

Abstract
Infiltrating monocytes and macrophages contribute to the initiation and perpetuation of mucosal inflammation characteristic for human inflammatory bowel disease (IBD). Peripheral blood monocytes expressing the low-affinity Fcγ receptor CD16 have been identified previously as a major proinflammatory cell population, based on their unique cytokine secretion profile. However, the contribution of these cells to the pathogenesis of inflammatory bowel disease remains to be elucidated. Thus, in this study we investigated whether the peripheral CD16+ monocyte count correlates with common IBD disease parameters, and whether these cells infiltrate the intestinal mucosa under inflammatory conditions. We observed that CD16+ peripheral blood monocytes are increased significantly in active Crohn's disease, particularly in patients with high Crohn's disease activity index and colonic involvement. Furthermore, we found that CD16+ cells are a major contributor to the inflammatory infiltrate in Crohn's disease mucosa, although their spontaneous migration through primary human intestinal endothelial cells is limited. Our data suggest that lamina propria, but not peripheral blood, CD16+ monocytes are a crucial proinflammatory cell population in IBD, and a potential target for anti-inflammatory therapy.
Keywords: CD14, CD16, Crohn's disease, inflammatory bowel disease, monocytes
Introduction
The two major human inflammatory bowel diseases (IBD), Crohn's disease (CD) and ulcerative colitis (UC), are characterized by chronic recurring inflammation of the intestinal mucosa. Despite intensive research, the aetiology of IBD remains incompletely understood; however, a prominent feature is the infiltration of highly activated lymphocytes and leucocytes into the lamina propria, which contribute to the development and perpetuation of intestinal inflammation [1,2].
A subset of blood monocytes expressing the lipopolysaccharide (LPS) co-receptor CD14 and the low-affinity Fcγ receptor CD16, referred hereto as CD14+ CD16+ cells, has been identified previously as a major proinflammatory cell population (reviewed in [3]). CD14+ CD16+ monocytes exhibit a distinct cytokine secretion pattern, with low production of interleukin (IL)-10, but high levels of tumour necrosis factor (TNF)-α, IL-1 and IL-12. In addition, it has been shown that these cells are more efficient antigen-presenting cells than regular CD16- blood monocytes [3,4]. Based on these observations, it has been suggested that CD14+ CD16+ cells play an important role in the pathogenesis of inflammatory disorders. Indeed, an expansion of this cell population has been observed in a variety of conditions, including bacterial sepsis [5], atherosclerosis [6] and rheumatoid arthritis [7], among others. Recently, Hanai et al. reported an increase of CD14+ CD16+ peripheral blood monocytes in patients with active CD [8]. In addition, the authors showed that these cells were removed readily from the circulation by leucocyte apheresis. Because it has also been shown in a previous report that CD14+ CD16+ blood monocytes are depleted selectively by glucocorticoids in vivo[9], it has been suggested that these cells may represent a therapeutic target for the treatment of IBD.
In this study, we sought to investigate the contribution of CD14+ CD16+ monocytes to the inflammatory infiltrate in Crohn's disease mucosa, and to evaluate the rationale of targeting proinflammatory blood monocytes in order to ameliorate intestinal inflammation. Consistent with a previous report, we observed that the CD14+ CD16+ peripheral blood monocyte population was enhanced significantly in patients with active CD, but not quiescent disease or ulcerative colitis. Additionally, we found that there was a dramatic increase of proinflammatory monocytes in actively inflamed lamina propria specimens, and a trend towards higher CD14+ CD16+ cell numbers with increasing Crohn's disease activity index (CDAI) and colonic involvement. In contrast, we observed that these cells showed a very low spontaneous migration through intestinal endothelial cells, which was increased moderately under inflammatory conditions. Based on these results, we propose that peripheral blood CD14+ CD16+ monocytes are an indicator of, rather than a contributor to, intestinal inflammation. However, our data also indicate that proinflammatory monocytes in the inflamed mucosa may be a valid therapeutic target, which might explain the higher efficacy of glucocorticoid treatment compared to leucocyte apheresis.
Materials and methods
Study participants
All experiments involving human subjects and tissues were approved by the Ethics Committee at the University Hospital of Muenster, Muenster, Germany (to A.L. and T.K.) and were conducted following informed consent. Sixty-six patients with Crohn's disease, 10 patients with ulcerative colitis and 10 healthy volunteers were enrolled this study (Table 1). All UC patients were in clinical remission when samples were obtained. CDAI at the time of sample acquisition was calculated for CD patients according to the criteria by Best et al. [10], with quiescent disease defined as CDAI<150 and active CD as CDAI≥150. Further disease parameters of CD patients are summarized in Table 2. Mucosal tissue samples for immunofluorescence microscopy were obtained from patients with IBD or non-IBD pathologies undergoing scheduled colectomy at the Department of General Surgery, University Hospital of Muenster. Mucosal inflammation was assessed by a trained gastrointestinal (GI) pathologist (A.L. or J.H.), and samples were snap-frozen in liquid nitrogen for further processing. Tissue samples for leucocyte isolation studies were acquired by endoscopic resection during routine colonoscopy from patients with active CD on non-IBD conditions.
Table 1.
Information on the healthy volunteers and inflammatory bowel disease (IBD) patients enrolled in this study.
| Healthy control | Crohn's disease | Ulcerative colitis | |
|---|---|---|---|
| Group size | 10 | 66 | 10 |
| Median age (range) | 30·1 (25–34) | 30·5 (21–71) | 31·2 (22–42) |
| Gender ratio (male/female) | 6/4 | 30/36 | 4/6 |
Table 2.
Disease parameters of the Crohn's disease patients enrolled in this study.
| Median CDAI (range) | 114(0–385) |
| Median C-reactive protein (mg/dl) (range) | 3·5(0·1–9·7) |
| Median leucocyte count (×109/l) (range) | 7·6(3·2–17·9) |
| Disease manifestation | |
| •Colon | 46/66 |
| •Ileum | 34/66 |
| •Other | 5/66 |
| Medication | |
| •Glucocorticoids | 32/66 |
| •Azathioprine/6-mercaptopurine | 11/66 |
| •Other | 2/66 |
CDAI: Crohn's disease activity index.
Peripheral blood mononuclear cells (PBMC) isolation and flow cytometry
Peripheral blood was collected using Vacutainer CPT tubes with sodium citrate as anticoagulant (Becton Dickinson, Heidelberg, Germany). Mononuclear cells were isolated by centrifugation over a Ficoll-Hipaque gradient. Cells were then washed with phosphate-buffered saline (PBS) without calcium and magnesium supplemented with 13 mM sodium citrate (Merck, Darmstadt, Germany) and 1% fetal calf serum (PAA Laboratories, Coelbe, Germany). Cells were stained with primary labelled antibodies against CD16 [fluorescein isothiocyanate (FITC), CD14 phycoerythin (PE) and CD36 allophycocyanin (APC); all from BD Pharmingen, Heidelberg, Germany] for 1 h at room temperature, and analysed on a FACSCalibur fluorescence-activated cell sorter (BD) with CellQuest and WinMDI 2·8 analysis software. In some experiments, cells were stained additionally with 7-amino-actinomycin D to assess cell death, with <2% positive cells at any time. All dot plots in this paper depict 1000 cells.
Human intestinal microvascular endothelial cells (HIMEC) isolation
The isolation and culture of primary HIMEC was approved by the Ethics Committee at the University Hospital of Muenster (to J.H.). HIMEC were isolated from the colonic mucosa essentially as described previously [11]. In brief, tissue samples were washed with PBS containing 75 mM dithiothreitol, and then incubated repeatedly with 1 mM ethylenediamine tetraacetic acid (EDTA) in Hanks' buffer. The tissue was then digested with collagenase type II, and endothelial cells were released by gentle squeezing. After filtration, cells were seeded on collagen-coated tissue culture dishes, and colonies were transferred to new dishes after 2 weeks to obtain clonal populations of HIMEC. Endothelial identity was confirmed by characteristic cobblestone morphology and von Willebrand factor staining. Cells were maintained in endothelial cell growth medium (Promocell, Heidelberg, Germany) supplemented with 10% fetal calf serum (FCS) and antibiotics. HIMEC of passages 4–10 were used for experiments.
Transendothelial migration studies
The HIMEC were seeded on collagen-coated Transwell polycarbonate filter inserts with a pore size of 5 µm (Corning, Acton, MA, USA) and grown to confluence. Freshly isolated PBMC from healthy donors were resuspended in a 1 : 1 mix of endothelial cell growth medium and RPMI-1640 (Cambrex, Verviers, Belgium) and applied to the Transwell. After the indicated migration period, cells in the bottom and top wells were collected by vigorous pipetting. Cells adhering to the endothelial monolayer and the underside of the filter membrane were detached by brief incubation with Accutase solution (Millipore, Billerica, MA, USA). Cell recovery in all experiments was >95% and cell viability was >98%, as determined by trypan blue exclusion. Cells were then washed with PBS, and analysed by flow cytometry. The expression of intercellular cell adhesion molecule (ICAM)-1 and leucocyte function antigen (LFA)-1 was determined using primary labelled antibodies against CD54 and CD11a (BD Pharmingen), respectively. In some experiments, HIMEC monolayers were preactivated with human recombinant TNF-α (10 ng/ml) and IL-1β (5 ng/ml; both from Biosource, Solingen, Germany) for 1 h, and rinsed repeatedly with PBS to remove remaining cytokines prior to transmigration assays. Human umbilical vein endothelial cells (HUVEC) of passages 4–6 were used for select transmigration studies, as indicated. In some experiments, transmigration was performed in the presence of a blocking CD11a antibody (BioLegend, San Diego, CA, USA) or matched isotype control, at a concentration of 5 µg/ml.
Isolation of lamina propria leucocytes
Leucocytes were retrieved from colonic mucosal sections by repeated incubation with 1 mM EDTA in warm Hanks' buffer followed by rapid agitation. The tissue was then digested with DNase I (5 Kunitz-U/ml) and collagenase D (100 U/ml; both from Roche Diagnostics, Mannheim, Germany) in RPMI-1640 supplemented with 20% FCS for 1 h at 37°C. Leucocytes were purified by filtration and centrifugation over a non-continuous Percoll gradient, and analysed by flow cytometry.
Immunofluorescence microscopy
Cryosections of 7 µm thickness were cut from mucosal tissues embedded in Tissue-Tek OCT (Sakura Finetek, Staufen, Germany). Sections were fixed with 100% ethanol at −20°C for 20 min, washed with Hanks' buffer, and blocked with 2% wt/vol bovine serum albumin in Hanks' buffer with 0·1% Tween-20. Slides were then incubated at 4°C overnight with the following primary antibodies: mouse anti-CD14 (BioLegend, San Diego, CA, USA), rat anti-CD16 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit anti-CD36 (Novus Biologicals, Littleton, CO, USA). For the detection of endogenous TNF-α, specimens were incubated with a rabbit anti-TNF-α antibody (Novus Biologicals), instead of anti-CD36. Tissue was stained subsequently with AlexaFluor-labelled secondary antibodies (Invitrogen, Carlsbad, CA, USA), and nuclei were stained in dual-labelled samples using TOPRO-3 iodide (Invitrogen). Slides were embedded in p-phenylene, and analysed on an LSM510 confocal microscope equipped with a Plan-NEOFLUAR 40×/1·3 and 63×/1·4 oil objective (Carl Zeiss, Thornwood, NY, USA), using LSM5 software supplied by the vendor. TNF-α mean fluorescence per cell was determined using NIH ImageJ software.
Assessment of nucleotide-binding oligomerization domain containing 2 (NOD2) gene status
Genomic DNA was isolated from the peripheral blood using a QIAamp DNA Blood Kit (Qiagen, Hilden, Germany). Mutation of the NOD2 gene at the known IBD susceptibility loci 702, 908 and 1007 was determined by polymerase chain reaction (PCR), as described previously [12]. PCR products were separated by electrophoresis on a 12% horizontal polyacrylamide gel.
Statistics
Data were analysed using Prism 5 (GraphPad Software, La Jolla, CA, USA). Means were compared with Student's t-test or analysis of variance (anova). Correlation coefficients were determined by Spearman's rank-order analysis. P < 0·05 was considered statistically significant. Results are displayed as mean ± standard error of the means.
Results
CD14+ CD16+ monocytes are enhanced in active Crohn's disease
To investigate if the proinflammatory CD14+ CD16+ cell population is enhanced in the IBD patients enrolled in this study, we analysed peripheral blood samples by three-channel flow cytometry (Fig. 1a). Monocytes were identified by their characteristic forward- and side-scatter profile and expression of CD36, and separated by CD14/CD16 expression. Consistent with a previous study [13], CD16+ monocytes accounted for approximately 10% of all CD14+ cells in healthy volunteers. In contrast, we observed a marked increase of this population in Crohn's disease patients with quiescent (CDAI<150) and particularly active (CDAI>150) disease. Quantification of this difference revealed a significant enhancement of CD14+ CD16+ monocytes in CD patients compared to control (9·8 ± 1·1% versus 13·8 ± 1·2%; P = 0·01) (Fig. 1b). As our results suggested that the CD16+ population increases in acute inflammation, we examined CD patients based on disease activity (Fig. 1c). In agreement with a recent study [8], a significant increase of CD14+ CD16+ blood monocytes was observed in patients with active CD (18·9 ± 2·7%; P < 0·05 versus control), but not in quiescent CD (12·1 ± 1·1%) or ulcerative colitis in remission (9·8 ± 2·2%).
Fig. 1.
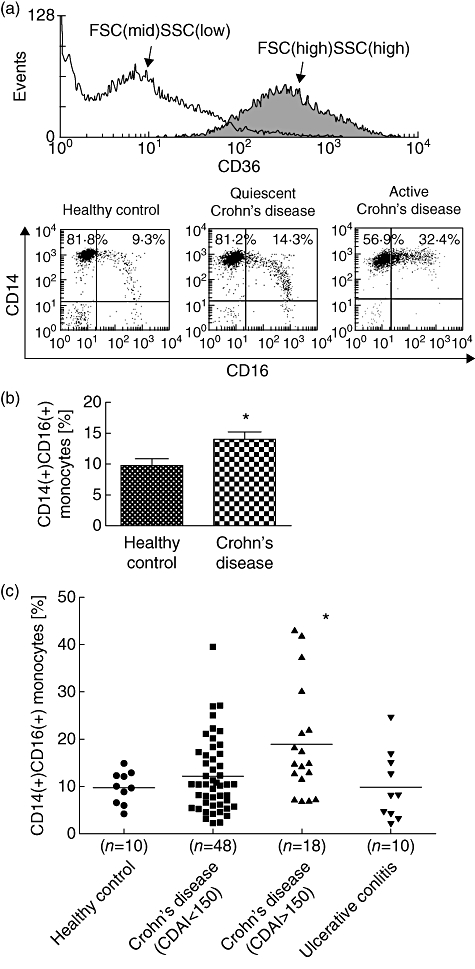
The peripheral blood CD14+ CD16+ monocyte population is increased in active Crohn's disease. (a) Peripheral blood mononuclear cells (PBMC) were stained for CD14, CD16 and CD36 and analysed by flow cytometry. Cells were pre-gated for high forward-scatter (FSC), side-scatter (SSC) and CD36 expression, and monocytes separated by CD14/CD16 expression. The lower panels show representative dot plots from a healthy volunteer, and Crohn's disease patients with quiescent [Crohn's disease activity index (CDAI)<150] and active (CDAI>150) inflammation. (b) The ratio of CD14+ CD16+ monocytes was increased significantly in Crohn's disease patients compared to healthy controls. *P = 0·01 (two-tailed Student's t-test with Welch's correction). (c) Further categorization of patients revealed that the CD14+ CD16+ population was increased in active Crohn's disease compared to healthy controls, but not quiescent disease or patients with ulcerative colitis. *P < 0·05 (Bonferroni's post-test following one-way analysis of variance).
Occurrence of CD14+ CD16+ blood monocytes does not correlate with common CD disease parameters
Because we and others have observed that the proinflammatory CD14+ CD16+ monocyte population is increased in active Crohn's disease, we next asked whether the ratio of these cells correlated with known CD disease parameters. No association with patient age, C-reactive protein levels and peripheral blood leucocyte count was observed in this study, suggesting that these factors are not relevant for the interpretation of the data (Fig. 2a–c). In contrast, there was a trend towards higher CD14+ CD16+ cell counts with increasing CDAI (Fig. 2d); however, this association did not reach statistical significance (R2 = 0·07; P > 0·05). Glucocorticoids are a potent therapeutic tool in the treatment of IBD. We therefore examined the occurrence of CD14+ CD16+ monocytes in patients receiving corticosteroids at the time of sample acquisition (Fig. 2e). CD16+ cells were reduced substantially in patients on glucocorticoid medication, when compared to the patient group receiving no or non-steroidal drugs (12·5 ± 1·8% versus 15·7 ± 1·5%; P = 0·14), consistent with a previous report showing selective depletion of these cells with methylprednisolone [9]. Remarkably, this reduction was observed despite a higher disease activity index in the patient group receiving corticosteroid medication (CDAI 123·8 ± 17·4 versus 99·8 ± 12·3), indicating that the actual effect of these drugs on CD14+ CD16+ cells may be masked by the more severe disease manifestation. Mutations in the NOD2 gene expressed by monocytes pathologically alter the response of phagocytes to bacterial exposure, and have been linked to the development of IBD [14]. Thus, we asked if mutation of NOD2 at known susceptibility loci (R702W, G908R, 1007fs) increased the ratio of CD14+ CD16+ monocytes (Fig. 2f). No difference was observed between the two groups in our study population (normal: 13·5 ± 1·8%; mutated: 14·9 ± 1·6%), suggesting that NOD2 is not involved in the occurrence of these cells. Finally, we investigated if disease manifestation may affect the number of CD14+ CD16+ monocytes (Fig. 2g). We observed that CD16+ cells were lowest in the patient group with ileal involvement (11·7 ± 1·7%), and markedly higher in patients with colonic CD (15·6 ± 2·2%). The greatest number of proinflammatory monocytes was observed in patients with combined colonic and ileal CD (16·9 ± 3·1%), which may reflect the extent of disease.
Fig. 2.
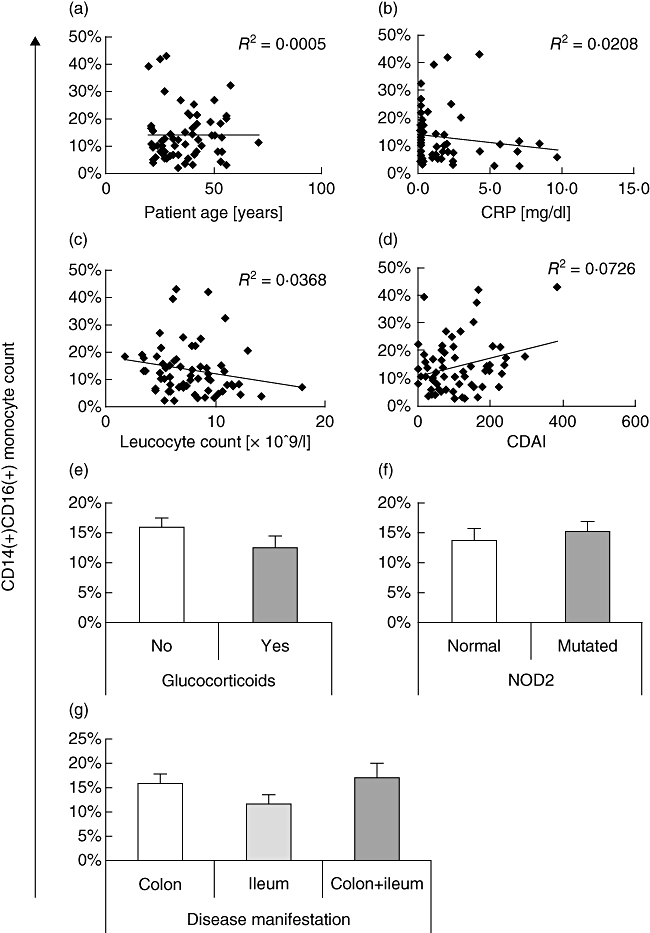
Correlation of CD14+ CD16+ peripheral blood mononuclear cells (PBMC) with Crohn's disease parameters. The ratio of CD14+ CD16+ cells was plotted against (a) patient age; (b) C-reactive protein levels; (c) peripheral blood leucocyte count; and (d) Crohn's disease activity index (CDAI). No significant correlation was observed for any of these factors, although there was a trend towards higher CD14+ CD16+ counts with increasing CDAI. (e) Extended treatment with glucocorticoid medication reduced the number of CD14+ CD16+ monocytes. (f) Mutations in the nucleotide-binding oligomerization domain containing 2 (NOD2) gene (R702W, G908R, 1007fs or combination thereof) did not change the ratio of CD14+ CD16+ cells. (g) Patients with colonic or combined colonic and ileal disease manifestation exhibited an increased ratio of CD14+ CD16+ monocytes.
CD14+ CD16+ monocytes are increased significantly in the inflamed intestinal mucosa
It is feasible that peripheral CD14+ CD16+ monocytes contribute to the inflammatory infiltrate found in the lamina propria of IBD patients. We therefore investigated the occurrence of these cells in the mucosa of Crohn's disease patients. Monocytic origin of intestinal CD14+ CD16+ cells was confirmed by three-colour immunofluorescence microscopy (Fig. 3a). CD14+ CD16- and CD14- CD16+ cells exhibited limited staining for CD36. In contrast, CD14+ CD16+ cells showed strong immunoreactivity for CD36. We next assessed CD14+ CD16+ monocytes in representative mucosal sections from non-IBD control patients, non-inflamed Crohn's disease mucosa, actively inflamed CD tissue and non-inflamed ulcerative colitis specimens (Fig. 3b). We observed that these cells were rare in non-IBD controls, but were increased massively in mucosal sections from CD patients. Quantification of these results revealed a highly significant increase of proinflammatory monocytes in non-inflamed CD mucosa compared to controls [7·5 ± 0·6 versus 1·4 ± 0·2 cells/high-power field (HPF); P < 0·001] (Fig. 3c). This increase was enhanced further in actively inflamed tissue samples (11·2 ± 0·8 cells/HPF; P < 0·001 versus IBD non-inflamed). Interestingly, the number of proinflammatory monocytes was only slightly elevated in UC tissue from patients in remission (3·9 ± 0·2 cells/HPF). We additionally investigated the ratio of CD14+ CD16+ cells among all CD14+ leucocytes in these mucosal specimens (Fig. 3d). The percentage of CD14+ CD16+ monocytes was increased dramatically in non-inflamed (43·0 ± 2·4%) and inflamed IBD tissue (49·1 ± 2·5%) compared to non-IBD control (12·8 ± 2·1%; P < 0·001), but not in ulcerative colitis samples (23·5 ± 1·6%). Although our results suggest that proinflammatory monocytes may play a more important role in the pathobiology of Crohn's disease, a potential contribution of these cells to the pathology of severe active UC remains to be investigated in future studies. To confirm that the above observations were not the result of observer bias, we isolated total lamina propria leucocytes from endoscopic mucosal samples from CD patients and non-IBD controls, and analysed these cells by flow cytometry (Fig. 3e). In good agreement with the microscopy data, we saw a large population of CD14+ CD16+ monocytes in IBD specimens, but not in non-IBD controls. Finally, as peripheral CD14+ CD16+ cells have been characterized as highly proinflammatory based on their cytokine secretion profile, we sought to confirm this phenotype in mucosal monocytes by assessing their expression of TNF-α (Fig. 3f). Indeed, within the same field of vision, CD16+ cells consistently exhibited a stronger expression of TNF-α than CD16- monocytes [mean fluorescence intensity (MFI) 24·7 ± 2·7 versus 18·5 ± 2·1; P < 0·001]. Taken together, these results indicate that CD14+ CD16+ cells are a major proinflammatory cell population in the mucosal infiltrate seen in inflammatory bowel disease.
Fig. 3.
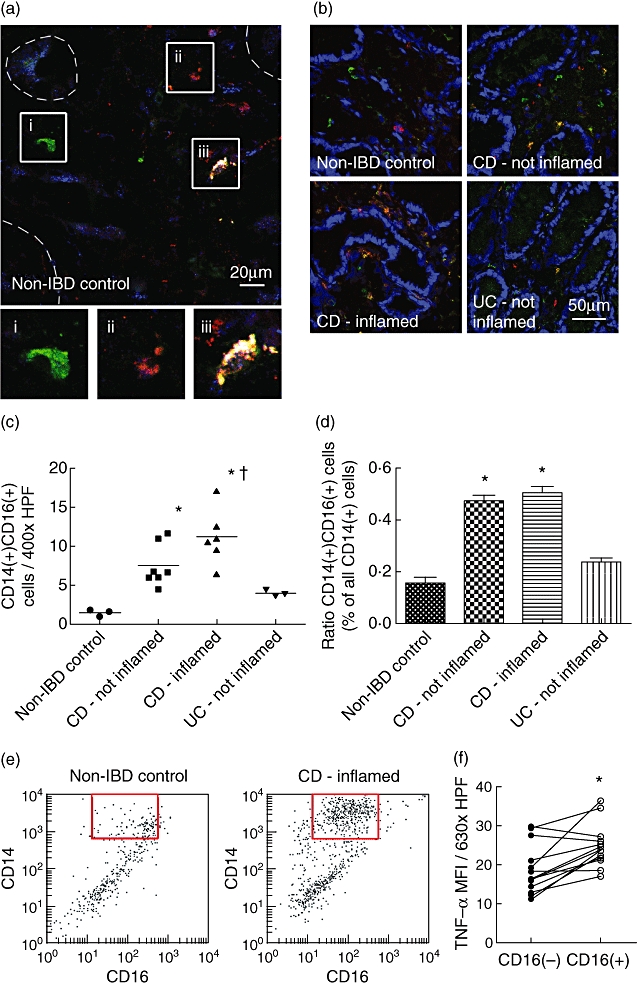
The number of CD14+ CD16+ monocytes is enhanced significantly in the lamina propria of Crohn's disease patients. (a) Monocytic lineage of CD14+ CD16+ cells in the colonic mucosa was confirmed by three-channel immunofluorescence microscopy for CD14 (green), CD16 (red) and CD36 (blue). No or limited CD36 staining was observed in CD14+ CD16– cells (i) and CD14– CD16+ cells (ii). In contrast, CD14+ CD16+ cells stained strongly for CD36 (iii, white pseudocolour). Colonic crypts are indicated with dashed lines. Lower panels, magnification of the boxed cells shown above. (b) Representative images of colonic mucosal sections from non-inflammatory bowel disease (IBD) control patients, non-inflamed and inflamed tissue from Crohn's disease patients, and non-inflamed ulcerative colitis samples. CD14+ CD16+ monocytes are shown in yellow pseudocolour, with nuclei in blue. (c) The number of CD14+ CD16+ cells per high-power field (HPF) was significantly increased in Crohn's disease mucosa, and enhanced further in actively inflamed tissue. Each data point represents the average of five or more HPF per patient sample. *P < 0·001 versus control, †P < 0·001 versus IBD – not inflamed [Bonferroni's post-test following one-way analysis of variance (anova)]. (d) The ratio of CD14+ CD16+ cells was increased significantly in Crohn's disease patients compared to controls. *P < 0·001 (Bonferroni's post-test following one-way anova). (e) The increase in total CD14+ CD16+ monocytes was confirmed by flow cytometry. Mononuclear cells were isolated from control and Crohn's disease colonic mucosa as described in the Materials and methods, pre-gated for forward-scatter (high), side-scatter (high) CD36+, and separated by CD14/CD16 expression. A dramatic increase of CD14+ CD16+ monocytes was seen in IBD tissue (red box). Plots are representative of three patient samples per group. (f) The tumour necrosis factor (TNF)-α mean fluorescence intensity (MFI, arbitrary units) of CD14+ CD16+ monocytes was significantly higher compared to CD14+ CD16– cells in the same field of vision. The graph shows the result from 14 HPF taken from three Crohn's disease patient samples. *P < 0·001 (two-tailed Student's t-test).
CD14+ CD16+ monocytes migrate poorly through intestinal endothelial cells
Two independent mechanisms may contribute to the expansion of inflammatory CD14+ CD16+ monocytes in IBD mucosa: an increased extravasation of these cells in response to inflammatory stimuli, and the de novo expression of CD16 on CD14+ CD16- cells. To investigate the former hypothesis, we employed an in vitro transmigration model of freshly isolated peripheral blood mononuclear cells (PBMC) through a confluent monolayer of primary human microvascular endothelial cells (HIMEC) that is depicted schematically in Fig. 4a. Preliminary studies revealed that leucocyte attachment and migration was most pronounced at 2–4 h, and no further extravasation was seen after 8 h (data not shown). Thus, after 4 h of migration, non-adherent, adherent and transmigrated PBMC were collected as described in the Materials and methods, and analysed by flow cytometry (Fig. 4a). We observed that significantly fewer CD16+ monocytes than CD16- cells had migrated through the endothelial monolayer at this time-point (4·7 ± 0·4% versus 18·0 ± 2·0%; P < 0·01). In contrast, considerably more CD16+ cells were found attached to the endothelial cells (27·5 ± 7·1% versus 14·5 ± 3·7), consistent with a previous report [15]. Interestingly, the reduced migration was reflected in control conditions with empty Transwell filters (migrated CD16- cells: 76·5 ± 3·2%; migrated CD16+ cells: 37·6 ± 6·8%), suggesting that CD14+ CD16+ monocytes exhibit strong adherence, but reduced migration and detachment properties compared to CD16- cells. We next investigated if prestimulation of endothelial cells with inflammatory cytokines enhanced the extravasation of proinflammatory monocytes. Preincubation of HIMEC with TNF-α and IL-1β resulted in a small decrease in attachment to, and corresponding increase in migration through endothelial cells, which was more pronounced in CD16+ monocytes. To study this effect more closely, we repeated these experiments with a greater sample size and longer transmigration time (Fig. 4b). Prestimulation of HIMEC resulted in a significantly increased transendothelial migration of CD16+ monocytes, but not CD16- cells, after 8 h (1·7 ± 0·1 versus 0·9 ± 0·0-fold of unstimulated; P < 0·001). However, the net migration of proinflammatory monocytes was still markedly lower under these conditions. To investigate potential cell adhesion molecules responsible for the strong attachment but poor migration of CD14+ CD16+ monocytes, we examined the expression of intercellular adhesion molecule-1 (ICAM-1/CD54) and leucocyte function-associated antigen-1 α-chain (LFA-1/CD11a) on peripheral blood monocytes. Consistent with a previous study [16], we observed an increased expression of CD54 on CD16+ monocytes, compared to CD16- cells (mean fluorescence intensity 1711 ± 297 versus 1087 ± 206; P < 0·001) (Fig. 4c). Conversely, high levels of CD11a were detected only on cells with a high expression of CD14, which correspond mainly to the CD14+ CD16- monocyte population (Fig. 4d). Analysis of migrated cells revealed that these cells were almost exclusively CD14high, with a dramatic enrichment in the CD14high CD11ahigh subset of monocytes. Indeed, further examination of CD14high CD11a+ cells showed that the majority of these cells had migrated through, or were attached to, the endothelial monolayer after 4 h (migrated: 45·4 ± 3·7%; adherent: 28·8 ± 1·6%). To confirm the critical importance of CD11a for leucocyte extravasation, monocytes were allowed to migrate through TNF-α/IL-1β-stimulated HUVEC cells for 4 h, in the presence of a blocking CD11a antibody or isotype control (Fig. 4e). As can be seen, inhibition of CD11a substantially reduced the migration of CD16- (0·5 ± 0·04-fold of control; P = 0·01) and CD16+ monocytes (0·09 ± 0·4-fold of control; P = 0·30). Similar results were obtained with unstimulated endothelial cells (not shown). These data support that LFA-1 is critical for transendothelial migration, and suggest that the failure of CD16+ monocytes to travel across the monolayer may be explained by a reduced expression of this cell adhesion molecule.
Fig. 4.
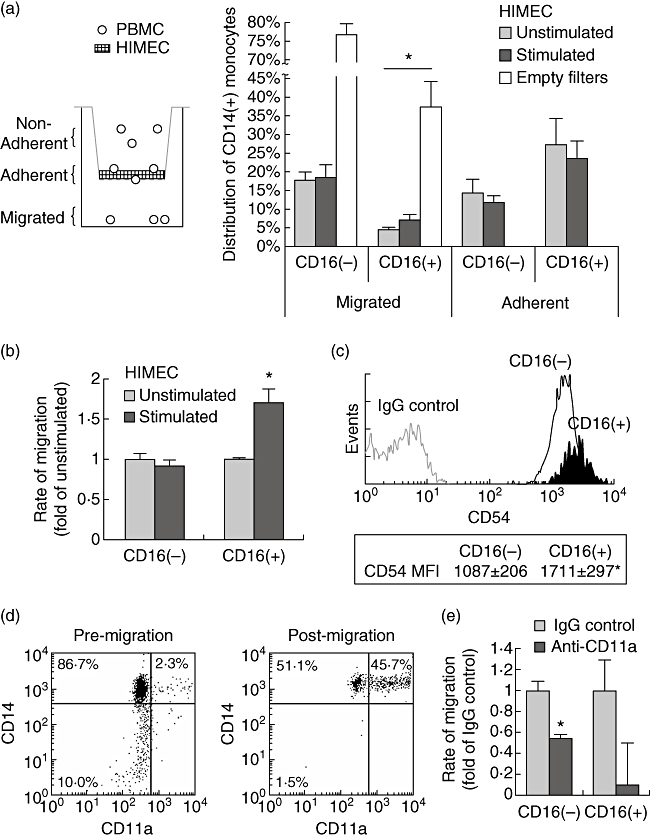
Stimulation of intestinal endothelial cells increases the extravasation of CD14+ CD16+ monocytes. (a) Freshly isolated peripheral blood mononuclear cells (PBMC) were allowed to migrate through primary human microvascular endothelial cells (HIMEC) for 4 h. CD16+ cells were attached preferentially to endothelial cells, but transmigrated less efficiently than CD16– cells. Prestimulation of HIMEC with tumour necrosis factor (TNF)-α (10 ng/ml) and interleukin (IL)-1β (5 ng/ml) increased the extravasation of CD16+ monocytes. *P < 0·05 versus CD16–[Bonferroni's post-test following one-way analysis of variance (anova)]. (b) PBMC were allowed to migrate through HIMEC for 8 h. Prestimulation of endothelial cells increased the transmigration of CD16+ monocytes, but not CD16– cells. *P < 0·001 versus CD16– (two-tailed Student's t-test). (c) CD16+ monocytes exhibited a higher CD54 mean fluorescence intensity (MFI) compared to CD16– cells. *P < 0·001 versus CD16– (two-tailed Student's t-test). (d) PBMC were allowed to migrate through unstimulated HIMEC for 4 h. Transmigrated cells were strongly enriched for CD14high CD11ahigh monocytes. (e) Monocyte transmigration through human umbilical vein endothelial cells (HUVEC) cells was inhibited strongly in the presence of a blocking CD11a antibody. *P = 0·01 versus immunoglobulin G (IgG) control (two-tailed Student's t-test).
Discussion
A hallmark of human inflammatory bowel disease is chronic recurring mucosal inflammation, which is perpetuated mainly by infiltrating lymphocytes and leucocytes. The CD16+ subset of peripheral blood monocytes has been identified as a major proinflammatory cell population, and its expansion has been reported in patients with active IBD [3,8]. It has thus been proposed that these peripheral CD14+ CD16+ monocytes contribute to the inflammatory mucosal infiltrate, and that their depletion from the circulation may alleviate IBD symptoms [8,9,17].
However, based on the transendothelial migration and correlation studies performed here, together with previously published data from other groups, it appears unlikely that the peripheral blood is a major reservoir for migrating proinflammatory CD16+ monocytes. We now propose the following model, which is illustrated in Fig. 5. At the onset of inflammation, CD16- monocytes extravasate into the affected tissue, whereas the majority of CD16+ cells fails to traverse the endothelial barrier (Fig. 5a). This is supported by the transmigration data presented in this study, as well as by a previous publication showing reduced CD16+ migration through HUVEC in the presence of CCL2, which was attributed to a lack of CCR2 on these cells [18]. Similarly, it has been shown that CX3CL1 increases the adhesion of these cells to HUVEC and brain endothelial cells, but decreases their transendothelial migration [15]. In additional support of our model, in vivo studies in mice revealed that Ly-6C (low) blood monocytes, which correspond to human CD14+ CD16+ cells, failed to infiltrate sites of inflammation in a model of bacterial infection [19]. In this context, it is important to point out that the gene expression profile of intestinal endothelial cells differs markedly from that of HUVEC and skin endothelial cells (J.H., unpublished data), which are used commonly for extravasation studies. In fact, it has been shown that the expression of cell adhesion molecules in response to cytokines differs markedly between HIMEC and HUVEC [20,21], which highlights the physiological relevance of this study. Additionally, the critical role of cell adhesion molecules was highlighted by our observation that a gradient of serum or monocyte chemotactic protein-1 did not increase the extravasation of CD14+ CD16+ cells (data not shown).
Fig. 5.
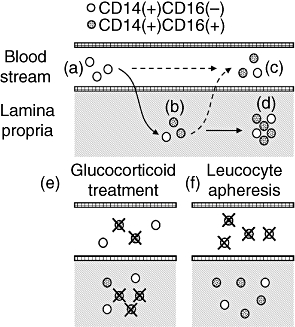
Proposed model of CD16+ monocyte enrichment during intestinal inflammation. Hypotheses not investigated in this manuscript are indicated by dashed arrows. (a) CD14+ CD16– extravasate into the mucosa in response to inflammatory stimuli. (b) In the lamina propria, inflammatory monocytes undergo a phenotype switch to express CD16. (c) Some CD16+ monocytes reverse-transmigrate back into the blood stream. (d) CD16– and proinflammatory CD16+ monocytes perpetuate intestinal inflammation through secretion of inflammatory cytokines. (e) The efficacy of glucocorticoids may be conveyed in part by their ability to deplete CD16+ cells in the bloodstream and the lamina propria. (f) In contrast, leucocyte apheresis only removes immune cells from the peripheral blood, thereby reducing the reservoir of infiltrating leucocytes.
In light of these observations, it is reasonable to assume that the majority of CD14+ CD16+ monocytes in IBD mucosa identified here are derived from immature CD16- cells (Fig. 5b). Indeed, it has been reported that CD16 expression on phagocytes can be induced by a variety of cytokines and chemokines found in inflammatory disorders, including macrophage-colony-stimulating factor (M-CSF), IL-10 and transforming growth factor (TGF)-β[22–24]. Recently, Paulsson et al. showed that monocytes that had migrated into skin blisters acquired a phenotype with striking resemblance of peripheral CD14+ CD16+ cells [25], which strongly supports our working model. Interestingly, it has been shown previously that CD16+ monocytes can reverse-transmigrate spontaneously back into the bloodstream, and differentiate into dendritic cells with high antigen-presenting potential [26]. This is concurrent with the notion that CD16+ blood cell represent a more mature monocyte subset, based on the expression of chemokines and chemokine receptors on these cells, and a reduction of CD11b and CD33 similar to mature macrophages [16,27]. Although we have not investigated this hypothesis in the current study, it is possible in light of these observations that at least some peripheral CD14+ CD16+ monocytes are, in fact, recruited from tissue reservoirs, rather than vice versa (Fig. 5c). The elevated levels of these cells observed in IBD and other inflammatory disorders may thus reflect the grade of tissue inflammation, which is suggested by our observation that CD14+ CD16+ correlate with Crohn's disease activity. Alternatively, it is conceivable that chemokines and cytokines in the blood may drive the differentiation of CD16- cells during inflammation. We suggest that these cells, while in the periphery, are not an important therapeutic target due to their limited migratory potential. However, based on the data presented in this study, it is likely that mucosal CD14+ CD16+ cells are a critical contributor to the perpetuation of intestinal inflammation, and a potential target for the treatment of inflammatory bowel disease (Fig. 5d).
Glucocortoids and leucocyte apheresis have both been shown to efficiently deplete the peripheral pool of proinflammatory monocytes [8,9,17], but while corticosteroid preparations have consistently proven to be a potent tool for treating IBD, large-scale studies confirming the efficacy the leucocyte apheresis are still unavailable [28]. With regard to the cell populations investigated here, this difference in efficacy may be explained by the ability of corticosteroid drugs to act on cells in the lamina propria. Thus, it is feasible that these drugs preferentially induce apoptosis in CD16+ monocytes in the peripheral blood [9,29], as well as in the intestinal mucosa (Fig. 5e), although further studies will be required to assess if mucosal CD14+ CD16+ cells are susceptible to corticosteroid treatment. In contrast, due to the nature of leucocyte apheresis, immune cells are depleted only from the peripheral blood, while mucosal leucocytes are unaffected (Fig. 5f). This may reduce the pool of monocytes which replenishes the mucosal inflammatory infiltrate, and thus ameliorate chronic, but not acute inflammation.
In summary, in the current study we identify mucosal CD14+ CD16+ monocytes as a major proinflammatory immune cell population in Crohn's disease, and discuss the role of peripheral CD14+ CD16+ cells as potential disease indicators and drug targets for anti-inflammatory treatment. Interestingly, a recent report showed that macrophages derived from peripheral blood monocytes of CD patients secrete significantly fewer cytokines after bacterial challenge than cells from healthy controls [30]. As cytokine concentrations in CD mucosa are elevated, it is possible that the phenotypical switch of CD16- monocytes that we propose here coincides with an activation of these cells. This hypothesis remains to be evaluated in future studies.
Acknowledgments
The authors would like to thank Mrs Sonja Dufentester and Elke Weber for technical assistance, and Drs Christian Krieglstein and Matthias Bruewer for help with the acquisition of surgical specimens. S.K. is supported by a Research Fellowship Award from the Crohn's and Colitis Foundation of America.
Disclosure
The authors declare no conflict of interest.
References
- 1.Mahida YR. The key role of macrophages in the immunopathogenesis of inflammatory bowel disease. Inflamm Bowel Dis. 2000;6:21–33. doi: 10.1097/00054725-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Selby WS, Janossy G, Bofill M, Jewell DP. Intestinal lymphocyte subpopulations in inflammatory bowel disease: an analysis by immunohistological and cell isolation techniques. Gut. 1984;25:32–40. doi: 10.1136/gut.25.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol. 2007;81:584–92. doi: 10.1189/jlb.0806510. [DOI] [PubMed] [Google Scholar]
- 4.Grage-Griebenow E, Zawatzky R, Kahlert H, Brade L, Flad H, Ernst M. Identification of a novel dendritic cell-like subset of CD64(+)/CD16(+) blood monocytes. Eur J Immunol. 2001;31:48–56. doi: 10.1002/1521-4141(200101)31:1<48::aid-immu48>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Fingerle G, Pforte A, Passlick B, Blumenstein M, Strobel M, Ziegler-Heitbrock HW. The novel subset of CD14+/CD16+ blood monocytes is expanded in sepsis patients. Blood. 1993;82:3170–6. [PubMed] [Google Scholar]
- 6.Hakkinen T, Karkola K, Yla-Herttuala S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Colocalization with epitopes of oxidized low-density lipoprotein, scavenger receptor, and CD16 (Fc gammaRIII) Virchows Arch. 2000;437:396–405. doi: 10.1007/s004280000239. [DOI] [PubMed] [Google Scholar]
- 7.Kawanaka N, Yamamura M, Aita T, et al. CD14+, CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002;46:2578–86. doi: 10.1002/art.10545. [DOI] [PubMed] [Google Scholar]
- 8.Hanai H, Iida T, Takeuchi K, et al. Adsorptive depletion of elevated proinflammatory CD14+CD16+DR++ monocytes in patients with inflammatory bowel disease. Am J Gastroenterol. 2008;103:1210–16. doi: 10.1111/j.1572-0241.2007.01714.x. [DOI] [PubMed] [Google Scholar]
- 9.Fingerle-Rowson G, Angstwurm M, Andreesen R, Ziegler-Heitbrock HW. Selective depletion of CD14+ CD16+ monocytes by glucocorticoid therapy. Clin Exp Immunol. 1998;112:501–6. doi: 10.1046/j.1365-2249.1998.00617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Best WR, Becktel JM, Singleton JW, Kern F., Jr Development of a Crohn's disease activity index. National Cooperative Crohn's Disease Study. Gastroenterology. 1976;70:439–44. [PubMed] [Google Scholar]
- 11.Haraldsen G, Rugtveit J, Kvale D, et al. Isolation and longterm culture of human intestinal microvascular endothelial cells. Gut. 1995;37:225–34. doi: 10.1136/gut.37.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Helio T, Halme L, Lappalainen M, et al. CARD15/NOD2 gene variants are associated with familially occurring and complicated forms of Crohn's disease. Gut. 2003;52:558–62. doi: 10.1136/gut.52.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- 14.Strober W, Kitani A, Fuss I, Asano N, Watanabe T. The molecular basis of NOD2 susceptibility mutations in Crohn's disease. Mucosal Immunol. 2008;1(Suppl 1):S5–9. doi: 10.1038/mi.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ancuta P, Moses A, Gabuzda D. Transendothelial migration of CD16+ monocytes in response to fractalkine under constitutive and inflammatory conditions. Immunobiology. 2004;209:11–20. doi: 10.1016/j.imbio.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Ziegler-Heitbrock HW, Fingerle G, Strobel M, et al. The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur J Immunol. 1993;23:2053–8. doi: 10.1002/eji.1830230902. [DOI] [PubMed] [Google Scholar]
- 17.Kanai T, Makita S, Kawamura T, et al. Extracorporeal elimination of TNF-alpha-producing CD14(dull) CD16(+) monocytes in leukocytapheresis therapy for ulcerative colitis. Inflamm Bowel Dis. 2007;13:284–90. doi: 10.1002/ibd.20017. [DOI] [PubMed] [Google Scholar]
- 18.Weber C, Belge KU, von Hundelshausen P, et al. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. doi: 10.1002/jlb.67.5.699. [DOI] [PubMed] [Google Scholar]
- 19.Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–17. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 20.Haraldsen G, Kvale D, Lien B, Farstad IN, Brandtzaeg P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J Immunol. 1996;156:2558–65. [PubMed] [Google Scholar]
- 21.Ogawa H, Binion DG, Heidemann J, et al. Mechanisms of MAdCAM-1 gene expression in human intestinal microvascular endothelial cells. Am J Physiol Cell Physiol. 2005;288:C272–81. doi: 10.1152/ajpcell.00406.2003. [DOI] [PubMed] [Google Scholar]
- 22.Iwahashi M, Yamamura M, Aita T, et al. Expression of Toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 2004;50:1457–67. doi: 10.1002/art.20219. [DOI] [PubMed] [Google Scholar]
- 23.Wahl SM, Allen JB, Welch GR, Wong HL. Transforming growth factor-beta in synovial fluids modulates Fc gamma RII (CD16) expression on mononuclear phagocytes. J Immunol. 1992;148:485–90. [PubMed] [Google Scholar]
- 24.Sanchez-Torres C, Garcia-Romo GS, Cornejo-Cortes MA, Rivas-Carvalho A, Sanchez-Schmitz G. CD16+ and CD16– human blood monocyte subsets differentiate in vitro to dendritic cells with different abilities to stimulate CD4+ T cells. Int Immunol. 2001;13:1571–81. doi: 10.1093/intimm/13.12.1571. [DOI] [PubMed] [Google Scholar]
- 25.Paulsson JM, Held C, Jacobson SH, Lundahl J. In vivo extravasated human monocytes have an altered expression of CD16, HLA-DR, CD86, CD36 and CX(3)CR1. Scand J Immunol. 2009;70:368–76. doi: 10.1111/j.1365-3083.2009.02306.x. [DOI] [PubMed] [Google Scholar]
- 26.Randolph GJ, Sanchez-Schmitz G, Liebman RM, Schakel K. The CD16(+) (FcgammaRIII(+)) subset of human monocytes preferentially becomes migratory dendritic cells in a model tissue setting. J Exp Med. 2002;196:517–27. doi: 10.1084/jem.20011608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grip O, Bredberg A, Lindgren S, Henriksson G. Increased subpopulations of CD16(+) and CD56(+) blood monocytes in patients with active Crohn's disease. Inflamm Bowel Dis. 2007;13:566–72. doi: 10.1002/ibd.20025. [DOI] [PubMed] [Google Scholar]
- 28.Abreu MT, Plevy S, Sands BE, Weinstein R. Selective leukocyte apheresis for the treatment of inflammatory bowel disease. J Clin Gastroenterol. 2007;41:874–88. doi: 10.1097/MCG.0b013e3180479435. [DOI] [PubMed] [Google Scholar]
- 29.Dayyani F, Belge KU, Frankenberger M, Mack M, Berki T, Ziegler-Heitbrock L. Mechanism of glucocorticoid-induced depletion of human CD14+CD16+ monocytes. J Leukoc Biol. 2003;74:33–9. doi: 10.1189/jlb.1202612. [DOI] [PubMed] [Google Scholar]
- 30.Smith AM, Rahman FZ, Hayee B, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med. 2009;206:1883–97. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]