

Abstract
Anti-neutrophil cytoplasmic antibodies (ANCA) to proteinase 3 (PR3) are found in patients with small-vessel vasculitis. PR3-ANCA bind strongly to membrane PR3 (mPR3) that is presented by the NB1 receptor. We performed high-throughput screening using a small molecule library to identify compounds that inhibit PR3-NB1 binding. We established a human embryonic kidney (HEK293) cell-based system, where approximately 95 ± 2% of the NB1-transfected cells expressed the NB1 receptor on the cell surface. Addition of 0·1 µg/ml human PR3 to 104 NB1-expressing HEK293 cells resulted in PR3 binding that was detected by immunofluorescence using a fluorescence plate reader assay. We identified 13 of 20 000 molecules that inhibited PR3 binding by >70%. Seven of 13 substances showed reproducible inhibition in four additional validation experiments. Two selected compounds (27519 and 27549) demonstrated a dose-dependent inhibition over a range from 6·25 to 100 µM as measured by the plate reader assay. We used flow cytometry as a second assay, and found that both compounds reproducibly inhibited PR3 binding to NB1-transfected HEK293 cells at 50 µM (inhibition to 42 ± 4% with compound 27519 and to 47 ± 6% with compound 27549 compared to the dimethylsulphoxide control). Furthermore, compounds 27519 and 27549 also inhibited binding of exogenous PR3 to human neutrophils. In contrast, the compounds did not decrease mPR3 expression on resting neutrophils, but reduced the tumour necrosis factor-α-mediated mPR3 increase on NB1pos neutrophils when present continuously during the assay. The findings suggest that small inhibitory compounds provide a potential therapeutic tool to reduce mPR3 by preventing its binding to NB1.
Keywords: ANCA, compound screening, NB1, neutrophils, proteinase 3
Introduction
The neutrophil serine protease proteinase 3 (PR3) is a major autoantigen for anti-neutrophil cytoplasmic autoantibodies (ANCA) in Wegener's granulomatosis [1]. Binding of PR3-ANCA to cytokine-primed neutrophils results in respiratory burst activity, increased adhesion, degranulation of proteolytic enzymes and dysregulated apoptosis in vitro[2,3]. Animal models established that ANCA-neutrophil interactions provide a key effector system for the disease [4–7]. We have demonstrated previously that the glycosylphosphatidylinositol (GPI)-anchored receptor NB1 (CD177) presents PR3 on a neutrophil subset [8,9]. NB1 and PR3 are expressed on the membrane of the same neutrophil subset and the percentage of neutrophils that are membrane-positive for both molecules ranges from 0 to 100% [8,10]. A higher percentage of mPR3/NB1-positive neutrophils is associated with an increased risk for and worse outcome of ANCA vasculitis [11–14]. The mPR3/NB1 phenotype may have implications above and beyond vasculitis as shown for rheumatoid arthritis, transfusion-related acute lung injury (TRALI) and for patients with infectious diseases [15,16].
PR3 is stored mainly in primary granules, but also in secondary and secretory vesicles [17–21], while NB1 resides in secondary and tertiary vesicles [9,22]. Membrane PR3 expression is increased with cytokine priming during adhesion in the presence of PR3-ANCA, and as a result of apoptosis [23–27]. The phospholipidscramblase-1 controls the latter process [28]. The exact mechanisms that mediate activation-induced mPR3 up-regulation on NB1-expressing cells are not yet understood. Recently, a hydrophobic patch of the PR3 molecule was shown to be important for its NB1 binding [29]. Conceivably, intracellular NB1 and PR3 translocate independently from the same or different granules and associate on the membrane. PR3 and NB1 could also translocate to the membrane as an already preformed complex from intracellular pools. Alternatively, PR3 could be secreted and extracellular PR3 would then rebind to the NB1 receptor, as suggested in a recent review by Witko-Sarsat et al.[30]. Preventing the association of PR3 and NB1, or disrupting established complexes, would have obvious therapeutic implications for PR3-ANCA-mediated neutrophil activation. We performed a high-throughput screening with 20 000 small synthetic organic molecules to inhibit the PR3-NB1 interaction. We identified compounds that abrogated binding of exogenous PR3 to membrane-expressed NB1. The compounds did not displace PR3 from the membrane of NB1pos resting neutrophils, but reduced the tumour necrosis factor (TNF)-α-mediated mPR3 increase on NB1pos neutrophils when present continuously during the assay. Small inhibitory compounds may provide a potential therapeutic tool to reduce mPR3 by preventing its binding to NB1.
Methods
PR3 was detected using monoclonal antibodies (mAbs) 12·8 (CLB, Amsterdam, the Netherlands) and 81·3.3 (Biogenes, Berlin, Germany); the NB1 antibody was clone MEM166 (BD Pharmingen and Biolegend, San Diego, CA, USA), the isotype control immunoglobulin G1 (IgG1)kappa and secondary fluorescein isothiocyanate (FITC)-conjugated F(ab) fragments were from Dako (Hamburg, Germany). TNF-α was from Genzyme (Rüsselsheim, Germany). Poly-L-lysine was from Sigma (Munich, Germany).
Preparation of human neutrophils and cell culture conditions
Heparinized whole blood was drawn from healthy human donors after due written informed consent was obtained according to the requirements of our internal review board (AA3/00/44). Neutrophils were isolated by red blood cell sedimentation with dextran 1%, followed by Ficoll-Hypaque density gradient centrifugation. Erythrocytes were lysed by incubation with hypotonic saline for 15 s, and neutrophils were spun down (200 g, 10 min) and reconstituted in Hanks' balanced salt solution (HBSS) with calcium and magnesium. The final cell concentration was 5 × 106 cells per ml. The cell viability was detected in every cell preparation by trypan blue exclusion and found to be greater than 99%. The percentage of neutrophils after isolation was >95% by Wright–Giemsa staining and light microscopy.
Culture and transfection of human embryonic kidney (HEK293) cells
HEK293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1% glutamine, and 1% penicillin/streptomycin and subcultured thrice weekly (Biochrom, Berlin, Germany). HEK293 cells were seeded in six wells the day before and were transfected with 4 µg NB1 using Fugene HD transfection reagent (Roche, Mannheim, Germany). Additionally, β-galactosidase (β-Gal) transfection served as a control. Cells were trypsinized the following day and seeded into poly-L-lysine-coated 384 microtitre plates, 10 000 cells per well, respectively. Cells were allowed to adhere for 1 more day. Transfection efficiency was 78–95% starting from day 2 after transfection for more than 6 days. Plates were washed with phosphate-buffered saline (PBS) using a cell washer from Biotek (Winooski, VT, USA) to remove medium.
Vectors, cloning and sequencing
NB1 was removed from clone pCMV-SPORT6-CD177 (RZPD, Berlin, Germany) by digestion with EcoRI and NotI (NEB, Frankfurt, Germany) and inserted into pcDNA4 (Invitrogen, Karlsruhe, Germany). pcDNA4–β-galactosidase (Invitrogen) was used for control transfection. All plasmids were sequenced using ABI PRISM 377 DNA sequencer (Applied Biosystems, Darmstadt, Germany) prior to use. Plasmids were amplified in Escherichia coli One Shot Top (Invitrogen) and purified by endotoxin-free Qiagen Maxi-Prep kit (Qiagen, Hilden, Germany).
Compound screening method
The FMP-20 ChemBioNet library containing 20 000 small organic molecules was purchased from ChemDiv (San Diego, CA, USA). Compounds were added to the cells using a robot from Caliper Life Science (Hopkinton, MA, USA). The final concentration used was 50 µM. Dimethylsulphoxide (DMSO) served as a control on NB1-transfected cells. After incubation for 10 min at room temperature, purified PR3 (0·1 µg/ml) was added to each well and was allowed to bind for 2 h. Plates were washed thereafter to remove all unbound PR3 from the cells; anti-PR3 (1 µg/ml) was added for 20 min, followed by FITC IgG anti-mouse for a further 20 min. Plates were washed thoroughly with the cell washer to remove free FITC and fluorescence intensity was measured using the plate reader Tecan Safire II (Tecan, Maennedorf, Switzerland). Fluorescence intensity was measured in each well; if the amount of inhibition was more than 70% the compound was picked again and tested for further verifications and in different concentrations.
Flow cytometry
Flow cytometry was used to evaluate the membrane expression of NB1 and PR3. If indicated, cells were stimulated with 2 ng/ml TNF-α for 20 min at 37°C. Samples were incubated on ice for antibody binding followed by a secondary FITC-conjugated F(ab)2-fragment of goat anti-mouse IgG. Cells were washed and flow cytometry was performed using a fluorescence activated cell sorter (FACScan) (Becton Dickinson, Heidelberg, Germany); 10 000 events per sample were collected and analysed with CellQuest Pro software (Becton Dickinson).
Statistics
Statistics were calculated using StatView version 4·5 (Abacus Concepts, Berkeley, CA, USA). Correlations are given with 95% confidence interval. Values are given with standard error of the mean (s.e.m.). P-values less than 0.05 were considered significant and are indicated by asterisks.
Results
Compounds 27519 and 27549 prevent binding of exogenous PR3 to NB1-transfected HEK293 in a dose-dependent manner
In order to identify small synthetic organic molecules that inhibit PR3 binding to membrane-expressed NB1 (mNB1), we established a cell-based system for a high-throughput screening assay. Cells for these experiments needed to remain adherent in 384-well plates after multiple washing steps and to show high mNB1 expression after NB1 transfection. Neutrophils did not meet these criteria, but our screening experiments suggested that HEK293 would be useful for these studies. After optimizing the experimental conditions, we observed a reproducible high mNB1 expression after 48 h when 4 µg NB1 plasmid was used. The transfection efficacy remained at 95 ± 2% for the first 4 days and decreased to 78 ± 10% at day 6 (Fig. 1a). Transfection with a β-Gal plasmid served as a control and showed no significant mNB1 staining (8 ± 2% on day 2 to 4 ± 0 on day 6, n = 2). A typical flow cytometry experiment is shown in Fig. 1b. Based on these data we used transfected HEK293 cells from days 2 to 4 for further experiments.
Fig. 1.
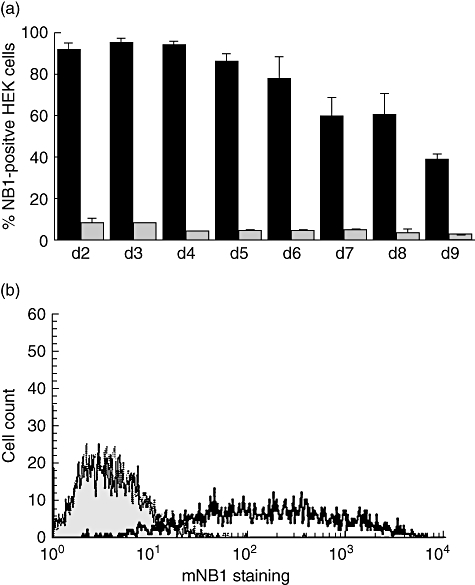
Time–course of the NB1 transfection efficiency in human embryonic kidney (HEK293) cells; (a) 6 × 105 HEK293 cells were seeded into six-well plates and were transfected with 4 µg/ml NB1 using Fugene HD transfection reagent on the following day. Membrane-expressed NB1 (mNB1) expression was evaluated from days 2 to 9 by flow cytometry (n = 3). (b) A typical fluorescence activated cell sorter (FACS) flow experiment from day 2 using NB1-transfected HEK293 cells is depicted. Staining with the isotype control is shown in grey and with the anti-NB1 monoclonal antibody (mAb) with the bold line. The dotted line represents the β-Gal-transfected control stained with the anti-NB1 mAb and is basically identical with the isotype control in NB1-transfected cells.
HEK293 cells were trypsinized approximately 24 h after NB1 transfection and seeded into 384 microtitre plates precoated with poly-L-lysine to enhance adherence during cell washes. Using this cell-based system and a fluorescence plate reader, titration experiments established the optimal concentration of exogenously added human PR3, anti-PR3 antibody and secondary FITC-labelled F(ab)2 fragments. When we added 0·1 µg/ml PR3 to 10 000 HEK293 cells per well, used the primary mAb to PR3 at 1 : 200 (1 µg/ml final concentration), and the secondary FITC-labelled antibody at a 1 : 100 dilution, we observed an excellent mPR3 staining after NB1 transfection that was not seen with β-Gal control transfection. The mPR3 staining obtained in a fluorescence plate reader at 530 nm was 4972 arbitrary units (AU) after NB1 and 1895 AU after β-Gal transfection. After optimal assay conditions were established, cells were preincubated with 20 000 small organic compounds (50 µM) from the FMP-20 compound library before adding PR3 and assessing PR3 binding to the NB1-expressing HEK293 cells in the fluorescence plate reader. Thirteen of 20 000 compounds inhibited PR3 binding by more than 70% in the first screen. The inhibitory effect of these compounds was assessed in four more validation experiments. Seven of these 13 compounds inhibited PR3 binding reproducibly to NB1-expressing HEK293 cells. Two compounds had to be excluded because of cell toxicity as assayed by the trypan blue exclusion test (data not shown). The results of a total of five independent experiments for the remaining five compounds are shown in Fig. 2a.
Fig. 2.
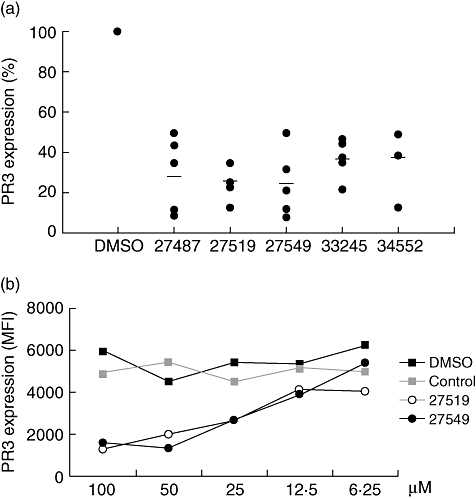
Small compounds inhibit proteinase 3 (PR3) binding to NB1-transfected human embryonic kidney (HEK293) cells; (a) 1 × 104 NB1-transfected HEK293 cells were seeded into 384-well plates and five validated compounds were tested at 50 µM for their inhibitory effect on PR3 binding to NB1 using the fluorescence plate reader (n = 5). The horizontal line shows the mean inhibition for each compound. PR3 binding to NB1-expressing HEK293 cells preincubated with dimethylsulphoxide (DMSO) control was set at 100%; (b) 1 × 104 NB1-transfected HEK293 cells were incubated in 384-well plates with increasing concentrations of compounds 27519 and 27549. The non-inhibitory compound 39679 (control) and DMSO were used as controls. Exogenous PR3 (0.1 µg/ml) was added thereafter for 2 h and PR3 membrane expression was assessed in a fluorescence plate reader (n = 2).
Based on the data shown in Fig. 2a, the compounds 27519 and 27549 showed the strongest inhibition and were selected for a dose–response study using the fluorescence plate reader assay. NB1-transfected HEK293 were incubated with increasing compound concentrations over a range from 6·25 to 100 µM. The non-inhibitory compound 39679 served as a control. Both compounds 27519 and 27549 inhibited mPR3 binding to NB1-transfected HEK293 in a dose-dependent manner (Fig. 2b, n = 2). In contrast, DMSO and a non-inhibitory control compound had no effect. Toxicity of the compounds was ruled out by the trypan blue test. More than 95% of the cells incubated with the highest compound concentration excluded the dye.
Inhibition of PR3 binding to NB1 by compounds 27519 and 27549 is genuine and not merely a consequence of masking a certain PR3
The structure of compounds 27519 and 27549 and the control compound 39679 is depicted in Fig. 3a. Both hit structures were checked for chemically related screening compounds in the ChemBioNet library. A structure was defined as chemically similar if the fingerprint-based Tanimoto coefficient was higher than 0·6. We found that compound 27519 has only four chemically related compounds within the screening library. Comparing these structures, only compound 27519 has substituted the central phenyl-tetrahydrochinoline by a hydroxyl group in combination with a carboxylic function. According to our definition of chemical similarity, compound 27549 was identified as a singleton. Nevertheless, this structure shares some structural features with 27519. The central scaffold of both structures is very similar, and they are substituted additionally by carboxylic groups. Both hit structures fulfil the Lipinski rule of five, indicating a reasonable bioavailability.
Fig. 3.
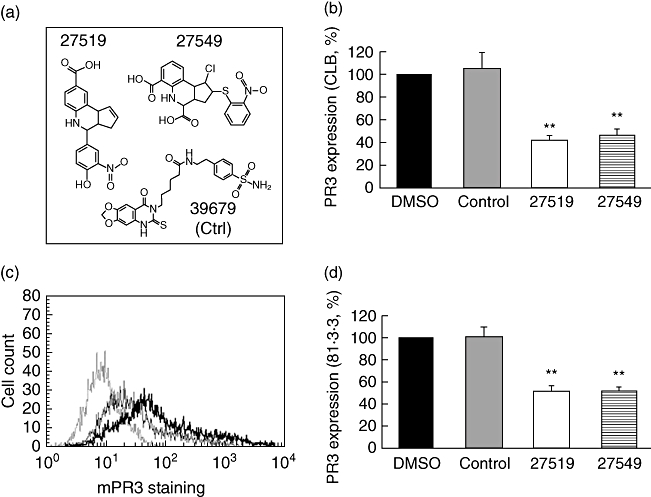
Compounds 27519 and 27549 inhibit binding of exogenous proteinase 3 (PR3) to NB1-transfected human embryonic kidney (HEK293) cells by flow cytometry. (a) The structure of the inhibitory compounds 27519 and 27549 and the control compound 39679 is depicted; (b) 1 × 105 NB1-transfected HEK293 cells were incubated with 50 µM of compounds 27519 and 27549 for 30 min before the addition of 1 µg/ml PR3 for 2 h. The compound 39679 (control) served as a non-inhibitory control and dimethylsulphoxide was used as the buffer control. PR3 membrane expression was assessed by flow cytometry using the monoclonal CLB anti-PR3 antibody (n = 5). (c) A typical experiment is depicted with the isotype control (thin dotted line) and PR3 staining after treatment of cells with PR3 in the presence of the control compound (thick line) or in the presence of compound 27519 (thin line). (d) Experiments were carried out as in (b) except that a different monoclonal 81.3.3 anti-PR3 antibody was used for detection of PR3 membrane staining (n = 3). **P < 0·01.
Flow cytometry was used as a second assay to further evaluate compounds 27519 and 27549 together with a control substance. The flow cytometry data demonstrate that preincubation with compounds 27519 and 27549, but not with the control compound or buffer control, abrogated PR3 binding significantly to NB1-expressing HEK293 cells (Fig. 3b, n = 5, P < 0·01). In these experiments, the monoclonal CLB antibody to PR3 was used and a typical flow cytometry result is shown in Fig. 3c. To exclude the possibility that the compounds only mask the PR3 epitope to which the mAb anti-PR3 antibody binds, a different monoclonal anti-PR3 antibody (81·3.3) was used for detection of PR3 staining. Our data show similar compound-mediated inhibition of PR3 membrane binding when the monoclonal 81·3.3 anti-PR3 antibody was used for detection (Fig. 3d, n = 3, P < 0·01). To gain further insight as to whether the compounds interacted rather with the NB1 receptor or with PR3, we used two approaches. First, we tested the effect of washing steps between compound incubation and the addition of exogenous PR3. We observed that when the cells were washed after 30 min preincubation with compound 27519, treatment with 1 µg PR3 reduced mPR3 staining to 46 ± 4% compared to DMSO-treated cells (P < 0·01). This inhibition was similar to results shown in Fig. 3b that were obtained without introducing a washing step (inhibition to 42 ± 4%). In the second set of experiments, compound 27519 was preincubated with purified PR3 for 30 min and then added to NB1-transfected cells. In these experiments, we found an even stronger inhibition of exogenous PR3 binding to the HEK293 cell membrane PR3, suggesting additional compound binding to PR3 (33 ± 2%, P < 0·05, n = 3).
Compounds 27519 and 27549 prevent binding of exogenous PR3 to human neutrophils, do not inhibit constitutive PR3 membrane expression, but abrogate TNF-α-induced PR3 translocation when present during all staining steps.
After having characterized the PR3-NB1 inhibitory properties of compounds 27519 and 27549 in HEK293 cells, we studied the neutrophil. We reasoned that secreted PR3 would resemble closely the setting of adding exogenous PR3 to NB1-expressing HEK293 cells, where the compounds were shown to be inhibitory. To substantiate this assumption, we incubated resting neutrophils with compounds 27519 and 27549 or appropriate controls, respectively. After 30 min 1 µg PR3 was added for 90 min. In preliminary experiments we performed a time–course study, showing that 90 min of incubation with exogenous PR3 caused a significant gain on PR3 membrane expression (data not shown). To prevent increasing neutrophil activation, experiments were performed with the 90-min time-point and on ice to decrease cell activation and therefore granule translocation. Using flow cytometry, we observed almost complete abrogation of PR3 binding to the membrane of NB1-positive neutrophils with both compounds (Fig. 4a, n = 5). In contrast, DMSO control or the control compound 39679 had no effect. A typical experiment for compound 27519 is depicted in Fig. 4b. Similar to the HEK293 cell experiments, we tested the effect of washing between compound incubation and addition of exogenous PR3 and obtained similar inhibition with both conditions (inhibition by 98 ± 4% without and by 97 ± 3% with washing using compound 27519, P < 0·01, n = 3).
Fig. 4.
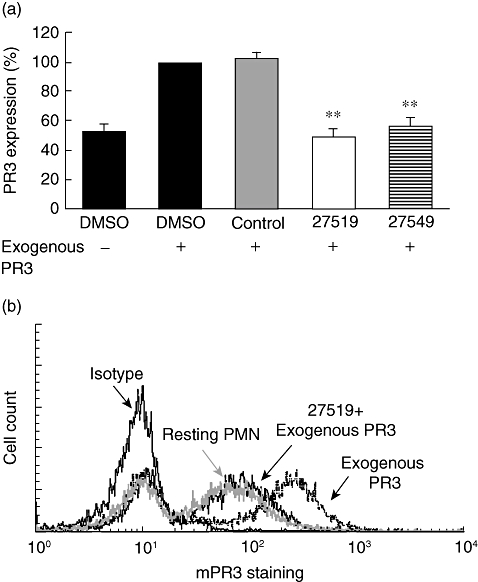
Effect of compounds 27519 and 27549 on exogenous proteinase 3 (PR3) binding to neutrophils; (a) 1 × 106 neutrophils were treated for 30 min with buffer, 50 µM compound 27519, compound 27549 or the control compound 39679 (control), respectively. Cells were then incubated with 1 µg PR3 for 90 min. Membrane PR3 expression was assessed by flow cytometry using the CLB anti-PR3 monoclonal antibody (n = 5). (b) A typical flow experiment is depicted for compound 27519. **P < 0·01.
In the last set of experiments we tested whether or not the compounds would inhibit constitutive mPR3 expression on the NB1-positive neutrophil subset or prevent the up-regulation of mPR3 in response to TNF-α treatment. Thus, we incubated the cells with compounds 27519 and 27549 or the control compound for 30 min, respectively. Cells were then treated with buffer or were activated with 2 ng/ml TNF-α to induce PR3 translocation from intracellular granules to the cell surface. Our results demonstrate that even with the highest compound concentration (100 µM), neither PR3 expression in resting neutrophils (buffer incubation) nor TNF-α-induced up-regulation of PR3 was abrogated. The data obtained with compound 27519 are shown in Fig. 5a (n = 8). In contrast, when compound 27519 (100 µM) was present continuously during the entire staining process, we observed a significant inhibition of TNF-α-induced PR3 up-regulation to approximately 60%, while the control compound had no effect (Fig. 5b, n = 7). Furthermore, when we used 50 µM of compound 27519 we still observed a significant reduction in PR3 expression to 70 ± 7% (n = 4, P < 0·01). Thus, our data suggest that the compounds were not able to disrupt an established PR3-NB1 complex on the (resting) neutrophil membrane, but diminished the TNF-α-induced increase of mPR3 when present during the entire translocation and staining process.
Fig. 5.
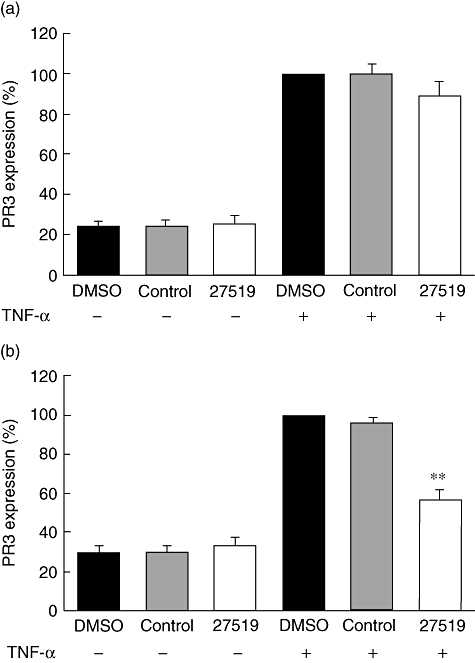
Effect of compound 27519 on membrane proteinase 3 (mPR3) expression in resting and tumour necrosis factor (TNF)-α-stimulated neutrophils; (a) 1 × 106 neutrophils were incubated for 30 min with buffer, 100 µM compound 27519 or the control 39679 compound (control), respectively. Cells were then treated with buffer or 2 ng/ml TNF-α. Membrane PR3 expression was assessed by flow cytometry using the CLB monoclonal antibody to PR3 (n = 8). (b) Cells were treated as in (a), but compound was re-added during all staining steps (n = 7). **P < 0·01.
Discussion
We used high-throughput screening with 20 000 compounds from a defined FMP-20 library to identify compounds that block the interaction between PR3 and its receptor NB1. We identified five compounds that prevented binding of PR3 to the NB1 receptor on NB1-transfected HEK 293 cells. We selected two compounds to confirm their inhibitory effect on human neutrophils. The compounds blocked binding of extracellular PR3 to the neutrophil and diminished mPR3 up-regulation in TNF-α-stimulated cells when present continuously, presumably by binding to recirculating NB1 and/or PR3. The compounds did not displace PR3 from the membrane of resting neutrophils.
We and others have described the existence of two neutrophil subsets with respect to mPR3 expression [11,13,31]. Moreover, neutrophil-specific NB1 (CD177) was shown to present PR3 on the neutrophil membrane and allows PR3-ANCA binding to the NB1pos/mPR3high subset [8,10]. The functional importance of this finding was underscored by the fact that PR3-ANCA activate PI3K/Akt, and subsequently superoxide release and degranulation, only in the mPR3high-expressing neutrophil subset [32]. Interestingly, Hu et al. recently found no difference in hydrogen peroxide generation between PR3high/mNB1pos- and PR3low/mNB1neg-expressing neutrophils [14]. In an ongoing study, we performed head-to-head comparisons and observed that PR3high/mNB1pos do respond with higher generation of extracellular superoxide, whereas the intracellular hydrogen peroxide production was similar to the PR3low/mNB1neg subset (data not shown). Thus, there is only an apparent discrepancy between Hu et al. and our data and the data on degranulation and superoxide clearly establish the significance of the mPR3 phenotype for PR3-ANCA-mediated neutrophil activation. The reasons that explain the difference between superoxide and hydrogen peroxide will be elucidated in future studies.
Better understanding of how PR3 membrane expression is controlled has mechanistic, but also therapeutic, aspects. PR3 as well as NB1 lacks a transmembrane domain. Thus, several questions remain, including further characterization of the mNB1 signalling complex, the causes for the disparate NB1 expression in neutrophil subsets and the mechanisms of how NB1–PR3 complexes are formed. In addition, prevention or, even better, disruption of existing NB1–PR3 membrane complexes would have potentially therapeutic implications.
To address some of the issues mentioned above, we used high-throughput compound screening employing small synthetic organic molecules of 300–400 kDa. This method has been used increasingly during recent years and has broad applications. Compound screens carried out in our Institute for Molecular Pharmacology (JPK) identified molecules that block the active site of mycobacterial P450 enzyme CYP130, the sterol 14α-demethylase CYP51, and inhibited the protein tyrosine phosphatase Shp2 [33–35]. Using the same chemical library, we identified small molecules that prevented PR3 binding to NB1. The strength of our results is that we demonstrate the feasibility of such an approach in a cell-based system, where cells were transfected to express the NB1 receptor on their membrane. Our intention was that using a cell system and membrane-expressed NB1 would resemble the in vivo situation more closely than coating recombinant NB1 on the surface of a plastic dish. With this strategy, we identified compounds that inhibited the binding of human PR3 to NB1 expressed on HEK293 cells. Importantly, the data were confirmed in human neutrophils. The fact that the compounds block PR3 binding to membrane-expressed NB1 effectively gave us the opportunity to investigate whether or not rebinding of extracellularly released PR3 to the mNB1pos neutrophil subset occurs. We also reasoned that the compounds may displace PR3 from its binding to mNB1. However, the compounds did not displace PR3 from the membrane of NB1-positive resting neutrophils, but reduced the TNF-α-mediated mPR3 increase on NB1pos neutrophils when present continuously during the assay. The latter finding may be explained by the results from Bauer et al., who suggested that mPR3 and mNB1 undergo rapid internalization and recirculation [10].
Future modification of the compounds' structure is needed to render them suitable as therapeutics in PR3-ANCA-associated vasculitis. In our experiments. these original compounds from a chemical screening library were used at rather high concentrations. Further modifications may allow employment of nanomolar concentrations that are then suitable for in vivo experiments. It would be helpful if the three-dimensional structure of NB1 were known. Protein–protein interaction studies and computational modelling could then be applied to elucidate the NB1–PR3 binding site. This strategy would allow the design of specific compounds that would compete for important interaction sites between the two molecules. However, in contrast to NB1, the PR3 structure is known, and a different approach would be to map the PR3 region(s) that are pivotal for NB1 binding and then target these sites by designer molecules. In this regard, important information was obtained recently when Hajjar et al. used molecular simulation to identify a hydrophobic patch in PR3 that is essential for its membrane anchorage [36]. Elegant experiments by Korkmaz et al. using gibbon/human PR3 hybrids mapped a PR3 region of closely clustered hydrophobic residues Phe166, Trp218 and Leu223, and showed its importance for the binding to human NB1 [29]. Furthermore, α1-protease inhibitor (α1-PI) was shown to trigger dissociation of PR3 from the NB1 receptor, most probably by conformational distortion of the hydrophobic loop on PR3. However, α1-PI is no suitable therapeutic compound for the purpose of disrupting NB1–PR3, because it would bind covalently to all serine proteases and therefore exhibit effects that go beyond the PR3–NB1 interaction.
In summary, we identified compounds that prevent binding of exogenous PR3 to mNB1 using a cell-based expression system and high-throughput screening. The compounds did not displace PR3 from the membrane of NB1pos resting neutrophils, but reduced the TNF-α-mediated mPR3 increase on NB1pos neutrophils when present continuously during the assay. After further optimization, small inhibitory compounds may provide a potential therapeutic tool to reduce mPR3 by preventing its binding to NB1.
Acknowledgments
The Deutsche Forschungsgemeinschaft (DFG: KE 576/7-1) and the Experimental and Clinical Research Center supported the study.
Disclosure
RK states that his work was supported by grants from the Deutsche Forschungsgemeinschaft and Clinical Research Center. All other authors have declared that they have no conflicts of interest.
References
- 1.Niles JL, McCluskey RT, Ahmad MF, Arnaout MA. Wegener's granulomatosis autoantigen is a novel neutrophil serine proteinase. Blood. 1989;74:1888–93. [PubMed] [Google Scholar]
- 2.Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–19. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harper L, Ren Y, Savill J, Adu D, Savage CO. Antineutrophil cytoplasmic antibodies induce reactive oxygen-dependent dysregulation of primed neutrophil apoptosis and clearance by macrophages. Am J Pathol. 2000;157:211–20. doi: 10.1016/S0002-9440(10)64532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–63. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Little MA, Smyth L, Salama AD, et al. Experimental autoimmune vasculitis: an animal model of anti-neutrophil cytoplasmic autoantibody-associated systemic vasculitis. Am J Pathol. 2009;174:1212–20. doi: 10.2353/ajpath.2009.080458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreiber A, Xiao H, Falk RJ, Jennette JC. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. 2006;17:3355–64. doi: 10.1681/ASN.2006070718. [DOI] [PubMed] [Google Scholar]
- 7.Xiao H, Heeringa P, Liu Z, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167:39–45. doi: 10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Vietinghoff S, Tunnemann G, Eulenberg C, et al. NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils. Blood. 2007;109:4487–93. doi: 10.1182/blood-2006-10-055327. [DOI] [PubMed] [Google Scholar]
- 9.von Vietinghoff S, Eulenberg C, Wellner M, Luft FC, Kettritz R. Neutrophil surface presentation of the anti-neutrophil cytoplasmic antibody-antigen proteinase 3 depends on N-terminal processing. Clin Exp Immunol. 2008;152:508–16. doi: 10.1111/j.1365-2249.2008.03663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer S, Abdgawad M, Gunnarsson L, Segelmark M, Tapper H, Hellmark T. Proteinase 3 and CD177 are expressed on the plasma membrane of the same subset of neutrophils. J Leukoc Biol. 2007;81:458–64. doi: 10.1189/jlb.0806514. [DOI] [PubMed] [Google Scholar]
- 11.Witko-Sarsat V, Lesavre P, Lopez S, et al. A large subset of neutrophils expressing membrane proteinase 3 is a risk factor for vasculitis and rheumatoid arthritis. J Am Soc Nephrol. 1999;10:1224–33. doi: 10.1681/ASN.V1061224. [DOI] [PubMed] [Google Scholar]
- 12.Schreiber A, Otto B, Ju X, et al. Membrane proteinase 3 expression in patients with Wegener's granulomatosis and in human hematopoietic stem cell-derived neutrophils. J Am Soc Nephrol. 2005;16:2216–24. doi: 10.1681/ASN.2004070609. [DOI] [PubMed] [Google Scholar]
- 13.Rarok AA, Stegeman CA, Limburg PC, Kallenberg CGM. Neutrophil membrane expression of proteinase 3 (PR3) is related to relapse in PR3-ANCA-associated vasculitis. J Am Soc Nephrol. 2002;13:2232–8. doi: 10.1097/01.asn.0000028642.26222.00. [DOI] [PubMed] [Google Scholar]
- 14.Hu N, Westra J, Huitema MG, et al. Coexpression of CD177 and membrane proteinase 3 on neutrophils in antineutrophil cytoplasmic autoantibody-associated systemic vasculitis: anti-proteinase 3-mediated neutrophil activation is independent of the role of CD177-expressing neutrophils. Arthritis Rheum. 2009;60:1548–57. doi: 10.1002/art.24442. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto T, Kaneko T, Wada H, et al. Proteinase 3 expression on neutrophil membranes from patients with infectious disease. Shock. 2006;26:128–33. doi: 10.1097/01.shk.0000223122.11147.5a. [DOI] [PubMed] [Google Scholar]
- 16.Bux J, Becker F, Seeger W, Kilpatrick D, Chapman J, Waters A. Transfusion-related acute lung injury due to HLA-A2-specific antibodies in recipient and NB1-specific antibodies in donor blood. Br J Haematol. 1996;93:707–13. doi: 10.1046/j.1365-2141.1996.d01-1703.x. [DOI] [PubMed] [Google Scholar]
- 17.Borregaard N, Cowland JB. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood. 1997;89:3503–21. [PubMed] [Google Scholar]
- 18.Calafat J, Goldschmeding R, Ringeling PL, Janssen H, van der Schoot CE. In situ localization by double-labeling immunoelectron microscopy of anti-neutrophil cytoplasmic autoantibodies in neutrophils and monocytes. Blood. 1990;75:242–50. [PubMed] [Google Scholar]
- 19.Csernok E, Ludemann J, Gross WL, Bainton DF. Ultrastructural localization of proteinase 3, the target antigen of anti-cytoplasmic antibodies circulating in Wegener's granulomatosis. Am J Pathol. 1990;137:1113–20. [PMC free article] [PubMed] [Google Scholar]
- 20.Lominadze G, Powell DW, Luerman GC, Link AJ, Ward RA, McLeish KR. Proteomic analysis of human neutrophil granules. Mol Cell Proteomics. 2005;4:1503–21. doi: 10.1074/mcp.M500143-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Witko-Sarsat V, Cramer EM, Hieblot C, et al. Presence of proteinase 3 in secretory vesicles: evidence of a novel, highly mobilizable intracellular pool distinct from azurophil granules. Blood. 1999;94:2487–96. [PubMed] [Google Scholar]
- 22.Goldschmeding R, van Dalen CM, Faber N, et al. Further characterization of the NB 1 antigen as a variably expressed 56–62 kD GPI-linked glycoprotein of plasma membranes and specific granules of neutrophils. Br J Haematol. 1992;81:336–45. doi: 10.1111/j.1365-2141.1992.tb08237.x. [DOI] [PubMed] [Google Scholar]
- 23.Brachemi S, Mambole A, Fakhouri F, et al. Increased membrane expression of proteinase 3 during neutrophil adhesion in the presence of anti proteinase 3 antibodies. J Am Soc Nephrol. 2007;18:2330–9. doi: 10.1681/ASN.2006121309. [DOI] [PubMed] [Google Scholar]
- 24.Durant S, Pederzoli M, Lepelletier Y, et al. Apoptosis-induced proteinase 3 membrane expression is independent from degranulation. J Leukoc Biol. 2004;75:87–98. doi: 10.1189/jlb.0203079. [DOI] [PubMed] [Google Scholar]
- 25.Harper L, Cockwell P, Adu D, Savage CO. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int. 2001;59:1729–38. doi: 10.1046/j.1523-1755.2001.0590051729.x. [DOI] [PubMed] [Google Scholar]
- 26.Hellmich B, Csernok E, Trabandt A, Gross WL, Ernst M. Granulocyte–macrophage colony-stimulating factor (GM-CSF) but not granulocyte colony-stimulating factor (G-CSF) induces plasma membrane expression of proteinase 3 (PR3) on neutrophils in vitro. Clin Exp Immunol. 2000;120:392–8. doi: 10.1046/j.1365-2249.2000.01205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kettritz R, Scheumann J, Xu Y, Luft FC, Haller H. TNF-alpha-accelerated apoptosis abrogates ANCA-mediated neutrophil respiratory burst by a caspase-dependent mechanism. Kidney Int. 2002;61:502–15. doi: 10.1046/j.1523-1755.2002.00161.x. [DOI] [PubMed] [Google Scholar]
- 28.Kantari C, Pederzoli-Ribeil M, Amir-Moazami O, et al. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood. 2007;110:4086–95. doi: 10.1182/blood-2007-03-080457. [DOI] [PubMed] [Google Scholar]
- 29.Korkmaz B, Kuhl A, Bayat B, Santoso S, Jenne DE. A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J Biol Chem. 2008;283:35976–82. doi: 10.1074/jbc.M806754200. [DOI] [PubMed] [Google Scholar]
- 30.Witko-Sarsat V, Reuter N, Mouthon L. Interaction of proteinase 3 with its associated partners: implications in the pathogenesis of Wegener's granulomatosis. Curr Opin Rheumatol. 2010;22:1–7. doi: 10.1097/BOR.0b013e3283331594. [DOI] [PubMed] [Google Scholar]
- 31.Schreiber A, Busjahn A, Luft FC, Kettritz R. Membrane expression of proteinase 3 is genetically determined. J Am Soc Nephrol. 2003;14:68–75. doi: 10.1097/01.asn.0000040751.83734.d1. [DOI] [PubMed] [Google Scholar]
- 32.Schreiber A, Luft FC, Kettritz R. Membrane proteinase 3 expression and ANCA-induced neutrophil activation. Kidney Int. 2004;65:2172–83. doi: 10.1111/j.1523-1755.2004.00640.x. [DOI] [PubMed] [Google Scholar]
- 33.Eddine AN, von Kries JP, Podust MV, Warrier T, Kaufmann SH, Podust LM. X-ray structure of 4,4′-dihydroxybenzophenone mimicking sterol substrate in the active site of sterol 14alpha-demethylase (CYP51) J Biol Chem. 2008;283:15152–9. doi: 10.1074/jbc.M801145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hellmuth K, Grosskopf S, Lum CT, et al. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc Natl Acad Sci USA. 2008;105:7275–80. doi: 10.1073/pnas.0710468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Podust LM, Ouellet H, von Kries JP, de Montellano PR. Interaction of Mycobacterium tuberculosis CYP130 with heterocyclic arylamines. J Biol Chem. 2009;284:25211–19. doi: 10.1074/jbc.M109.017632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hajjar E, Mihajlovic M, Witko-Sarsat V, Lazaridis T, Reuter N. Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins. 2008;71:1655–69. doi: 10.1002/prot.21853. [DOI] [PubMed] [Google Scholar]