et al. Critical role for asparagine endopeptidase in endocytic toll-like receptor signaling in dendritic cells. Immunity. 2009;31:737–48. doi: 10.1016/j.immuni.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 178.Latz E, Verma A, Visintin A, et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–9. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- 179.Yao M, Liu X, Li D, Chen T, Cai Z, Cao X. Late endosome/lysosome-localized Rab7b suppresses TLR9-initiated proinflammatory cytokine and type I IFN production in macrophages. J Immunol. 2009;183:1751–8. doi: 10.4049/jimmunol.0900249. [DOI] [PubMed] [Google Scholar]
- 180.Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–31. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]