

Abstract
Recent studies have tested genetic variation at the C1QA, C1QB and C1QC (complement component 1, q subcomponent, A chain, complement component 1, q subcomponent, B chain and complement component 1, q subcomponent, c chain) loci in relation to systemic lupus erythematosus (SLE) risk. Evidence for a significant effect of C1Q locus gene polymorphisms on SLE predisposition remains unclear. We aimed to identify associations between common C1Q polymorphisms and SLE risk and serum C1q, C3 and C4 levels. We performed family-based association tests in 295 nuclear families with one affected proband. Tag-single nucleotide polymorphisms (SNPs) ranging from 35·4 kb upstream of the C1QA gene to 28 kb downstream of the C1QB gene were selected to represent the entire C1Q gene locus. We performed transmission disequilibrium tests for affectation status and continuous traits, including C1q, C3 and C4 levels using family-based association tests (FBAT). There was no evidence for a significant role of C1Q locus gene polymorphisms in SLE risk predisposition. The strongest association was observed with a variant in the 3′UTR region of the C1QB gene (rs294223, P = 0·06). We found nominally significant associations with a second variant (rs7549888) in the 3′UTR region of the C1QB gene and C1q (P = 0·01), C3 (P = 0·004) and C4 levels (P = 0·01). In a large family-based association study of C1Q gene cluster polymorphisms no evidence for a genetic role of C1Q locus SNP in SLE risk predisposition was obtained in patients of European ancestry. This is in contrast to other cohorts, in which single variants associated with C1Q, C3 and C4 levels and nephritis have been studied and shown associations.
Keywords: C1QA, C1QB, C1QC, complement concentrations, SLE
Introduction
The complement proteins are known to be involved in the pathogenesis of systemic lupus erythematosus (SLE). Initial observations confirmed that during active disease, complement proteins, and in particular C3, are deposited at the site of inflammation in various organs, including the spleen, heart and kidney [1]. This, in turn, leads to reduced serum levels of complement as it is rapidly consumed and bound with immune complexes in such patients. These early studies suggested that inadvertent activation of complement during autoimmunity was proinflammatory, and played a similar role as it does in host defence against infectious disease [2]. However, studies on patients with homozygous deficiencies for complement proteins have revealed that complement deficiencies promote rather than inhibit the development of SLE [3–14]. This has led to the ‘paradoxical hypothesis’ being proposed by Walport and colleagues [15] that complement activation, although primarily a proinflammatory immune mechanism, is actually anti-inflammatory during the development of certain types of autoimmunity. This may be due to the anti-inflammatory functions of C1q, namely its ability to help solubilize immune complexes and aid in the clearance of apoptotic debris [16], which is defective in SLE patients [17].
Despite homozygous complement gene deficiencies being rare, the absences of complement genes are very powerful markers of disease susceptibility. A hierarchical grade of susceptibility exists in which 93%, 75%, ∼10%, 13% and >5% of individuals will develop lupus-like illness if they are deficient in C1q, C4, C2, C3 or C5–9, respectively [18] The rarity of these genetic deficiencies has led to further sequence analysis of the A, B and C chains of C1q to identify single nucleotide polymorphisms (SNP) that may account for defective C1q function, C1q antigenicity or decreased concentration in the serum of SLE sufferers [19–22]. However, these studies have been small, or have examined only a small number of variants. Here we extend analysis from previous studies to study associations of common haplotype tagging SNPs across the C1Q locus and SLE affectation status, C1Q, C3 and C4 levels. We used HapMap Build 35 information to select tag-SNPs across an 87·8 kb region in a sample of 295 trios.
Methods
Patients and samples
Details of samples used in this analysis are given in Table 1. The cohort investigated here included 295 nuclear families (including unaffected parents and a single affected proband). All the participants included in this analysis are of European ancestry [23] and had provided informed consent. The patient recruitment and samples were approved by the London Multi-Centre Research Ethics Committee (MREC) and the study protocol approved further by Exeter MREC 06/Q2102/56. Details of patient recruitment have been reported previously [24]. Briefly, patients were recruited via attendance at rheumatology and/or renal clinics at the Hammersmith Hospital or responded to advertisements in the media and then their disease status checked by medical questionnaire of their symptoms and through their clinical records by collaborating physicians in the United Kingdom. Each patient gave written permission to obtain medical record information. The SLE status of each patient was determined according to the American College of Rheumatology (ACR) 1982 and 199 revised criteria for SLE [25,26]. As far as possible, the initial onset date for the initial diagnosis of SLE was recorded in 118 of 295 subjects. C1q, C3 and C4 were measured in a subset of 166 parents and 84 affected SLE probands by commercial enzyme-linked immunosorbent assay (ELISA), according to the manufacturer's instructions.
Table 1.
Characteristics of subjects and clinical parameters presented as mean (standard deviation).
| Number of participants† | 295 nuclear participants, 997 individuals | Normal range of complement proteins [29] |
|---|---|---|
| Age of probands | 38·50 (9.53) | |
| Age of parents | 66·20 (9.99) | |
| Age at diagnosis (n = 118) | 23·30 (9.61) | |
| C1q levels probands (n = 84) | 192·10 mg/l (50·21) | 50–250 mg/l |
| C1q levels parents (n = 166) | 214·08 mg/l (52·46) | |
| Complement C3 probands (n = 84) | 944·26 mg/l (322·83) | 750–1650 mg/l |
| Complement C3 parents (n = 166) | 1178·94 mg/l (357·82) | |
| Complement C4 probands (n = 84) | 163·48 mg/l (96·10) | 140–540 mg/l |
| Complement C4 parents (n = 166) | 238·15 mg/l (83·39) |
All patients were Caucasian and residents of the United Kingdom. All subjects were recruited while attending the rheumatology and/or renal clinics of the Hammersmith Hospital, London or referred from collaborating physicians in the United Kingdom.
Tag SNP selection
A total of 23 tagging polymorphisms across the C1QA, C1QB and C1QC loci were selected to capture all common SNPs in the CEU samples of the Hapmap project [minor allele frequency (MAF) >5%] at r2 > 0·8. These SNPs extended from 35·4 kb upstream of the C1QA gene to 28 kb downstream of the C1QB gene (Fig. 1). Tag SNPs were selected using the tagger SNPs selection algorithm available in Haploview (http://www.broad.mit.edu/mpg/haploview/).
Fig. 1.
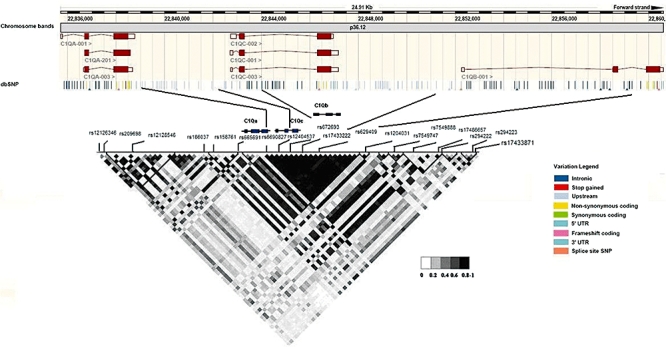
Positions of the 18 common gene variants studied are marked along the linkage disequilibrium plot of the C1Q locus. Positions of these variants along the actual genetic co-ordinates according to the dbSNP database are marked in the upper half of the diagram.
Genotyping and quality control
The polymorphisms were all typed by MALDI-TOF (matrix-assisted laser desorption ionization time-of-flight) mass spectrometry (Sequenom, Hamburg, Germany), using the standard conditions described for the iPLEX kit. All polymerase chain reactions (PCRs) were carried out on DNA Engine Tetrads (MJ Research Incorporated, Waltham, MA, USA) in either a 96- or 384-well format. The details of all amplification and extension primers are available from the authors. Of the 23 variants selected to capture the majority of common variation (MAF >5%) across the C1q locus, based on HapMap Phase II Build 35 data, 18 SNPs were acceptable for analysis following quality control (QC). This included removing SNPs with recorded Mendelian error rates >1%, SNPs with Hardy–Weinberg equilibrium (HWE) P-values below 0·01 significance level in the unaffected parent samples and SNPs with call rates <85% in the trios.
Statistics
We used the transmission disequilibrium test (TDT), as implemented in the FBAT program, to assess the evidence that alleles were transmitted differentially from parents to SLE-affected probands. We used the quantitative-trait disequilibrium test (QTDT) test to study associations with C1q, C3 and C4 levels as continuous traits. The strength of association in FBAT is specified by the significance of the P-value and the standardized FBAT ‘U-statistic’, represented as Z. ‘U’ is based on a linear combination of genotypes and traits [27] and is calculated as the difference between expectation of the product of offspring genotypes and traits and the product of coded traits values and offspring genotypes. Power calculations were performed using quanto http://hydra.usc.edu/GxE/. This study had 88% power to detect a genetic effect size of odds ratio (OR) = 1·5, at α = 0·05 for a SNP with MAF = 0·25. For a more modest effect size equivalent to an OR of 1·2 we had 28% power to detect associations at α = 0·05.
Results
Of the 23 variants included after genetic coverage analysis, 18 variants passed quality control analysis and were included in association tests (Fig. 1). The selected variants across the C1QA, C1QC and C1QB genes captured 30% and 75% of variation based on HapMap Phase II Build 36 data at r2 > 0·8 and r2 > 0·5, respectively. Extending this region to 20 kb either side of the C1Q locus allowed us to capture 38% and 73% of variation at r2 > 0·8 and r2 > 0·5, respectively.
No evidence that C1Q SNPs are associated with SLE
We performed TDT analysis in 292 nuclear families to test for association of SNPs residing in the C1Q locus and SLE affectation status. Of the 18 variants included in single marker association tests, none of these were associated with affectation status in TDT analysis (Table 2). The lowest observed P-value did not reach nominal significance (rs294223, P = 0·06, Z = −1·88). Similarly, no evidence for association was found when transmission of haplotypes formed by allelic combinations of SNPs ranging from 5′ transcription start site of the C1QA gene to the 3′ transcription stop site of the C1QB gene were studied (Table 3). The two most common haplotypes were observed at frequencies of 53% and 16%, and these were marked by the common and rare alleles of five SNPs which covered the C1Q locus, respectively (Table 3).
Table 2.
Association of informative single nucleotide polymorphisms (SNPs) with systemic lupus erythematosus disease status.
| SNP ID | Major allele | Minor allele | Location | P-value | Z-statistic for minor alleles | MAF parents | MAF probands |
|---|---|---|---|---|---|---|---|
| rs12126436 | G | A | 22672978 | 0·27 | 1·10 | 0·12 | 0·14 |
| rs209698 | T | C | 22674119 | 0·17 | 1·35 | 0·33 | 0·35 |
| rs12128546 | T | G | 22682400 | 0·94 | −0·07 | 0·21 | 0·21 |
| rs186037 | T | C | 22697692 | 0·86 | −0·17 | 0·13 | 0·13 |
| rs158761 | G | T | 22699678 | 0·36 | −0·91 | 0·41 | 0·40 |
| rs665691 | G | C | 22705660 | 0·94 | −0·06 | 0·44 | 0·44 |
| rs6690827 | G | A | 22712802 | 0·89 | 0·14 | 0·27 | 0·28 |
| rs12404537 | C | T | 22714674 | 0·66 | −0·43 | 0·19 | 0·18 |
| rs672693 | G | A | 22716752 | 0·72 | −0·35 | 0·33 | 0·32 |
| rs294180 | G | T | 22720100 | 0·33 | −0·96 | 0·38 | 0·36 |
| rs17433222 | G | A | 22723952 | 0·69 | −0·39 | 0·26 | 0·25 |
| rs12040131 | C | G | 22734004 | 0·17 | −1·35 | 0·21 | 0·19 |
| rs7549747 | T | C | 22741877 | 0·96 | −0·06 | 0·43 | 0·44 |
| rs7549888 | G | T | 22749325 | 0·74 | 0·32 | 0·36 | 0·36 |
| rs17486657 | C | T | 22752481 | 0·37 | −0·90 | 0·29 | 0·29 |
| rs294222 | G | G | 22752866 | 1·00 | 0·00 | 0·22 | 0·23 |
| rs294223 | A | C | 22761312 | 0·13 | −1·45 | 0·25 | 0·23 |
| rs17433871 | T | G | 22761409 | 0·77 | −0·38 | 0·25 | 0·25 |
MAF: minor allele frequency.
Table 3.
Haplotype structure resulting from single nucleotide polymorphisms (SNPs) across the C1q locus and 20 kb either side; association analysis with affectation status.
| Haplotype | rs6690827 | rs12404537 | rs672693 | rs294180 | rs17433222 | Haplotype frequency | Haplotype specific association P-value |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | 1 | 1 | 0·53 | 0·16 |
| 2 | 2 | 2 | 2 | 2 | 2 | 0·16 | 0·80 |
| 3 | 1 | 1 | 2 | 2 | 1 | 0·09 | 0·06 |
C1Q SNPs and associations with C1Q, C3 or C4 levels
We tested the 18 variants analysed for association with disease status for further associations with C1q, C3 and C4 levels, which were measured in a subset of the probands (n = 84) and parents (n = 116) (Table 4). These tests therefore included both SLE patients and their predominantly unaffected parents, but it is important to note that genetic associations cannot be confounded by, or secondary to, disease processes and that the use of both unaffected and affected SLE patients is unlikely to result in false positive associations between C1Q variants and circulating protein levels. The minor allele of a variant (rs7549888) in the 3′UTR region of the C1QB locus was associated with lower C1q levels (P = 0·01) and higher C3 (P = 0·004) and C4 levels (P = 0·01). The variant associated most strongly with SLE status in this study was in moderate linkage disequilibrium with rs7549888 (r2 = 0·52), although this variant itself was not associated with disease status (P = 0·81, Z = −0·14).
Table 4.
Association of single nucleotide polymorphisms (SNPs) 20 kb either side of C1Q locus with C1q, C3 and C4 levels.
| SNP ID | Alleles | Location | P-value for association with C1q levels | Z-statistic for minor allele | P-value for association with C3 levels | Z-statistic for minor allele | P-value for association with C4 levels | Z-statistic for minor allele |
|---|---|---|---|---|---|---|---|---|
| rs12126436 | G/A | 22672978 | 0·22 | 1·20 | 0·07 | −1·83 | 0·16 | 1·38 |
| rs209698 | T/C | 22674119 | 0·08 | 1·72 | 0·18 | 1·33 | 0·08 | 1·71 |
| rs12128546 | T/G | 22682400 | 0·59 | −0·52 | 0·47 | −0·71 | 0·42 | −0·79 |
| rs186037 | T/C | 22697692 | 0·73 | 0·34 | 0·32 | 0·98 | 0·18 | 1·35 |
| rs158761 | G/T | 22699678 | 0·66 | 0·43 | 0·62 | 0·49 | 0·53 | 0·62 |
| rs665691 | G/C | 22705660 | 0·96 | 0·05 | 0·92 | −0·09 | 0·81 | −0·24 |
| rs6690827 | G/A | 22712802 | 0·61 | −0·51 | 0·81 | −0·23 | 0·70 | −0·38 |
| rs12404537 | C/T | 22714674 | 0·02 | −2·30 | 0·01 | −2·01 | 0·01 | −2·50 |
| rs672693 | G/A | 22716752 | 0·83 | −0·20 | 0·94 | 0·08 | 0·84 | 0·19 |
| rs294180 | G/T | 22720100 | 0·61 | −0·50 | 0·55 | −0·60 | 0·79 | −0·26 |
| rs17433222 | G/A | 22723952 | 0·04 | −2·28 | 0·01 | −2·35 | 0·01 | −2·50 |
| rs12040131 | C/G | 22734004 | 0·16 | −1·85 | 0·07 | −1·80 | 0·05 | −1·91 |
| rs7549747 | T/C | 22741877 | 0·69 | 0·39 | 0·65 | 0·45 | 0·47 | 0·72 |
| rs7549888 | G/T | 22749325 | 0·01 | −2·33 | 0·004 | 2·84 | 0·01 | 2·40 |
| rs17486657 | C/T | 22752481 | 0·33 | −0·97 | 0·17 | −1·38 | 0·34 | −0·94 |
| rs294222 | G/C | 22752866 | 0·71 | 0·36 | 0·44 | 0·77 | 0·29 | 1·04 |
| rs294223 | A/G | 22761312 | 0·29 | −1·05 | 0·37 | −0·90 | 0·38 | −0·94 |
| rs17433871 | T/C | 22761409 | 0·18 | −1·35 | 0·14 | −1·48 | 0·41 | −0·82 |
In a recent study of 103 SLE cases and 195 family members weak associations of two tag-SNPs at the C1Q locus and C1q levels were described. The strongest association in this study was observed with the variant rs587585 (P = 0·051) [20]. We tagged this SNP with a proxy at r2 = 0·43. No association of this proxy variant (rs186037) with C1q levels was observed in our study (P = 0·32). The association between the rs7549888 variant and C1q levels is the strongest association identified between a C1Q locus SNP and serum C1q levels, although this association does not survive a conservative Bonferroni correction for 18 SNPs tested (P = 0·14). Further studies are required to confirm this association.
Previous studies have analysed associations of two further candidate SNPs in the exonic regions [19,21,22] (C1QA-GLY70 (rs172378) (G/A) and C1QC-Pro14 (rs15940) (T/C)) of the C1QA and the C1QC genes and SLE affectation status. Neither of these polymorphisms have been genotyped in Caucasian participants of the HapMap project. We identified a tag-SNP (rs292001) for the C1QA-GLY70 polymorphism from the Seattle SNPs project at r2 > 0·8. This SNP is in strong linkage disequilibrium (LD) with a SNP (rs665691, r2 = 0·90) that we genotyped for association analysis. This SNP was not associated with C1q levels (P = 0·92) and SLE status (P = 0·67).
Discussion
The C1Q locus is an important candidate locus for association analysis with SLE. Here we report association analysis of 18 SNPs spread across the C1q encoding locus and the regulatory regions of these genes. We report a novel association of a variant in the 5′UTR region of the C1Q locus and C1q levels, but replication studies are required to increase the evidence for these associations. In our association screen of selected variants across 295 nuclear families, we failed to identify any strong associations between C1Q locus variants and SLE status. Our results do not provide any evidence that common variants at the C1Q locus confer high risk of SLE (OR ≥1·5), at least in our European population, although we may have missed weaker associations (OR ≤1·2) in our study, given the limited power and lack of complete coverage. Identification of C1Q variants associated with SLE would improve understanding of the role of C1Q locus in SLE predisposition. In addition, a recent study by Namjou et al. identified specific SNPs in the C1q gene (rs665691 and rs172378) that correlated with kidney nephritis, but this was specific to African American patients [28]. The SNP rs172378 is also known to be associated with lower levels of C1q in European ancestry patients with subacute cutaneous lupus [22]. In our study, we measured C1q titres in our European ancestry families with SLE and there was no difference in C1q titres compared with normal levels of C1q. These different associations with a single SNP of the C1QA gene appear to be important in different subsets of lupus patients. This might go some way in explaining the complex results and lack of correlation between C1q SNPs and association with SLE in mixed cohorts of patents. It would appear that the lupus cohort plays an influential role between complement genetics and phenotypic expression of disease activity.
In conclusion, we have performed a comprehensive analysis of common genetic variation at the C1q protein encoding loci. We found no evidence to suggest a causal role of common C1Q locus polymorphisms in SLE risk predisposition in a cohort of European ancestry.
Acknowledgments
This work was supported by research grants from the Arthritis Research Campaign, grants E0521, E0543, 17231 and 16537, and Lupus UK and the support of the Peninsula NIHR Clinical Research facility. We thank Mrs Susan Westoby for secretarial assistance.
Disclosure
The authors have no financial conflicts of interest.
References
- 1.Lachmann PJ, Muller-Eberhard HJ, Kunkel HG, Paronetto F. The localization of in vivo bound complement in tissue section. J Exp Med. 1962;115:63–82. doi: 10.1084/jem.115.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schur PH. Systemic lupus erythematosus and complement. Nephrologie. 1988;9:53–60. [PubMed] [Google Scholar]
- 3.Botto M. C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp Clin Immunogenet. 1998;15:231–4. doi: 10.1159/000019076. [DOI] [PubMed] [Google Scholar]
- 4.Botto M, Dell’Agnola C, Bygrave AE, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 5.Bowness P, Davies KA, Norsworthy PJ, et al. Hereditary C1q deficiency and systemic lupus erythematosus. QJM. 1994;87:455–64. [PubMed] [Google Scholar]
- 6.Clemenceau S, Castellano F, Montes de Oca M, Kaplan C, Danon F, Levy M. C4 null alleles in childhood onset systemic lupus erythematosus. Is there any relationship with renal disease? Pediatr Nephrol. 1990;4:207–12. doi: 10.1007/BF00857655. [DOI] [PubMed] [Google Scholar]
- 7.Fan Q, Uring-Lambert B, Weill B, Gautreau C, Menkes CJ, Delpech M. Complement component C4 deficiencies and gene alterations in patients with systemic lupus erythematosus. Eur J Immunogenet. 1993;20:11–21. doi: 10.1111/j.1744-313x.1993.tb00091.x. [DOI] [PubMed] [Google Scholar]
- 8.Lindqvist AK, Alarcon-Riquelme ME. The genetics of systemic lupus erythematosus. Scand J Immunol. 1999;50:562–71. doi: 10.1046/j.1365-3083.1999.00664.x. [DOI] [PubMed] [Google Scholar]
- 9.Petri M, Watson R, Winkelstein JA, McLean RH. Clinical expression of systemic lupus erythematosus in patients with C4A deficiency. Medicine. 1993;72:236–44. doi: 10.1097/00005792-199307000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Segurado OG, Arnaiz-Villena AA, Iglesias-Casarrubios P, et al. Combined total deficiency of C7 and C4B with systemic lupus erythematosus (SLE) Clin Exp Immunol. 1992;87:410–14. doi: 10.1111/j.1365-2249.1992.tb03011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan KE, Kim NA, Goldman D, Petri MA. C4A deficiency due to a 2 bp insertion is increased in patients with systemic lupus erythematosus. J Rheumatol. 1999;26:2144–7. [PubMed] [Google Scholar]
- 12.Traustadottir KH, Sigfusson A, Steinsson K, Erlendsson K. C4A deficiency and elevated level of immune complexes: the mechanism behind increased susceptibility to systemic lupus erythematosus. J Rheumatol. 2002;29:2359–66. [PubMed] [Google Scholar]
- 13.Truedsson L, Sturfelt G, Nived O. Prevalence of the type I complement C2 deficiency gene in Swedish systemic lupus erythematosus patients. Lupus. 1993;2:325–7. doi: 10.1177/096120339300200509. [DOI] [PubMed] [Google Scholar]
- 14.Wilson WA. Complement C4 levels in SLE in relation to null allele status. J Rheumatol. 1994;21:577–8. [PubMed] [Google Scholar]
- 15.Walport MJ, Davies KA, Morley BJ, Botto M. Complement deficiency and autoimmunity. Ann NY Acad Sci. 1997;815:267–81. doi: 10.1111/j.1749-6632.1997.tb52069.x. [DOI] [PubMed] [Google Scholar]
- 16.Sontheimer RD, Racila E, Racila DM. C1q: its functions within the innate and adaptive immune responses and its role in lupus autoimmunity. J Invest Dermatol. 2005;125:14–23. doi: 10.1111/j.0022-202X.2005.23673.x. [DOI] [PubMed] [Google Scholar]
- 17.Donnelly S, Roake W, Brown S, et al. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 2006;54:1543–56. doi: 10.1002/art.21783. [DOI] [PubMed] [Google Scholar]
- 18.Lewis MJ, Botto M. Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity. 2006;39:367–78. doi: 10.1080/08916930600739233. [DOI] [PubMed] [Google Scholar]
- 19.Chew CH, Chua KH, Lian LH, Puah SM, Tan SY. PCR–RFLP genotyping of C1q mutations and single nucleotide polymorphisms in Malaysian patients with systemic lupus erythematosus. Hum Biol. 2008;80:83–93. doi: 10.3378/1534-6617(2008)80[83:PGOCMA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 20.Martens HA, Zuurman MW, de Lange AH, et al. 1q polymorphisms suggests association with SLE, serum C1q and CH50 levels and disease severity. Ann Rheum Dis. 2009;68:715–20. doi: 10.1136/ard.2007.085688. [DOI] [PubMed] [Google Scholar]
- 21.Petry F, Loos M. Common silent mutations in all types of hereditary complement C1q deficiencies. Immunogenetics. 2005;57:566–71. doi: 10.1007/s00251-005-0023-z. [DOI] [PubMed] [Google Scholar]
- 22.Racila DM, Sontheimer CJ, Sheffield A, Wisnieski JJ, Racila E, Sontheimer RD. Homozygous single nucleotide polymorphism of the complement C1QA gene is associated with decreased levels of C1q in patients with subacute cutaneous lupus erythematosus. Lupus. 2003;12:124–32. doi: 10.1191/0961203303lu329oa. [DOI] [PubMed] [Google Scholar]
- 23.Russell AI, Cunninghame Graham DS, Shepherd C, et al. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Hum Mol Genet. 2004;13:137–47. doi: 10.1093/hmg/ddh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunninghame Graham DS, Manku H, Wagner S, et al. Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum Mol Genet. 2007;16:579–91. doi: 10.1093/hmg/ddl469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–7. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 26.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 27.Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 28.Namjou B, Gray-McGuire C, Sestak AL, et al. Evaluation of C1q genomic region in minority racial groups of lupus. Genes Immun. 2009;10:517–24. doi: 10.1038/gene.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milford Ward A, Sheldon J, Rowbottom A. 9th edn. Sheffield: 2007. Handbook of clinical immunochemistry. Protein reference units publications. [Google Scholar]