

Abstract
Gap junctions (GJs) allow direct communication between cells. In the heart, GJs mediate the electrical coupling of cardiomyocytes and as such dictate the speed and direction of cardiac conduction. A prominent feature of acquired structural heart disease is remodeling of GJ protein expression and localization concomitant with increased susceptibility to lethal arrhythmias, leading many to hypothesize that the two are causally linked. Detailed understanding of the cellular mechanisms that regulate GJ localization and function within cardiomyocytes may therefore uncover potential therapeutic strategies for a significant clinical problem. This review will outline our current understanding of GJ cell biology with the intent of highlighting cellular mechanisms responsible for GJ remodeling associated with cardiac disease.
Keywords: gap junctions, connexin 43, protein trafficking, structural heart disease, arrhythmia
INTRODUCTION
Disturbances in electrical propagation are a hallmark of many acquired heart diseases, such as ischemic cardiomyopathy and heart failure, and are associated with an increased prevalence of cardiac arrhythmias, resulting in sudden cardiac death. A common electrophysiological feature of failing myocardium is a decrease in conduction velocity (CV) that can serve as a substrate for reentry, causing potentially lethal ventricular arrhythmias. Gap junctions (GJs) are specialized cell–cell contacts that mediate conduction in the heart. The tight correlation between decreased CV and remodeling of GJ protein expression, post-translational modifications and localization with cardiac disease, has led many to hypothesize that GJ remodeling plays a causative role in the decrease in CV and the generation of arrhythmias. Consistent with this hypothesis are studies that demonstrate a high incidence of lethal cardiac arrhythmias upon genetic disruption of connexin 43 (Cx43), the primary GJ-forming protein of the working myocardium.1–3 To fully understand the role that GJ remodeling plays in diseased hearts, it is essential to understand the molecular and cellular mechanisms by which intercellular communication through GJs is regulated.
This review will outline key features of GJ remodeling associated with cardiac disease, followed by a detailed discussion of our current mechanistic understanding of the GJ life cycle. We will begin with a description of GJ structure and function followed by a brief discussion of the relationship between changes in GJ function and structural heart disease. We will then provide an outline of the steps involved in the assembly and targeting, as well as internalization and degradation of GJs. Particular emphasis will be placed on Cx43 because it is the most widely studied and the primary connexin in ventricular myocardium.
GJs form low-resistance pores between cells and are composed of protein subunits called connexins of which there are up to 21 different types in humans (Fig. 1A). In addition to Cx43, at least four other connexins are present in the heart, Cx30.2, Cx37, Cx40, and Cx45. These different connexins exhibit distinct regional, cell type–specific, and heart chamber–specific expression patterns including endothelial cells and specialized conduction tissues. Connexins are transmembrane proteins with four membrane-spanning helices (Fig. 1B) and can oligomerize into hexameric channels called connexons. Connexons composed of a single type of connexin are called homomeric connexons, whereas those made of more than one type of connexin are called heteromeric connexons (Fig. 1C). GJ channels are formed when two apposing cells each contribute a single connexon that dock together in the extracellular space.4 GJ channels formed from two identical connexons are called homotypic channels, whereas those formed by two different connexons are called heterotypic channels (Fig. 1C). GJ channels cluster together into semicrystalline arrays ranging from tens to thousands of channels, forming GJ plaques (Fig. 1D). Once formed, GJs allow the exchange of ions and small molecules between the coupled cells. Depending on the isoforms of connexin forming the GJ channels, the conductance and selectivity may vary.5 Furthermore, GJ channels can open and close in response to a variety of physiological stimuli including voltage, ionic concentrations, pH, and local lipid and protein interactions, with different connexins exhibiting distinct gating properties. The enormous diversity in GJ channel function allows for the possibility of fine-tuning intercellular coupling.
FIGURE 1.

GJ structure. A, List of all expressed human connexins. B, Topological arrangement of connexin protein monomers. The cylinders represent transmembrane domains and the blue-boxed area represents the highly conserved region among different connexins, whereas the red-boxed area represents the highly divergent region. C, Connexon and GJ channel types. D, Topological arrangement of GJ channels. The GJ channel schematic is superimposed on a transmission electron micrograph of a GJ from canine cardiac tissue. Scale bar = 20 nm.
A remarkable feature of GJs is the extremely rapid time course with which they progress through their life cycle relative to most integral membrane proteins. Most connexins have half-lives on the order of 1–3 hours, and such rapid kinetics necessitate highly coordinated and tightly regulated trafficking mechanisms.6 The constitutive GJ life cycle involves multiple steps, including connexin synthesis and oligomerization, hemichannel translocation to the plasma membrane (PM) for incorporation into GJs, and subsequent removal from the PM and degradation with continuous intercellular coupling being maintained.
GJ REMODELING IN DISEASED HEARTS
A wide variety of cardiac diseases are associated with remodeling of GJs.7 Although the etiologies of the cardiac diseases associated with GJ remodeling are diverse, the nature of the remodeling among them shares common features. In healthy cardiac tissue, GJs are typically located at the region of contact between cardiomyocyte cell ends, called the intercalated disk (ID), with only occasional connexin expression occurring at cardiomyocyte lateral membranes. It is this preferential formation of GJs at the ID that is the basis for anisotropic conduction in the heart. A common immuno-histochemical finding in diseased myocardium is a redistribution of Cx43 protein to the lateral border of cardiomyocytes (referred to as lateralized GJs) (Fig. 2). Often accompanying this redistribution is an overall decrease in total tissue Cx43 expression levels. It is the combined decrease in Cx43 expression and redistribution of Cx43 localization to cardiomyocyte lateral membranes that has been hypothesized to directly contribute to the reduced ventricular CV associated with structural heart disease.8,9 Furthermore, heterogeneities in Cx43 expression across the ventricular wall have been demonstrated to contribute to the development of reentrant arrhythmias in failing hearts.10,11
FIGURE 2.
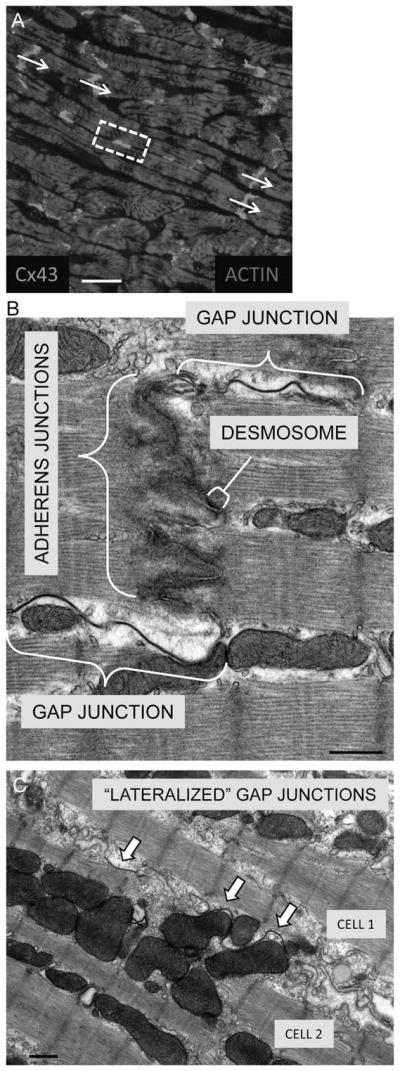
Localization of GJs in cardiac tissue. A, Immuno-histochemical staining of canine ventricular myocardium for Cx43. Cx43 appears white predominantly at cell ends, the myocytes are diffusely stained by actin. The arrows indicate the direction of tissue fiber orientation through Cx43 staining at ID. The boxed region indicates representative Cx43 staining between cardiomyocyte lateral membranes, which is referred to as lateralized Cx43. B, Transmission electron microscopy of normal canine ventricular tissue depicting a close-up view of a region within an ID between 2 myocytes. The 3 main types of cardiomyocyte cell–cell connections are depicted: fascia adherens (composed of adherens junctions), desmosomes, and GJs. C, Localization of “lateralized” GJs within failing canine ventricular tissue. Scale bars: A = 20 μm; B, C = 0.5 μm.
Moreover, decreased ventricular CV is a key electro-physiological feature of diseased myocardium that increases the susceptibility to the development of ventricular arrhythmias associated with sudden cardiac death. In the next section, we describe essential features of the GJ life cycle from a cell biological perspective. For a more complete description of GJ remodeling as it relates specifically to heart disease, readers are urged to consult recent excellent reviews on the subject.7,12
GJ ASSEMBLY: ENDOPLASMIC RETICULUM TO GOLGI APPARATUS
Like most integral membrane proteins, connexins are cotranslationally inserted into the endoplasmic reticulum (ER) membrane for transport through the secretory pathway. To form hemichannels, connexins must oligomerize into hexamers at a particular stage along the secretory pathway, and isoform-specific differences exist in the stage at which oligomerization occurs. For example, Cx32 will oligomerize in the ER and/or the ER–Golgi intermediate compartment, whereas Cx43 oligomerizes later in the secretory pathway in the trans-Golgi network (TGN).13–15 The absence of Cx43 oligomerization in the ER is consistent with the presence of a quality control mechanism that prevents oligomerization before the TGN. There is also evidence that specific structural determinants exist within connexins that determine the stage at which they will oligomerize and define their compatibility for oligomerization with other connexins.15 The oligomerization of connexins is essential for normal functional expression because disease-causing mutations have been identified that prevent the oligomerization of connexins, thus causing their destruction by the ER-associated degradation (ERAD) pathway.16–18
The mechanisms that regulate the transport of connexins early in the secretory pathway are virtually unknown. Only a few proteins have been identified that interact with connexins to regulate their transport within the secretory pathway. A novel Cx43-interacting protein (CIP75) was identified in a yeast two-hybrid screen using the Cx43 C-terminus (CT) as bait.19 CIP75 is a ubiquitin-like/ubiquitin-associated domain-containing Cx43-interacting protein with a molecular weight of 75 kDa that regulates the turnover of Cx43 early in the secretory pathway via ERAD (see section on GJ Internalization and Degradation below for further discussion of CIP75). Another recent study has suggested that the small guanosine triphosphatase (GTPase) Rab20 is a potential regulator of Cx43 trafficking between the ER and Golgi apparatus.20 In a mammalian cell–based expression screen, Rab20 was demonstrated to rescue aberrant ER exit and oligomerization of Cx43 containing a dibasic ER retention motif. Over-expression of this ER-retained Cx43 mutant resulted in saturation of the retention mechanism, resulting in ER exit and Cx43 localization within a perinuclear Golgi compartment. Overexpression of Rab20 was able to prevent this Golgi localization and revert Cx43 back to a primarily ER localization. Furthermore, in cells expressing wild-type Cx43, overexpression of Rab20 resulted in ER retention and prevention of trafficking to the PM. This study suggests that Rab20 plays a role in the quality control pathway, regulating ER exit and forward trafficking of Cx43. Whether or not other mechanisms exist that control the rate of flux through the secretory pathway, as well as how these mechanisms might be regulated in different cellular conditions, requires further study.
GJ ASSEMBLY: GOLGI APPARATUS TO THE PLASMA MEMBRANE
Connexins are transported through the Golgi apparatus and ultimately packaged into transport vesicles in the TGN for delivery to the PM. The mechanisms regulating the appropriate packaging of connexins into transport vesicles, as well as connexin vesicle delivery, and dynamics at the PM are incompletely understood. Studies have demonstrated the formation of pleomorphic post–Golgi vesicles, which deliver connexins to the PM.21–23 Cx43 has been shown to bind directly to microtubules,24,25 and pharmacological disruption of microtubules seems to disrupt the efficiency of Cx43 delivery to the PM but not inhibit the process altogether.26 A recent study by Shaw et al27 demonstrated the direct delivery of Cx43 vesicles to adherens junctions (AJs) via microtubules. The authors suggest a mechanism whereby Cx43 vesicles are “hooked” onto microtubules by a protein complex including the microtubule plus-end capping protein EB1. This protein complex is proposed to track Cx43 vesicles along a particular microtubular path that inserts at sites of AJ formation, which provide strong mechanical coupling between cells. This model contrasts with the prevailing model of connexin delivery to the PM, which proposes that connexons are delivered to regions of unapposed PM where they are free to diffuse in the plane of the membrane. Laterally diffusing connexons will then coalesce with the edges of existing GJs where they dock with connexons from opposing cells to form new GJ channels. It is possible that some combination of the two models exists.
There is mounting evidence that connexons acting as hemichannels in the PM mediate exchange between the cytoplasm and the extracellular space.28 For example, a widely studied role for hemichannels is the release of intracellular ATP, which can mediate both autocrine and paracrine signaling.29,30 Different mechanisms may therefore exist for delivery of connexons to the PM depending on whether they are to function as hemichannels or to be incorporated into GJs. Regardless of the mechanism of delivery, it seems that connexons are added to growing GJs around the plaque periphery. This phenomenon was elegantly shown by Gaietta et al31 using Cx43 with genetically encoded tetracysteine tags linked to the CT, which bind to the biarsenical fluorescent dyes FlAsH (green) and ReAsH (red). Using temporally separated dual color labeling and fluorescent microscopy, the authors were able to demonstrate the addition of newly synthesized Cx43 to the periphery of GJ plaques, whereas older Cx43 was confined to the center of GJ plaques and subsequently internalized into one cell.
GJ DYNAMICS AT THE PM: ROLE OF MECHANICAL JUNCTIONS
An emerging theme is the requirement for intact mechanical junctions (Table 1) between cells for proper GJ formation and maintenance. An important role has been demonstrated for the tight junction protein zonula-occludens 1 (ZO-1) in regulating GJ function. ZO-1 is a PDZ domain containing scaffolding protein originally identified as a component of tight junctions in polarized epithelial cells.32 Cx43 has been shown to bind directly to the second PDZ domain of ZO-1 via its CT PDZ binding motif.33,34 Studies have suggested that ZO-1 regulates the accrual of Cx43 into the periphery of GJ plaques and that the Cx43/ZO-1 interaction is important for regulating GJ size.35,36 Furthermore, the ZO-1/Cx43 interaction seems to be involved in the pathogenic remodeling of GJs associated with diseased hearts.37–40 In addition to Cx43, ZO-1 has been shown to bind and regulate the function of multiple other connexins, including Cx35,41 Cx36,42 Cx45,43 Cx47,44 and Cx50.45
TABLE 1.
Cell–Cell Junctions Between Cardiomyocytes and Associated Diseases
| Type | Definition | Function | Major Proteins | Human Disease |
|---|---|---|---|---|
| GJs | Intercellular channels | Intercellular passage of ions and small molecules | Connexins (Cx43) | Remodeled in structural heart disease; oculodentoldigital dysplasia |
| Adherens junctions | Sites of mechanical attachment linked to actin | Hold cells together | Cadherins (N-Cadherin) Catenins (β-catenin) |
— |
| Desmosomes | Sites of mechanical attachment linked to intermediate filaments | Hold cells together | Cadherins (desmoglein, desmocollin) Plakoglobin Plakophilin Desmoplakin |
ARVD/C |
AJ proteins, as well as AJs themselves, have been demonstrated to play a role in the formation of GJs. Application of Fab fragments of antibodies to N-cadherin, an AJ protein, inhibited the formation of GJs between cultured lens cells.46 Shaw et al showed that AJs were required for the targeted delivery of Cx43 to the PM. Disruption of beta-catenin or p150(Glued), another AJ protein, by small interfering RNA (siRNA) knockdown or blocking homophilic N-cadherin interactions with a blocking peptide resulted in reduced GJ formation in HeLa cells transfected with Cx43-YFP.27 Another study demonstrated inhibition of Cx43 trafficking to the cell surface upon siRNA knockdown of N-cadherin in NIH 3T3 cells47 and cardiac specific loss of N-cadherin resulted in arrhythmogenic sudden cardiac death due to conduction slowing and GJ remodeling.48,49 Cardiac-restricted deletion of vinculin, a protein that mediates actin anchoring to the PM and is involved in extracellular membrane adhesion, also resulted in remodeling of GJs and promoted lethal ventricular arrhythmias.50
Functioning desmosomes are required for efficient GJ formation. Knockdown of the desmosomal protein plakophilin-2 by RNA interference in cultured neonatal rat ventricular myocytes resulted in an inhibition of GJ formation and intercellular communication.51 Furthermore, a transgenic mouse model of a desmin-related cardiomyopathy exhibited impaired GJ formation and a significant decrease in ventricular CV.52 Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is a structural heart disease predominantly of the right ventricle with a high incidence of ventricular arrhythmias causing sudden cardiac death. ARVD/C is commonly caused by mutations in desmosomal proteins, such as plakoglobin, plakophilin, and desmoplakin,53 and altered desmosomal protein localization may in fact serve as a specific diagnostic of ARVD/C.54 Remodeling of GJs has been reported in ARVD/C and may in part explain the electrophysiological phenotype of this desmosomal disease.54–57
In addition to mechanical junctions stabilizing GJs at the PM, phosphorylated inositol lipids have recently been shown to be important for regulating the function of Cx43-based GJs.58 Phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis at the PM results in closure of GJs, thus explaining the mechanism by which phopholipase C beta (PLCβ) activation by Gαq protein–coupled receptors causes rapid inhibition of GJ communication. The authors demonstrate that ZO-1 is required for PIP2-associated GJ channel closure and propose that scaffolding by ZO-1 forms a complex between Cx43 (via the ZO-1 PDZ2 domain) and PLCβ3 (via the ZO-1 PDZ3 domain), thereby creating a local pool of PLCβ3 near Cx43 poised to mediate PIP2 hydrolysis. This model assumes an interaction (either direct or indirect) between PIP2 and the region in Cx43 responsible for GJ gating, although the nature of this interaction remains to be identified.
GJ INTERNALIZATION AND DEGRADATION
The rapid turnover kinetics of GJs implies a tightly regulated mechanism of GJ internalization and degradation. GJs are removed from the PM by a unique cellular process, whereby entire GJ plaques are engulfed into 1 of the 2 cells of communicating cell pairs (Fig. 3). GJ internalization requires significant membrane perturbations, which likely employ distinct cellular machinery and mechanisms from those involved in more classical modes of membrane endocytosis. Very little is known about the cellular machinery that orchestrates the internalization of GJs, although several mechanisms have been proposed.
FIGURE 3.

GJ internalization. A, Transmission electron micrograph of internalizing GJs in canine cardiac tissue. B, Internalized GJ, also known as an annular GJ. Scale bars: A = 0.2 μm; B = 0.1 μm.
An early ultrastructural study proposed a role for clathrin coats and actin microfilaments in the process of GJ internalization.59 More recent studies have supported a role for clathrin in GJ internalization using more direct methods.60–62 Still another study has provided more direct evidence for actin microfilaments and the unconventional myosin, myosin VI, playing a role in the dynamics of GJ internalization.60 Stabilization or depolymerization of actin, respectively, increased or decreased the velocity of internalized GJ movement within the cell. Furthermore, myosin VI was recruited to internalizing GJs, and overexpression of myosin VI increased the dynamics of internalization of GJs. Furthermore, in support of cytoskeletal actin regulating GJ dynamics is the identification of the actin-binding protein, drebrin, as a Cx43-interacting protein.63 However, siRNA depletion of drebrin resulted in an apparent increase in the internalization and degradation of GJs, suggesting that drebrin may play more of a role in actin-mediated stabilization of GJ plaques at the PM rather than in GJ internalization.
Recently, Src tyrosine kinase recruitment and ZO-1 displacement from GJ plaques have been implicated in the timing and directionality of GJ internalization.64–66 Gilleron et al demonstrated enhanced GJ internalization upon treatment of Sertoli cells with the nongenomic carcinogen hexachlorocyclohexane. They further showed that GJ internalization is associated with c-Src recruitment to GJ plaques resulting in ZO-1 displacement from Cx43. GJ internalization occurred preferentially into the cell from which c-Src was recruited and ZO-1 was displaced. This study is among the first to describe the molecular events involved in the directionality of GJ internalization.
Details regarding GJ degradation are not well understood. It has been reported that GJ degradation involves both the lysosome and the proteasome.67,68 The proteasome likely serves to degrade misfolded, and possibly membrane-extracted, connexins early in the secretory pathway via an ERAD mechanism.69 Consistent with this is a study by Musil et al,70 which demonstrated enhanced assembly of GJs in GJ- assembly defective cells upon proteasomal inhibition. Quite unexpectedly, though, inhibition of the proteasome also seems to stabilize GJs at the PM by decreasing their rate of internalization. The mechanism for this stabilization may be indirect, through regulation of the turnover of yet unidentified proteasomal substrates other than connexins, which may play a role in GJ internalization.71,72 GJ internalization and degradation has been suggested to be dependent upon short-lived proteins, consistent with an indirect mechanism of stabilization.73
Recent studies by Lau et al have identified two novel Cx43-interacting proteins involved in its degradation.19,74 Using a yeast two-hybrid screen, the authors identified Cx43-interacting proteins with molecular weights of 85 kDa (CIP85) and 75 kDa (CIP75) as novel binding partners of Cx43 at its CT domain. CIP85 is a previously uncharacterized protein with putative Rab-GAP activity, which was shown to bind to a proline-rich sequence within the Cx43 CT (P253LSP256). Interestingly, this amino acid sequence also possesses a mitogen-activated protein kinase (MAPK) phosphorylation site (Ser255) known to play a role in downregulation of GJ communication through plaque internalization.75,76 CIP85 is localized to GJ plaque regions in HeLa cells expressing Cx43 and was shown to regulate the degradation of GJs by a lysosomal pathway.74 The putative Rab-GAP activity of CIP85, although not yet demonstrated to modulate a Rab family GTPase directly, is intriguing in light of the recently described role for Rab20 in regulating Cx43 transport within the secretory pathway.20 CIP75 is a novel member of the ubiquitin-like/ubiquitin-associated domain protein family, which binds to the CT of Cx43 within a region spanning Lys264–Asp302. This amino acid region is of interest in that it also contains MAPK phosphorylation sites involved in GJ internalization (Ser279/282)75,76 and a previously described PY motif in conjunction with a tyrosine-based sorting signal (P283PGYKLV289).77 CIP75 is colocalized with Cx43 in the ER and regulates the turnover of Cx43 via a proteasomal pathway. The PY motif and associated tyrosine-based sorting signal have themselves been examined for their role in regulating Cx43 turnover. Steady state levels of Cx43 were increased by approximately 3.5-fold with mutation of valine at position 289 to aspartate (V289D), whereas a comparatively small increase of approximately 1.7-fold was observed with a P283L mutation.77 These results suggest that the tyrosine-based sorting signal plays more of a role in the steady state internalization of GJs, whereas the PY motif may be involved in regulated internalization upon stimulation. Consistent with this hypothesis is a study demonstrating interaction of the E3 ubiquitin ligase Nedd4 with the Cx43 PY motif via Nedd4 WW domains.78 The affinity of this interaction was moderately increased by phosphorylation of Cx43 amino acid residues Ser279/282. MAPK phosphorylation at Ser279/282 may therefore enhance the interaction of Nedd4 with Cx43 and increase the extent of Cx43 ubiquitination, thereby increasing the rate of GJ internalization (see Table 2).
TABLE 2.
Cx43-Interacting Partners
| Interacting Partner | Nature of Interaction | Proposed Function |
|---|---|---|
| CIP75 | Lys264–Asp302 | Regulates proteasomal mediated turnover of Cx43 |
| CIP85 | P253LSP256 | Regulates lysosomal mediated turnover of Cx43 |
| Rab20 | Undetermined | Regulates ER to Golgi traffic of Cx43 |
| Tubulin | Direct and/or via EB1 | Post-Golgi trafficking of Cx43 containing vesicles |
| EB1 | Undetermined | Attachment of Cx43 containing vesicles to microtubules |
| ZO-1 | PDZ binding motif (D379LEI382) | Regulates the delivery of Cx43 to the periphery of GJ plaques |
| PLCβ3 | Indirectly via ZO-1 | Closure of Cx43 GJs through hydrolysis of PIP2 |
| PIP2 | Undetermined | Maintains open GJs |
| Clathrin | Undetermined | Internalization of double membrane GJ plaques |
| Myosin VI | Undetermined | Regulates dynamics of internalizing/internalized GJ plaques |
| Drebrin | CT domain | Stabilization of GJ plaques at the PM |
| Src | Pro274-Pro284 | Phosphorylation of Cx43 mediating GJ closure |
| Nedd4 | PY motif (P283PGY286) | Ubiquitination of Cx43 |
| 14-3-3 | Phosphorylated Ser373 | Regulates forward trafficking of Cx43 |
The most plausible mechanism of GJ degradation after their internalization is delivery to lysosomes.67 The mechanisms mediating the delivery of internalized GJs to lysosomes have been the subject of recent study, although few details are understood. Leithe et al79 have described a process whereby internalized double-membrane GJs undergo progressive separation and involution to produce multivesicular endosomes with a single limiting membrane for subsequent fusion with lysosomes. Piehl et al60 also demonstrated morphological evidence of internalized GJs fragmenting into smaller vesicles before lysosomal degradation. Whether this process is mechanistically similar to the typical maturation of endosomes into multivesicular bodies via the endosomal sorting complex required for transport (ESCRT) pathway is unknown.
REGULATION OF GJS THROUGH CX43 PHOSPHORYLATION
Cx43 can be extensively phosphorylated at amino acid residues in its CT domain (Fig. 4). Cx43 phosphorylation has been shown to regulate many aspects of the GJ life cycle including assembly, gating, internalization, and degradation. Several excellent reviews have recently been written on the role of phosphorylation in GJ assembly,80 gating,81 internalization, and degradation,82 and therefore, the topic will be covered only briefly here with an emphasis placed on GJ trafficking.
FIGURE 4.

Structural motifs and Cx43 phosphorylation. The known and putative phosphorylation sites in the carboxyl-terminus are numbered and enclosed in boxes. The positioning of proposed structural features (Fig. 1) and protein–protein interaction sites are indicated by the labeled boxes.
Phosphorylation of Cx43 is not an absolute prerequisite for GJ channel function because Cx43 with the CT truncated removing all known Cx43 phosphorylation sites can still form functional GJs.83 Rather, phosphorylation of Cx43 can serve to regulate the formation and destruction of GJs under certain cellular conditions. Increased levels of intracellular cyclic adenosine monophosphate (cAMP) have been widely reported to enhance the forward trafficking and assembly of GJs, and this response is thought to be protein kinase A (PKA) dependent.84–87 The major sites of PKA-dependent phosphorylation are proposed to be serine residues within a series of three RXSS motifs located in the distal portion of the CT of Cx43 (Ser364/5, Ser368/9, and Ser372/3).80,88 Despite the PKA dependence of these phosphorylation events, it has not been established whether Cx43 is itself a substrate for PKA or whether the PKA effects are mediated through activation of other kinases or phosphatases. For example, Ser373 has recently been shown to be phosphorylated by the protein kinase Akt, which creates a binding site for 14-3-3θ.89,90 Phosphorylation-dependent binding of 14-3-3 has been shown to play a role in enhanced forward trafficking of various membrane proteins, often through masking dibasic ER retention motifs,91 although it remains to be elucidated whether this is the case for Cx43.
Protein kinase C (PKC) phosphorylates Cx43 at Ser368, and this has been shown to inhibit GJ communication by decreasing the unitary conductance of GJ channels.92 PKC-mediated phosphorylation at Ser368 mediates the GJ inhibition associated with the treatment of cells with phorbol esters, such as phorbol 12-myristate 13-acetate (PMA). Phosphorylation of Cx43 at Ser365 has recently been shown to modulate the potential for subsequent phosphorylation of Ser368 by PKC.93 Phosphorylation at Ser365 alters the structure of the Cx43 CT such that subsequent phosphorylation at Ser368 is inhibited and therefore prevents PKC-mediated closure of GJs. PKC-mediated closure of GJs is also correlated with enhanced internalization and degradation of GJs, although this may be the result of cross talk with MAPK signaling pathways.
It has long been known that growth factor stimulation, for example, by epidermal growth factor (EGF), disrupts GJ communication through phosphorylation of Cx43.94 The specific sites of phosphorylation on Cx43 after EGF stimulation were identified as Ser255/279/282, which were later shown to be phosphorylated by MAPK.75,76 The specific kinases that phosphorylate Cx43 at these sites in vivo have been the subject of some debate because there are many MAPK family members that may be activated upon EGF stimulation. For example, one study has demonstrated phosphorylation of Cx43 at Ser255 by ERK5, rather than ERK1/2 upon EGF stimulation,95 although this work has been directly challenged.96 Disruption of GJ communication has been demonstrated through stimulation with a wide variety of growth factors, including platelet-derived growth factor,97 vascular endothelial growth factor,98 fibroblast derived growth factor (FGF),99 and insulin,100 as well as phosphorylation of Cx43 with different members of the MAPK family.101,102 MAPK-dependent phosphorylation of Cx43 has most often been associated with enhanced internalization of GJ plaques. EGF stimulation was shown to induce monoubiquitination of Cx43 at the PM, followed by GJ internalization and degradation.103 It will be interesting to determine whether a common theme exists whereby pro–growth stimuli in cells result in MAPK-dependent ubiquitination of Cx43, causing internalization and degradation of GJs.
Nearly two decades ago, disruption of GJ communication was demonstrated with phosphorylation of tyrosine residues on Cx43, resulting from coexpression of the viral nonreceptor tyrosine kinases pp60v-Src (v-Src).104,105 Cx43 was shown to be directly phosphorylated at Tyr265 by v-Src,106 and an interaction between v-Src and Cx43 was shown to be dependent on SH2 and SH3 domains within v-Src, a proline-rich region within the Cx43 CT (Pro274-Pro284), as well as phosphorylation at Tyr265.107 More recent studies demonstrated that v-Src–phosphorylated Cx43 at both Tyr265 and Tyr247, which raised the possibility that tyrosine phosphorylation of Cx43 is processive, with Tyr265 phosphorylation causing enhanced binding of v-Src (via the v-Src SH2 domain) and subsequent phosphorylation at Tyr247.108 However, this model has been challenged by the work of Zhou et al109 who demonstrated in Xenopus oocytes, as well as NRK cells, that neither Tyr265 nor Tyr247 phosphorylation was responsible for the v-Src mediated inhibition of GJ communication but rather that the effect was dependent upon MAPK phosphorylation of Cx43. The reason for these disparate findings has not been resolved.
A role for the v-Src cellular homologue, c-Src, in inhibition of GJ communication has been demonstrated.110 Further study showed that Tyr265 of Cx43 was a direct substrate for phosphorylation by c-Src and that the two proteins interacted in vitro and in vivo.111 A role for altered c-Src phosphorylation of Cx43 has been demonstrated in a cardiomyopathy model in hamster,112 and the same authors demonstrated that in cardiomyocytes, c-Src interaction with Cx43 and phosphorylation at Tyr265 regulates the binding of Cx43 and ZO-1.113 This notion of reciprocal regulation of c-Src and ZO-1 binding to the CT of Cx43 has been further explored at the structural level using nuclear magnetic resonance (NMR) of the Cx43 CT114 and in cardiomyocytes in the context of intracellular acidification.65 Phosphorylation of Cx43 by c-Src has been implicated in a variety of extracellular stimuli regulating changes in GJ communication including endothelin-1,115 lipopolysaccharide (LPS),116 and tumor necrosis factor α.117
The mechanism by which Src phosphorylation of Cx43 inhibits GJ communication is presumed to be due to a decrease in channel open probability118; however, significant cross talk between the various signaling pathways involved suggests that MAPK phosphorylation of Cx43 and GJ internalization may also contribute to GJ inhibition.119
CONCLUSIONS
The ubiquity of GJ communication in multicellular organisms speaks to its fundamental importance in regulated cell–cell communication. The large number of connexin genes possessed by higher organisms, and the rich diversity in their expression patterns in different cell types and tissues, is testament to the many functions and robust regulatory potential that GJs provide. In the heart, for example, coordinated electrical activity across millions of cardiomyocytes is only possible due to the direct cell-cell connections provided by GJs. It is therefore not surprising that when GJ function is compromised, that this coordinated electrical activity can go awry, with potentially lethal consequences for the organism. Many questions remain to be answered with respect to GJ remodeling in heart disease. First, the degree to which GJ remodeling actually contributes to slowed conduction and arrhythmogenesis in the setting of human heart disease remains to be definitively shown. The precise cellular mechanisms responsible for GJ assembly, internalization, and degradation in cardiomyocytes also require further study. Specifically, how GJ assembly is spatially regulated to occur primarily at the ID and the mechanism and functional significance of GJ lateralization are important issues. Finally, the specific signaling events that regulate the formation and destruction of GJs in the heart are only partially understood and may prove to be viable therapeutic targets for conduction disturbances associated with disease.
Acknowledgments
The authors wish to acknowledge financial support from National Institute Health PO1 HL077180 and R33 HL 087345.
Footnotes
The authors report no conflicts of interest.
References
- 1.Guerrero PA, Schuessler RB, Davis LM, et al. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99:1991–1998. doi: 10.1172/JCI119367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reaume AG, de Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 3.Gutstein DE, Morley GE, Tamaddon H, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maeda S, Nakagawa S, Suga M, et al. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 5.Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- 6.Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Severs NJ, Bruce AF, Dupont E, et al. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Severs NJ. Gap junction remodeling and cardiac arrhythmogenesis: cause or coincidence? J Cell Mol Med. 2001;5:355–366. doi: 10.1111/j.1582-4934.2001.tb00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akar FG, Spragg DD, Tunin RS, et al. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95:717–725. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- 10.Poelzing S, Akar FG, Baron E, et al. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am J Physiol Heart Circ Physiol. 2004;286:H2001–H2009. doi: 10.1152/ajpheart.00987.2003. [DOI] [PubMed] [Google Scholar]
- 11.Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762–H1770. doi: 10.1152/ajpheart.00346.2004. [DOI] [PubMed] [Google Scholar]
- 12.Saffitz JE, Hames KY, Kanno S. Remodeling of gap junctions in ischemic and nonischemic forms of heart disease. J Membr Biol. 2007;218:65–71. doi: 10.1007/s00232-007-9031-2. [DOI] [PubMed] [Google Scholar]
- 13.Das Sarma J, Wang F, Koval M. Targeted gap junction protein constructs reveal connexin-specific differences in oligomerization. J Biol Chem. 2002;277:20911–20918. doi: 10.1074/jbc.M111498200. [DOI] [PubMed] [Google Scholar]
- 14.Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 15.Koval M. Pathways and control of connexin oligomerization. Trends Cell Biol. 2006;16:159–166. doi: 10.1016/j.tcb.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanSlyke JK, Deschenes SM, Musil LS. Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell. 2000;11:1933–1946. doi: 10.1091/mbc.11.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yum SW, Kleopa KA, Shumas S, et al. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11:43–52. doi: 10.1006/nbdi.2002.0545. [DOI] [PubMed] [Google Scholar]
- 18.Roscoe W, Veitch GI, Gong XQ, et al. Oculodentodigital dysplasia-causing connexin43 mutants are non-functional and exhibit dominant effects on wild-type connexin43. J Biol Chem. 2005;280:11458–466. doi: 10.1074/jbc.M409564200. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Su V, Kurata WE, et al. A novel connexin43-interacting protein, CIP75, which belongs to the UbL-UBA protein family, regulates the turnover of connexin43. J Biol Chem. 2008;283:5748–5759. doi: 10.1074/jbc.M709288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das Sarma J, Kaplan BE, Willemsen D, et al. Identification of rab20 as a potential regulator of connexin 43 trafficking. Cell Commun Adhes. 2008;15:65–74. doi: 10.1080/15419060802014305. [DOI] [PubMed] [Google Scholar]
- 21.Jordan K, Solan JL, Dominguez M, et al. Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol Biol Cell. 1999;10:2033–2050. doi: 10.1091/mbc.10.6.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lauf U, Giepmans BN, Lopez P, et al. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas T, Jordan K, Simek J, et al. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J Cell Sci. 2005;118:4451–4462. doi: 10.1242/jcs.02569. [DOI] [PubMed] [Google Scholar]
- 24.Giepmans BN, Verlaan I, Hengeveld T, et al. Gap junction protein connexin-43 interacts directly with microtubules. Curr Biol. 2001;11:1364–1368. doi: 10.1016/s0960-9822(01)00424-9. [DOI] [PubMed] [Google Scholar]
- 25.Giepmans BN, Verlaan I, Moolenaar WH. Connexin-43 interactions with ZO-1 and alpha- and beta-tubulin. Cell Commun Adhes. 2001;8:219–223. doi: 10.3109/15419060109080727. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RG, Meyer RA, Li XR, et al. Gap junctions assemble in the presence of cytoskeletal inhibitors, but enhanced assembly requires microtubules. Exp Cell Res. 2002;275:67–80. doi: 10.1006/excr.2002.5480. [DOI] [PubMed] [Google Scholar]
- 27.Shaw RM, Fay AJ, Puthenveedu MA, et al. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- 29.Pearson RA, Dale N, Llaudet E, et al. ATP released via gap junction hemichannels from the pigment epithelium regulates neural retinal progenitor proliferation. Neuron. 2005;46:731–744. doi: 10.1016/j.neuron.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 30.Stout CE, Costantin JL, Naus CC, et al. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 31.Gaietta G, Deerinck TJ, Adams SR, et al. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 32.Stevenson BR, Siliciano JD, Mooseker MS, et al. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103:755–766. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyofuku T, Yabuki M, Otsu K, et al. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–12731. doi: 10.1074/jbc.273.21.12725. [DOI] [PubMed] [Google Scholar]
- 34.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 35.Hunter AW, Barker RJ, Zhu C, et al. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunter AW, Jourdan J, Gourdie RG. Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun Adhes. 2003;10:211–214. doi: 10.1080/cac.10.4-6.211.214. [DOI] [PubMed] [Google Scholar]
- 37.Barker RJ, Price RL, Gourdie RG. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ Res. 2002;90:317–324. doi: 10.1161/hh0302.104471. [DOI] [PubMed] [Google Scholar]
- 38.Bruce AF, Rothery S, Dupont E, et al. Gap junction remodelling in human heart failure is associated with increased interaction of connexin43 with ZO-1. Cardiovasc Res. 2008;77:757–765. doi: 10.1093/cvr/cvm083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laing JG, Saffitz JE, Steinberg TH, et al. Diminished zonula occludens-1 expression in the failing human heart. Cardiovasc Pathol. 2007;16:159–164. doi: 10.1016/j.carpath.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Kieken F, Mutsaers N, Dolmatova E, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–1112. doi: 10.1161/CIRCRESAHA.108.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flores CE, Li X, Bennett MV, et al. Interaction between connexin35 and zonula occludens-1 and its potential role in the regulation of electrical synapses. Proc Natl Acad Sci U S A. 2008;105:12545–12550. doi: 10.1073/pnas.0804793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Olson C, Lu S, et al. Neuronal connexin36 association with zonula occludens-1 protein (ZO-1) in mouse brain and interaction with the first PDZ domain of ZO-1. Eur J Neurosci. 2004;19:2132–2146. doi: 10.1111/j.l460-9568.2004.03283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laing JG, Manley-Markowski RN, Koval M, et al. Connexin45 interacts with zonula occludens-1 and connexin43 in osteoblastic cells. J Biol Chem. 2001;276:23051–23055. doi: 10.1074/jbc.M100303200. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Ionescu AV, Lynn BD, et al. Connexin47, connexin29 and connexin32 co-expression in oligodendrocytes and Cx47 association with zonula occludens-1 (ZO-1) in mouse brain. Neuroscience. 2004;126:611–630. doi: 10.1016/j.neuroscience.2004.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen PA, Baruch A, Shestopalov VI, et al. Lens connexins alpha3Cx46 and alpha8Cx50 interact with zonula occludens protein-1 (ZO-1) Mol Biol Cell. 2003;14:2470–2481. doi: 10.1091/mbc.E02-10-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frenzel EM, Johnson RG. Gap junction formation between cultured embryonic lens cells is inhibited by antibody to N-cadherin. Dev Biol. 1996;179:1–16. doi: 10.1006/dbio.1996.0237. [DOI] [PubMed] [Google Scholar]
- 47.Wei CJ, Francis R, Xu X, et al. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J Biol Chem. 2005;280:19925–19936. doi: 10.1074/jbc.M412921200. [DOI] [PubMed] [Google Scholar]
- 48.Li J, Levin MD, Xiong Y, et al. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol. 2008;44:597–606. doi: 10.1016/j.yjmcc.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Patel VV, Kostetskii I, et al. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- 50.Zemljic-Harpf AE, Miller JC, Henderson SA, et al. Cardiacmyocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol Cell Biol. 2007;27:7522–7537. doi: 10.1128/MCB.00728-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oxford EM, Musa H, Maass K, et al. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101:703–711. doi: 10.1161/CIRCRESAHA.107.154252. [DOI] [PubMed] [Google Scholar]
- 52.Gard JJ, Yamada K, Green KG, et al. Remodeling of gap junctions and slow conduction in a mouse model of desmin-related cardiomyopathy. Cardiovasc Res. 2005;67:539–547. doi: 10.1016/j.cardiores.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 53.Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:258–267. doi: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 55.Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Oxford EM, Everitt M, Coombs W, et al. Molecular composition of the intercalated disc in a spontaneous canine animal model of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm. 2007;4:1196–1205. doi: 10.1016/j.hrthm.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joshi-Mukherjee R, Coombs W, Musa H, et al. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (ARVD/C)-related plakophilin-2 (PKP2) mutations. Heart Rhythm. 2008;5:1715–1723. doi: 10.1016/j.hrthm.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Zeijl L, Ponsioen B, Giepmans BN, et al. Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2007;177:881–891. doi: 10.1083/jcb.200610144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larsen WJ, Tung HN, Murray SA, et al. Evidence for the participation of actin microfilaments and bristle coats in the internalization of gap junction membrane. J Cell Biol. 1979;83:576–587. doi: 10.1083/jcb.83.3.576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piehl M, Lehmann C, Gumpert A, et al. Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol Biol Cell. 2007;18:337–347. doi: 10.1091/mbc.E06-06-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gumpert AM, Varco JS, Baker SM, et al. Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 2008;582:2887–2892. doi: 10.1016/j.febslet.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nickel BM, DeFranco BH, Gay VL, et al. Clathrin and Cx43 gap junction plaque endoexocytosis. Biochem Biophys Res Commun. 2008;374:679–682. doi: 10.1016/j.bbrc.2008.07.108. [DOI] [PubMed] [Google Scholar]
- 63.Butkevich E, Hulsmann S, Wenzel D, et al. Drebrin is a novel connexin-43 binding partner that links gap junctions to the submembrane cytoskeleton. Curr Biol. 2004;14:650–658. doi: 10.1016/j.cub.2004.03.063. [DOI] [PubMed] [Google Scholar]
- 64.Baker SM, Kim N, Gumpert AM, et al. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008;582:4039–4046. doi: 10.1016/j.febslet.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duffy HS, Ashton AW, O’Donnell P, et al. Regulation of connexin43 protein complexes by intracellular acidification. Circ Res. 2004;94:215–222. doi: 10.1161/01.RES.0000113924.06926.11. [DOI] [PubMed] [Google Scholar]
- 66.Gilleron J, Fiorini C, Carette D, et al. Molecular reorganization of Cx43, Zo-1 and Src complexes during the endocytosis of gap junction plaques in response to a non-genomic carcinogen. J Cell Sci. 2008;121:4069–4078. doi: 10.1242/jcs.033373. [DOI] [PubMed] [Google Scholar]
- 67.Laing JG, Tadros PN, Westphale EM, et al. Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp Cell Res. 1997;236:482–492. doi: 10.1006/excr.1997.3747. [DOI] [PubMed] [Google Scholar]
- 68.Laing JG, Beyer EC. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J Biol Chem. 1995;270:26399–26403. doi: 10.1074/jbc.270.44.26399. [DOI] [PubMed] [Google Scholar]
- 69.VanSlyke JK, Musil LS. Dislocation and degradation from the ER are regulated by cytosolic stress. J Cell Biol. 2002;157:381–394. doi: 10.1083/jcb.200111045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Musil LS, Le AC, VanSlyke JK, et al. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275:25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- 71.Qin H, Shao Q, Igdoura SA, et al. Lysosomal and proteasomal degradation play distinct roles in the life cycle of Cx43 in gap junctional intercellular communication-deficient and -competent breast tumor cells. J Biol Chem. 2003;278:30005–30014. doi: 10.1074/jbc.M300614200. [DOI] [PubMed] [Google Scholar]
- 72.VanSlyke JK, Musil LS. Cytosolic stress reduces degradation of connexin43 internalized from the cell surface and enhances gap junction formation and function. Mol Biol Cell. 2005;16:5247–5257. doi: 10.1091/mbc.E05-05-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.VanSlyke JK, Musil LS. Degradation of connexins from the plasma membrane is regulated by inhibitors of protein synthesis. Cell Commun Adhes. 2003;10:329–333. doi: 10.1080/cac.10.4-6.329.333. [DOI] [PubMed] [Google Scholar]
- 74.Lan Z, Kurata WE, Martyn KD, et al. Novel rab GAP-like protein, CIP85, interacts with connexin43 and induces its degradation. Biochemistry. 2005;44:2385–2396. doi: 10.1021/bi048306w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Warn-Cramer BJ, Cottrell GT, Burt JM, et al. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem. 1998;273:9188–9196. doi: 10.1074/jbc.273.15.9188. [DOI] [PubMed] [Google Scholar]
- 76.Warn-Cramer BJ, Lampe PD, Kurata WE, et al. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J Biol Chem. 1996;271:3779–3786. doi: 10.1074/jbc.271.7.3779. [DOI] [PubMed] [Google Scholar]
- 77.Thomas MA, Zosso N, Scerri I, et al. A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J Cell Sci. 2003;116:2213–2222. doi: 10.1242/jcs.00440. [DOI] [PubMed] [Google Scholar]
- 78.Leykauf K, Salek M, Bomke J, et al. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci. 2006;119:3634–3642. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- 79.Leithe E, Brech A, Rivedal E. Endocytic processing of connexin43 gap junctions: a morphological study. Biochem J. 2006;393:59–67. doi: 10.1042/BJ20050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 81.Moreno AP, Lau AF. Gap junction channel gating modulated through protein phosphorylation. Prog Biophys Mol Biol. 2007;94:107–119. doi: 10.1016/j.pbiomolbio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laird DW. Connexin phosphorylation as a regulatory event linked to gap junction internalization and degradation. Biochim Biophys Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 83.Maass K, Shibayama J, Chase SE, et al. C-terminal truncation of connexin43 changes number, size, and localization of cardiac gap junction plaques. Circ Res. 2007;101:1283–1291. doi: 10.1161/CIRCRESAHA.107.162818. [DOI] [PubMed] [Google Scholar]
- 84.Atkinson MM, Lampe PD, Lin HH, et al. Cyclic AMP modifies the cellular distribution of connexin43 and induces a persistent increase in the junctional permeability of mouse mammary tumor cells. J Cell Sci. 1995;108(pt 9):3079–3090. doi: 10.1242/jcs.108.9.3079. [DOI] [PubMed] [Google Scholar]
- 85.Paulson AF, Lampe PD, Meyer RA, et al. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci. 2000;113 (pt 17):3037–3049. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]
- 86.Holm I, Mikhailov A, Jillson T, et al. Dynamics of gap junctions observed in living cells with connexin43-GFP chimeric protein. Eur J Cell Biol. 1999;78:856–866. doi: 10.1016/S0171-9335(99)80087-9. [DOI] [PubMed] [Google Scholar]
- 87.Burghardt RC, Barhoumi R, Sewall TC, et al. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J Membr Biol. 1995;148:243–253. doi: 10.1007/BF00235042. [DOI] [PubMed] [Google Scholar]
- 88.Yogo K, Ogawa T, Akiyama M, et al. Identification and functional analysis of novel phosphorylation sites in Cx43 in rat primary granulosa cells. FEBS Lett. 2002;531:132–136. doi: 10.1016/s0014-5793(02)03441-5. [DOI] [PubMed] [Google Scholar]
- 89.Park DJ, Freitas TA, Wallick CJ, et al. Molecular dynamics and in vitro analysis of Connexin43: a new 14-3-3 mode-1 interacting protein. Protein Sci. 2006;15:2344–2355. doi: 10.1110/ps.062172506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Park DJ, Wallick CJ, Martyn KD, et al. Akt phosphorylates Connexin43 on Ser373, a “mode-1” binding site for 14-3-3. Cell Commun Adhes. 2007;14:211–226. doi: 10.1080/15419060701755958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mrowiec T, Schwappach B. 14-3-3 proteins in membrane protein transport. Biol Chem. 2006;387:1227–1236. doi: 10.1515/BC.2006.152. [DOI] [PubMed] [Google Scholar]
- 92.Lampe PD, TenBroek EM, Burt JM, et al. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solan JL, Marquez-Rosado L, Sorgen PL, et al. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J Cell Biol. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lau AF, Kanemitsu MY, Kurata WE, et al. Epidermal growth factor disrupts gap-junctional communication and induces phosphorylation of connexin43 on serine. Mol Biol Cell. 1992;3:865–874. doi: 10.1091/mbc.3.8.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cameron SJ, Malik S, Akaike M, et al. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase1/ERK5 but not ERK1/2 kinase activation. J Biol Chem. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- 96.Abdelmohsen K, Sauerbier E, Ale-Agha N, et al. Epidermal growth factor- and stress-induced loss of gap junctional communication is mediated by ERK-1/ERK-2 but not ERK-5 in rat liver epithelial cells. Biochem Biophys Res Commun. 2007;364:313–317. doi: 10.1016/j.bbrc.2007.09.132. [DOI] [PubMed] [Google Scholar]
- 97.Hossain MZ, Jagdale AB, Ao P, et al. Disruption of gap junctional communication by the platelet-derived growth factor is mediated via multiple signaling pathways. J Biol Chem. 1999;274:10489–10496. doi: 10.1074/jbc.274.15.10489. [DOI] [PubMed] [Google Scholar]
- 98.Suarez S, Ballmer-Hofer K. VEGF transiently disrupts gap junctional communication in endothelial cells. J Cell Sci. 2001;114:1229–1235. doi: 10.1242/jcs.114.6.1229. [DOI] [PubMed] [Google Scholar]
- 99.Doble BW, Chen Y, Bosc DG, et al. Fibroblast growth factor-2 decreases metabolic coupling and stimulates phosphorylation as well as masking of connexin43 epitopes in cardiac myocytes. Circ Res. 1996;79:647–658. doi: 10.1161/01.res.79.4.647. [DOI] [PubMed] [Google Scholar]
- 100.Homma N, Alvarado JL, Coombs W, et al. A particle-receptor model for the insulin-induced closure of connexin43 channels. Circ Res. 1998;83:27–32. doi: 10.1161/01.res.83.1.27. [DOI] [PubMed] [Google Scholar]
- 101.Ogawa T, Hayashi T, Kyoizumi S, et al. Anisomycin downregulates gap-junctional intercellular communication via the p38 MAP-kinase pathway. J Cell Sci. 2004;117:2087–2096. doi: 10.1242/jcs.01056. [DOI] [PubMed] [Google Scholar]
- 102.Petrich BG, Gong X, Lerner DL, et al. c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ Res. 2002;91:640–647. doi: 10.1161/01.res.0000035854.11082.01. [DOI] [PubMed] [Google Scholar]
- 103.Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- 104.Swenson KI, Piwnica-Worms H, McNamee H, et al. Tyrosine phosphorylation of the gap junction protein connexin43 is required for the pp60v-src-induced inhibition of communication. Cell Regul. 1990;1:989–1002. doi: 10.1091/mbc.1.13.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Crow DS, Beyer EC, Paul DL, et al. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol Cell Biol. 1990;10:1754–1763. doi: 10.1128/mcb.10.4.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Loo LW, Berestecky JM, Kanemitsu MY, et al. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J Biol Chem. 1995;270:12751–12761. doi: 10.1074/jbc.270.21.12751. [DOI] [PubMed] [Google Scholar]
- 107.Kanemitsu MY, Loo LW, Simon S, et al. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J Biol Chem. 1997;272:22824–22831. doi: 10.1074/jbc.272.36.22824. [DOI] [PubMed] [Google Scholar]
- 108.Lin R, Warn-Cramer BJ, Kurata WE, et al. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J Cell Biol. 2001;154:815–827. doi: 10.1083/jcb.200102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou L, Kasperek EM, Nicholson BJ. Dissection of the molecular basis of pp60(v-src) induced gating of connexin 43 gap junction channels. J Cell Biol. 1999;144:1033–1045. doi: 10.1083/jcb.144.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Azarnia R, Reddy S, Kmiecik TE, et al. The cellular src gene product regulates junctional cell-to-cell communication. Science. 1988;239:398–401. doi: 10.1126/science.2447651. [DOI] [PubMed] [Google Scholar]
- 111.Giepmans BN, Hengeveld T, Postma FR, et al. Interaction of c-Src with gap junction protein connexin-43. Role in the regulation of cell-cell communication. J Biol Chem. 2001;276:8544–8549. doi: 10.1074/jbc.M005847200. [DOI] [PubMed] [Google Scholar]
- 112.Toyofuku T, Yabuki M, Otsu K, et al. Functional role of c-Src in gap junctions of the cardiomyopathic heart. Circ Res. 1999;85:672–681. doi: 10.1161/01.res.85.8.672. [DOI] [PubMed] [Google Scholar]
- 113.Toyofuku T, Akamatsu Y, Zhang H, et al. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 114.Sorgen PL, Duffy HS, Sahoo P, et al. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 115.Postma FR, Hengeveld T, Alblas J, et al. Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol. 1998;140:1199–1209. doi: 10.1083/jcb.140.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lidington D, Tyml K, Ouellette Y. Lipopolysaccharide-induced reductions in cellular coupling correlate with tyrosine phosphorylation of connexin 43. J Cell Physiol. 2002;193:373–379. doi: 10.1002/jcp.10179. [DOI] [PubMed] [Google Scholar]
- 117.Huang S, Dudez T, Scerri I, et al. Defective activation of c-Src in cystic fibrosis airway epithelial cells results in loss of tumor necrosis factor-alpha-induced gap junction regulation. J Biol Chem. 2003;278:8326–8332. doi: 10.1074/jbc.M208264200. [DOI] [PubMed] [Google Scholar]
- 118.Cottrell GT, Lin R, Warn-Cramer BJ, et al. Mechanism of v-Src-and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol. 2003;284:C511–C520. doi: 10.1152/ajpcell.00214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Solan JL, Lampe PD. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes. 2008;15:75–84. doi: 10.1080/15419060802014016. [DOI] [PMC free article] [PubMed] [Google Scholar]