

Abstract
Hermansky Pudlak syndrome (HPS) is a heterogeneous recessive genetic disease with a tendency to develop lung fibrosis with aging. A mouse strain with two mutant HPS genes affecting separate vesicle trafficking pathways, C57BL/6-Hps1 ep-Ap3b1 pe, exhibits severe lung abnormalities at young ages, including enlarged alveolar type II (ATII) cells with giant lamellar bodies and foamy alveolar macrophages (AMs), which are readily identified histologically. In this study, the appearance of lung fibrosis in older animals was studied using classical histological and biochemical methods. The HPS double mutant mice, but not Chediak Higashi syndrome (C57BL/6-Lyst bg-J-J, CHS) or C57BL/6J black control (WT) mice, were found to develop lung fibrosis at about 17 months of age using Masson trichrome staining, which was confirmed by hydroxyproline analysis. TGF β1 levels were elevated in bronchial alveolar lavage samples at all ages tested in the double mutant, but not WT or CHS mice, indicative of a prefibrotic condition in this experimental strain; and AMs were highly positive for this cytokine using immunohistochemistry staining. Prosurfactant protein C staining for ATII cells showed redistribution and dysmorphism of these cells with aging, but there was no evidence for epithelial-mesenchymal transition of ATII cells by dual staining for prosurfactant C protein and α-smooth muscle actin. This investigation showed that the HPS double mutant mouse strain develops interstitial pneumonia (HPSIP) past 1 year of age, which may be initiated by abnormal ATII cells and exacerbated by AM activation. With prominent prefibrotic abnormalities, this double mutant may serve as a model for interventive therapy in HPS.
Keywords: Pulmonary fibrosis , Hermansky Pudlak syndrome , Alveolar macrophages, Alveolar type II cells, TGF β1
Introduction
Hermansky Pudlak syndrome (HPS) is a heterogeneous recessive genetic disease which affects vesicle trafficking in lysosome-related organelles, including melanosomes, platelet dense granules, and lamellar bodies in alveolar type II (ATII) cells (Bonifacino 2004; Wei 2006). Some patients with this disease develop pulmonary fibrosis, also called HPS interstitial pneumonia (HPSIP, Nakatani et al. 2000). HPSIP is characterized by giant lamellar bodies of ATII cells and their apparent degeneration, which is hypothesized to play a major role in the development of this fibrosing disorder.
There are some 14 mouse mutants that are animal models for HPS (Li et al. 2004). The mouse pale ear mutant, HPS1 ep, is the model for the most common HPS disease, HPS 1, while the pearl mutant, Ap3b1 pe, is the model for HPS 2. As for all HPS genes, the products HPS1 and HPS2 (a subunit of the AP-3 adaptor complex, Feng et al. 1999), respectively, are involved in vesicle trafficking and the secretory process, and known to influence separate pathways (Feng et al. 2002). The HPS mouse strain, C57BL/6-Hps1 ep-Ap3b1 pe, combines two mutant HPS genes into one mouse model for HPS (Feng et al. 2002).
Previous study of this strain showed that, at 1 year of age or younger, the lungs of these animals were filled with foamy and binucleate alveolar macrophages (AMs), exhibited emphysema, impaired lung ventilation, and giant lamellar bodies of ATII cells (Lyerla et al. 2003), that are not as pronounced in the individual mutant strains (Feng et al. 2002). Thus, the mutant effects of each gene on cellular secretion were exaggerated in this double homozygote condition, which resulted in abnormal perturbations of affected cell types, particularly ATII cells. Subsequent investigations showed levels of phospholipid and surfactant proteins B and C were highly elevated in lung homogenates, but decreased in bronchial alveolar lavages (BAL, Guttentag et al. 2005), indicating accumulated storage and blocked secretion for both surfactant lipids and proteins of the ATII cells that accounted for the giant lamellar bodies seen in these mutant cells.
The highly dysmorphic ATII cells in the HPS double mutants were similar to those observed in HPSIP patients (Nakatani et al. 2000), however, there was no evidence of lung fibrosis at these early ages (Lyerla et al. 2003). Studies of lung morphology at the cellular and histological levels in older ages, then, were needed to determine the possible development of pulmonary fibrosis with aging in this HPS double mutant, which could be attributed to the cellular defects in the secretory process caused by these mutations.
Abnormal lamellar bodies in ATII cells are also found in another mouse mutant, beige (Lyst bg , Lyst bg), which is an animal model for Chediak Higashi syndrome (CHS) (Chi et al. 1975). This mouse strain exhibits accumulation of surfactant in ATII cells, but shows normal secretion of surfactant phospholipids (Prueitt et al. 1978). Because of the shared dysmorphism of ATII cells in the HPS double mutant and CHS strains, the CHS mouse was also studied in these investigations.
TGF β1 is considered to be the most important cytokine for lung fibrosis because it links possible causative processes including inflammation and fibroblast growth in the formation of this disease. Previous studies found this cytokine is secreted from AMs in vitro (Nishimura et al. 2007) and stimulated the differentiation and growth of myofibroblasts (Gu et al. 2007), the collagen producing cells in lung fibrosis. TGF β1 administration can also induce epithelial–mesenchymal transition (EMT) (Kim et al. 2007; Liu 2004), a process that appears important for lung fibrosis (Kim et al. 2009). For these reasons, investigations of the time course of TGF β1 secretion and localization of this cytokine in vivo were made in conjunction with the histochemical and cellular analyses that assessed the development of pulmonary fibrosis in the HPS double mutant mouse strain.
Materials and methods
Mice
The breeding stock of C57BL/6-Hps1 ep-Ap3b1 pe, the double mutant strain homozygous for two Hermansky Pudlak Syndrome genes and referred to as the HPS double mutant, was kindly provided by Dr. Richard Swank of the Roswell Park Cancer Institute (Buffalo, NY, USA). C57BL/6J (wild type, or WT), and C57BL/6-Lyst bg-J-J, the beige mouse strain which is an animal model for Chediak Higashi Syndrome (CHS), were purchased from Jackson Laboratories (Bar Harbor, ME, USA). All experimental procedures were approved by the Institutional Animal Care and Use Committee and followed the NIH Guidelines for Laboratory. Mouse samples from the three strains were free of infectious disorders as determined by Assessment Plus (Charles River Laboratories, Wilmington, MA, USA).
For biochemical and histological analyses, mice of given ages were anesthetized with avertin, 1 mL of PBS bronchial alveolar lavage (BAL) fluid was transfused into cannulated lungs three times, and 80% BAL fluid was recovered. After the lavage, the right lobes were separated with surgical thread and snap frozen in liquid nitrogen for further analysis. The left lung was fully inflated and fixed in 4% buffered formalin, embedded in paraffin, sectioned at 5 μm, and mounted on positively charged slides. After centrifugation, the cell-free BAL supernatant was stored at −80°C for further studies.
Hydroxyproline analysis
The method of Lazo (1981) was used for hydroxyproline analyses from isolated right lobes. Briefly, the lobes were homogenized in 2 ml deionized water using either a Dounce homogenizer or Polytron tissue grinder (Brinkman, Lucern, Switzerland), and samples prepared from trichloroacetic acid precipitates were treated in 12 N HCl at 110°C for 16 h and dissolved in 2 ml deionized water. Samples and standards (50 μl each) were incubated with Chloramine T reagent for 20 min at room temperature, and with Ehrlich’s reagent for 15 min at 65°C. The final reaction solutions were measured for absorbance at 550 nm (UV/VIS spectrophotometer UV-1650PC, Shimadzu Scientific Instruments, Columbia, MD, USA) and hydroxyproline levels obtained as microgram per lung.
Alveolar size quantification
Mean linear intercept (Lm) was employed to assess for alveolar size according to Thurlbeck (1967). Briefly, photomicrographs of hematoxylin stained sections were divided into six identical squares by lines. Alveolar walls crossing each line were counted, the counts were summed for each print, and these were divided into the total length of the lines crossing the photos in order to provide the mean linear intercept value, Lm, for each photomicrograph. For this analysis, three photomicrographs were made from separate regions across each lung section at 400× magnification. Three mice were used from each of the strains at six different postnatal ages for a total of some 162 different photomicrographs (54 per strain). In order to eliminate a possible subjective bias in this study, four objective observers were employed to count the alveolar wall intercepts.
Immunohistochemistry (IHC)
IHC staining was done using a BenchMark® XT IHC/ISH staining apparatus (Ventana Medical Systems, Inc, Tucson, AZ, USA), according to the manufacturer’s detection XT iVIEW DAB Open Kit with manual adjustments of the program in order to accommodate mouse antigens. All antibodies were freshly diluted with 2.5% goat serum (Jackson ImmunoResearch Labs, Inc.). TGF β1 antibody (sc-146, Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used at 1:200 dilution, prosurfactant protein C polyclonal antibody (pro-spc, 20R-PR030, Fitzgerald Industries Int’l, Concord, MA, USA) for ATII cell labeling at 1:3,000, and biotinylated goat anti-rabbit secondary antibody (Jackson ImmunoResearch) at 1:500. Nonspecific staining was eliminated by pretreatment with 2.5% goat serum and endogenous biotin blocked by Blocker A/B reagents provided by Ventana Medical Systems. Slides were counterstained by hematoxylin followed by bluing reagent. Negative controls were treated the same, only without the primary antibodies.
Fluorescent dual stainings for surfactant C and α-smooth muscle actin (α-SMA) were performed by the core facility in the Departments of Oncology and Pathology at the Johns Hopkins Medical Institutions. Briefly, the sections were blocked in a mixture of Fab fragment of goat anti-mouse IgG (Jackson ImmunoResearch), protein block solution (Dako Cytomation, Carpinteria, CA, USA), normal goat serum (Fitzgerald Industries Int’l), and bovine serum albumin (Sigma Chemicals, St. Louis, MO, USA). The primary antibodies used were rabbit polyclonal anti-prosurfactant protein C (Fitzgerald Industries Int’l) and mouse monoclonal anti-smooth muscle actin (Dako Cytomation). Secondary antibodies used were anti-rabbit Alexa-488 and anti-mouse Alexa-610 (Molecular Probes Inc., Eugene, OR, USA). Slides were counterstained by DAPI (Sigma Chemical Co.).
ATII cell counting
Photomicrographs from prosurfactant protein C-stained slides were analyzed by Image J software for the numbers of ATII cells using the color deconvolution and analyze particles function. The size was set larger than 220 square pixels in order to avoid counting small non-cellular particles.
Masson trichrome staining
This was done using commercially provided Masson Trichrome Stain Kit (Richard-Allan Scientific, Kalamazoo, MI, USA) according to the manufacturer’s protocols.
ELISA analysis
Total TGF β1 levels in BAL supernatants were tested by ELISA assay (Promega, Madison, WI, USA), using acid activated samples according to the manufacturer’s protocol. Each sample was done in duplicate, and results reported as pg TGF β1 per milliliter BAL.
Statistical analyses
The statistical analyses of strain differences at each age and the age related trends of each mouse strain were performed separately by the Tukey HSD test, using JMP software (SAS Institute Inc., Cary, NC, USA). The criterion of significant difference was set at P < 0.05. Results are reported as averages ± standard deviations (±SD).
Results
Lung fibrosis in HPS double mutant mice
At 12 months of age, large areas of blue Masson trichrome stained collagen were observed in the alveolar epithelium in HPS (Fig. 1a). At 17 months, sections from HPS double mutants had intensely stained blue collagen in association with thickened alveolar walls and tissue patches, the classical histological presentation of lung fibrosis (Fig. 1b). In contrast, WT and CHS histological samples from all ages tested were devoid of the concentrated blue staining regions, indicative of the lack of collagen deposition in these two mice strains (Fig. 1c, d). At the ages from 2 to 9 months, there was no concentrated blue staining in the lung sections from the three mouse strains (data not shown).
Fig. 1.
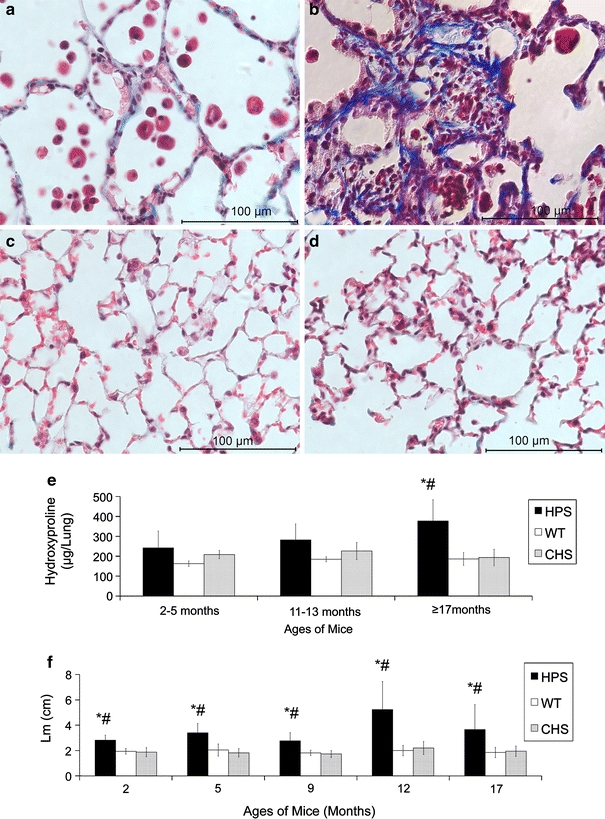
Histochemical and biochemical analyses of the lung fibrosis in aging HPS double mutant mice. Panels a–d are representative photomicrographs of Masson trichrome stained lung sections from a HPS at 12 months of age, b HPS at 17 months, c WT at 17 months, and d CHS mice at 17 months. Panel e shows the lung hydroxyproline levels (±SD) in HPS, WT, and CHS mice with aging; panel f shows the mean linear intercepts (Lm, ±SD) of the lungs from HPS, WT, and CHS with aging. Samples of 3–6 mice were used for each time category and strain. Mouse strains: HPS = HPS double mutant mouse, C57BL/6-Hps1 ep-Ap3b1 pe; CHS = CHS mouse, C57BL/6-Lyst bg-J-J; WT = wild type mouse, C57BL/6J. Significant difference P < 0.05: *HPS versus WT; #HPS versus CHS. No significant difference between CHS and WT was found at any age tested
The level of lung fibrosis was quantified by hydroxyproline analyses in the three strains from 2 to over 17 months of age (Fig. 1e). Results indicated that lung fibrosis in the HPS double mutant at 17 months of age was prominent, which was consistent with the observations from Masson trichrome stained lung sections.
HPS lungs also exhibited significantly larger alveoli compared with WT and CHS at all ages tested, indicative of emphysema (Fig. 1f) and consistent with previous studies of this strain (Lyerla et al. 2003). The enlargement of alveoli became more severe after 12 months of age, when lung fibrosis was observed.
Changes of distribution and morphology in AT II cells in HPS double mutant mice with aging
From the lung sections stained for prosurfactant C, ATII cells exhibited a homogeneous distribution in HPS double mutant mice at 2 months of age (Fig. 2a), where 1–3 ATII cells were found per alveolus. A change in this distribution was observed in samples from 5 month old mutants, where these cells appeared sparsely scattered and no longer present among all alveoli (Fig. 2b). At 9 months of age, ATII cell-rich regions were observed, and with some devoid of this cell type (Fig. 2c). Compared with these observations, ATII cells in WT and CHS samples at all ages tested were evenly distributed among all alveoli (Fig. 2d, e). The quantification of ATII cells showed that there was a significant cell number decrease at 5 months of age and a recovery of the cell number at the age of 9 months in the HPS strain (Fig. 2f). As the alveolar sizes were stable from 2 to 9 months of age in HPS (Fig. 1f), the change in ATII cell number represented an absolute reduction in the numbers of this cell type in 5 month old HPS lungs. Compared with HPS, ATII cell numbers in WT mice were constant at all ages tested. CHS ATII cells showed non-significant but a trend of decreasing in number with aging (Fig. 2g).
Fig. 2.
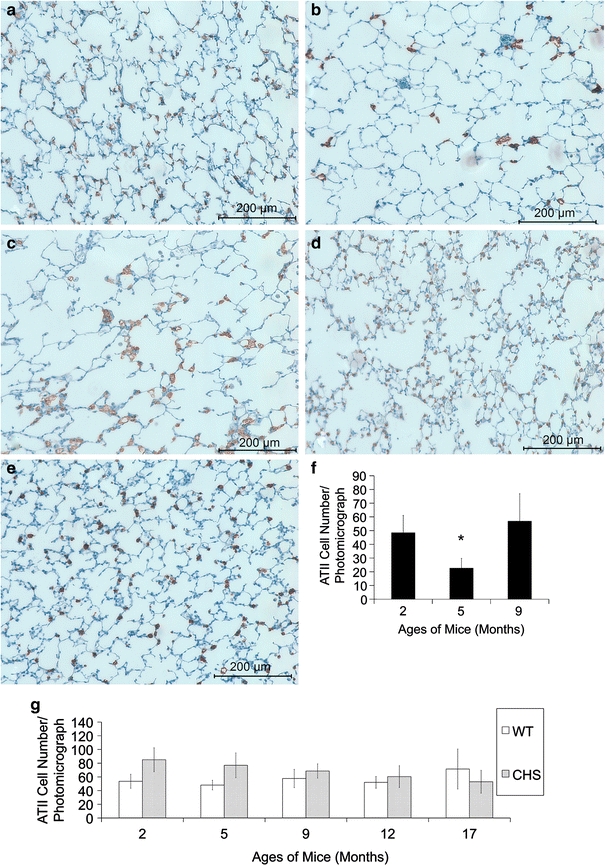
ATII cell distribution changes with aging in HPS double mutant mice. Representative prosurfactant C-stained (brown precipitate) lung sections using IHC from HPS mice at a 2 months, b 5 months, c 9 months, and d WT and e CHS at the age of 12 months. The ATII cells in HPS and CHS mutants are both enlarged compared with WT mice. Panel f shows the average ATII cell number per photomicrograph in HPS double mutant mice at the age of 2, 5, and 9 months. ATII cell number at 5 months of age is significantly lower than those at 2 and 9 months old (asterisk). Panel g shows the average ATII cell number per photomicrograph in WT and CHS mice from 2 months to 17 months of age. The cell density was constant in WT mice, but showed a non-significant decrease in CHS mice
In addition to their distribution, the morphology of ATII cells in HPS changed with aging. These cells were elongated along the alveolar walls in the double mutant at 1 year old and beyond (Fig. 3a, b), when it was not possible to assess the number of ATII cells/alveolus in HPS. ATII cells in WT and CHS lungs showed no obvious changes in shape (data not shown).
Fig. 3.
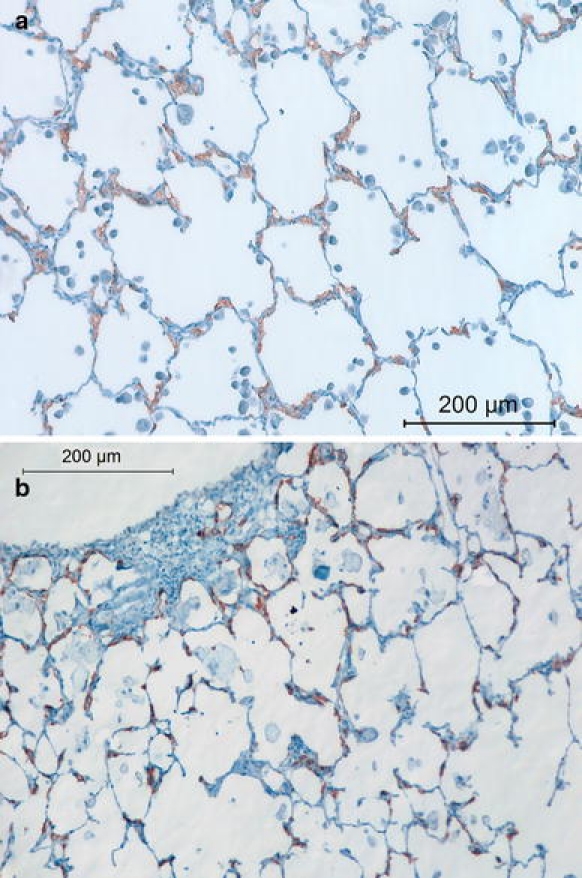
Representative prosurfactant protein C-stained lung sections from HPS mice at postnatal ages of a 12 and b 17 months. Note the aggregated and misshapen ATII cells in HPS double mutant mice seen at these older ages
The aggregation and dysmorphism of HPS ATII cells at later ages suggested an EMT process in this cell type. However, the dual stainings for ATII cell marker surfactant C and myofibroblast marker α-SMA did not exhibit co-staining (Fig. 4). Instead, the possible myofibroblasts with α-SMA staining and clusters of ATII cells both appeared around the thickened alveolar walls and fibrotic patches (Fig. 4a, b).
Fig. 4.
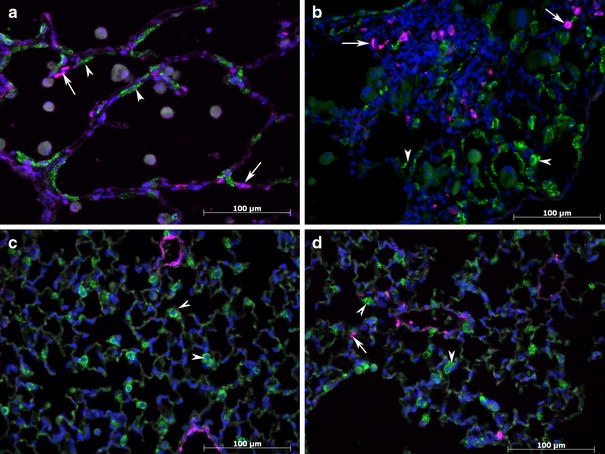
Representative prosurfactant protein C (strong green, shown by arrow heads) and α-SMA-stained (pink, shown by arrows) lung sections from HPS mice at postnatal ages of a 12, b 17 months with a fibrotic area, Lung sections of WT c and CHS d mice at 17 months of age. Arrow heads showed representative ATII cells which were stained strong green, and arrows showed representative myofibroblasts which were stained pink. Highly positive α-SMA cells were found in close proximity to aggregations of ATII cells at 12 months and 17 months of age in HPS double mutant mice, but this marker was not co-localized with that of prosurfactant C protein. Fewer myofibroblasts were found in WT and CHS. Walls of bronchioles and blood vessels were also stained with α-SMA marker
TGF β1 in HPS double mutant mice
As seen in Fig. 5a, significantly higher levels of TGF β1 in HPS BAL supernatants were found at all ages tested, compared with WT and CHS. Also, TGF β1 levels in HPS BAL were significantly higher at 17 months than those at 9 months of age. Dense staining using IHC for this cytokine was found in AMs of the HPS double mutants as well as WT control and CHS mouse strain throughout all ages tested. Dark staining was observed in cells of the alveolar walls in HPS mice at 12 and 17 months of age, but not in WT or CHS mice (Fig. 5b–e).
Fig. 5.
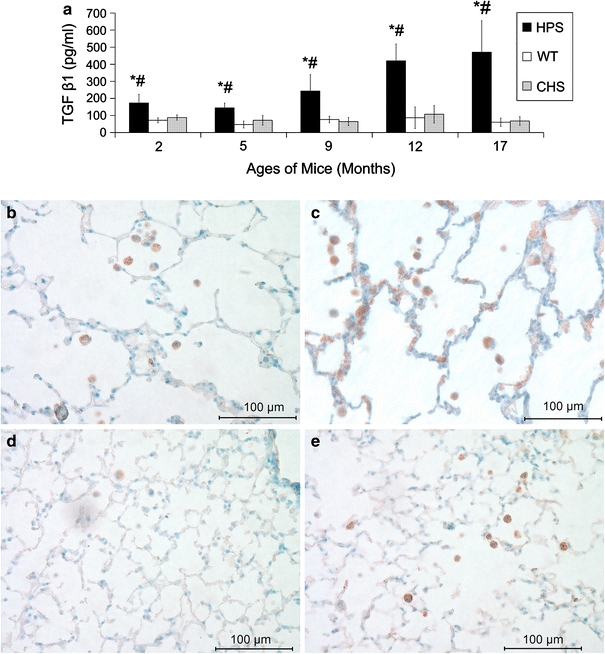
TGF β1 BAL levels and localization in lungs. Panel a shows average TGF β1 (±SD) levels in BAL samples of HPS, WT, and CHS mice from 2 to 17 months of age using ELISA. Samples of 4–10 mice were used for each time point and strain. Significant difference P < 0.05: *HPS versus WT; #HPS versus CHS. No significant difference between CHS and WT was found at any age tested. Panels b–e are representative TGF β1 IHC stained (brown precipitate) lung sections from HPS at (b) 9 and (c) 12 months of age and from (d) WT and (e) CHS at 12 months of age. AMs (intra-alveolar cells) were darkly stained in each of the mouse strains at all ages tested, and staining appeared in epithelial cells as well in older HPS mice but not in WT or CHS mice
Discussion
This research was undertaken in order to determine whether the HPS double mutant mice exhibited classical signs of lung fibrosis with aging beyond 1 year of age. Some 300 or more post-natal animals were examined for this study, including 131 HPS double mutants. Twenty-seven HPS mice 16 WT and 18 CHS were raised to 17 months of age or older. Among these, none of the WT control or CHS animals exhibited classical signs of fibrosis as evidenced by high levels of hydroxyproline or the presence of fibrotic patches, while 20 (74%) of the double mutant animals at 17 months and beyond had one or both of these symptoms. Most importantly, perhaps, the lung abnormalities in HPS mice, including lung fibrosis, are unlike those induced by drugs (Borzone et al. 2001), i.e., they are progressive until the end of the animals’ lives and do not heal with time.
Masson trichrome staining and hydroxyproline analysis both detected lung fibrosis in this double mutant at the age of 17 months. The histological method appeared to be more sensitive in detecting collagen deposition in lungs. That is, at 12 months of age, Masson trichrome staining showed obvious blue staining in the alveolar walls, which was not found in WT and CHS mouse strains or HPS at younger ages. Hydroxyproline levels, however, were not significantly different among these three strains at that age. This may be because pulmonary fibrosis starts as deposition in alveolar walls with insufficient amounts of collagen to affect the whole lung analysis of hydroxyproline levels. Also, hydroxyproline analysis can be affected by the increased levels of surfactant proteins in ATII cells (Lyerla et al. 2003), which explains the slightly higher levels seen in HPS and CHS mice at younger ages.
Many lung abnormalities including emphysema, dysmorphic and highly activated AMs (Lyerla et al. 2003), and higher levels of TGF β1 (this study), appeared much earlier than the overt fibrosis in the HPS double mutant animals. All of these abnormalities were more severe at 12 months of age, when the alveolar interstitium was thickened with collagen and lined with ATII cells. Thus, the lung abnormalities of HPS double mutant can be separated into two stages: the prefibrotic, in which early abnormalities are observed and persist to 9 months of age; and accelerated deterioration, after 9 months of age when these symptoms increase in severity as either the causes or effects of developing fibrosis.
The prefibrotic stage in the HPS double mutant may be useful for studies of early diagnosis and therapeutic interventions of HPSIP. For example, the changes of ATII cell distributions and decrease in number observed in HPS double mutant from 2 to 9 months of age were unique. This was not observed in earlier studies (Lyerla et al. 2003) and is presumably not detectable in bleomycin-induced animals due to the rapid nature of developing fibrosis in this model. The decrease of ATII cells in HPS lungs from 2 to 5 months may be due to the abnormal structure of this cell type with giant lamellar bodies. This might also be the reason for the trend of ATII cell decreasing in CHS mice with similar but less enlarged giant lamellar bodies. The increase of ATII cell numbers in HPS lungs at 9 months and beyond may be due to proliferation induced by mechanical stress as lung structure deteriorates. This phenomenon has been found in ATII cells of mice during compensatory regrowth after single lung pneumonectomy (Hoffman et al. 2010).
The persistent presence of abnormally high levels of total TGF β1 which was seen in BAL samples and localized in AMs by IHC staining, is another prefibrotic condition. This cytokine was mainly localized to AMs, suggesting this cell type is its major source, which is in keeping with other studies using chemically induced animal models (Chen et al. 2009 Nishimura et al. 2007). Previous studies have shown that the deficiency of surfactant protein B in mice promoted AM activation (Ikegami et al. 2005). Thus, the blocked secretion of surfactant proteins in HPS mice (Guttentag et al. 2005) may increase TGF β1 secretion by activation of AMs. This hypothesis is also supported by the studies of CHS mouse strain, which also possesses enlarged lamellar bodies in ATII cells but exhibits normal surfactant lipid secretion of ATII cells (Prueitt et al. 1978) and, in this study, showed no increased levels of TGF β1 in BAL samples with aging. On the basis of histological analyses, Tang et al. (2005) reported that CHS (Lyst bg) mice exhibited a more robust fibrosis than seen in the HPS1 mouse model, C57/B6 HPS1 ep. Our results do not confirm this observation regarding the appearance of pulmonary fibrosis in the CHS strain. Intra-strain variation and environmental factors may account for this discrepancy. However, the similar levels of TGF β1 and hydroxyproline observed in the lungs of CHS compared with WT mice in our laboratory were consistent with the histological assessment of the lack of fibrotic patches in the CHS animals at all ages tested. It is also consistent with CHS patients who are not at risk for lung fibrosis (Intone et al. 2009).
The appearance of TGF β1 in epithelial cells in lungs from HPS mice older than 1 year indicated a possible secondary source of this cytokine. This change coincided with an increase of TGF β1 in BAL which suggests that additional cell types contribute TGF β1 levels when HPS mice enter the accelerated deterioration period.
Although the role of TGF β1 in the development of lung fibrosis is well established, there are still questions about the importance of EMT in this process. For example, Kim et al. (2009) created a transgenic mouse strain that permanently expressed GFP in prosurfactant C positive cells, and found colocalization of α-SMA and GFP after bleomycin treatment. Königshoff et al. (2009) also found α-SMA expression in ATII primary cell cultures from bleomycin treated mice. In this study, when single DAB staining of α-SMA was used, some positively stained cells were observed with ATII cell morphology in HPS double mutant mice at old ages (data not shown). However, the fluorescent double staining method did not support EMT in ATII cells. This is similar to the work of Yamada et al. (2008) who showed no evidence of EMT using dual IHC staining of epithelial markers and myofibroblast markers, including α-SMA, in bleomycin treated mice. These inconsistencies may be because either the sensitivity of antibody staining is affected by double staining in animal tissues or that, in single staining, ATII cells from fibrotic lungs absorb the reactive antibody and cause false positives. In this investigation, the morphological changes observed in ATII cells of HPS mice with aging suggested that the cells underwent some conversion, but whether this involves EMT needs additional study. If this conversion is not due to EMT, these cells may differentiate into alveolar type I (ATI) cells in order to compensate for lung destruction. The abnormal prosurfactant C staining in HPS compared with WT mice may be due to difficulty in surfactant clearance in HPS ATII cells compared with WT. This hypothesis could be confirmed by double staining of prosurfactant C and ATI cell markers in our future studies.
The presence of fibrotic patches and elevated levels of hydroxyproline with aging, and the persistently high levels of TGF-β1 in alveoli of these mice, versus the absence of these features in the WT control and CHS strain provide evidence for progressive HPSIP in the double mutant strain. As this has not been reported for either of the single HPS mutant animals from which the HPS double mutant strain was derived, it appears that the combination of two vesicle trafficking disorders affecting separate pathways is especially injurious to long-term maintenance of lung homeostasis. The lung cell abnormalities that are known in this strain include the presence of giant lamellar bodies in ATII cells (Lyerla et al. 2003) resulting in the block to secretion of surfactant protein (Guttentag et al. 2005) and, from this study, the high levels of alveolar TGF-β1 mainly due to activated AMs. It appears that the blocked secretion of ATII cells induces AM activation and lung stresses such as alveolar destruction. The lung stresses may trigger a compensatory response including ATII cell regrowth. The activated AMs, which secrete high levels of TGF β1, and ATII cells, which show morphological cell conversion, lead the lung into progressive HPSIP in this strain.
Acknowledgments
Partial support for this research was provided by NIH grant R15 HL091503-01, and is gratefully acknowledged. We thank Nuclea Biotechnologies, LLC, for the use of their equipment, and Edmund Sosnoski for his advice and assistance with automated histological processing. We are also indebted to the anonymous reviewers for their suggested corrections and changes in the original manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Bonifacino JS. Insights into the biogenesis of lysosome-related organelles from the study of Hermanksy-Pudlak syndrome. Ann NY Acad Sci. 2004;1038:103–114. doi: 10.1196/annals.1315.018. [DOI] [PubMed] [Google Scholar]
- Borzone G, Moreno R, Urrea R, Meneses M, Oyarzun M, Lisboa C. Bleomycin-induced chronic lung damage does not resemble human idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;163:1648–1653. doi: 10.1164/ajrccm.163.7.2006132. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang X, Weng D, Tao S, Lv L, Chen J. A TSP-1 functional fragment inhibits activation of latent transforming growth factor-beta1 derived from rat alveolar macrophage after bleomycin treatment. Exp Toxicol Pathol. 2009;61(1):67–73. doi: 10.1016/j.etp.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Chi EY, Prueitt JL, Lagunoff D. Abnormal lamellar bodies in type II pneumocytes and increased lung surface active material in the beige mouse. J Histochem Cytochem. 1975;23:863–869. doi: 10.1177/23.11.1242724. [DOI] [PubMed] [Google Scholar]
- Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT. The beta-3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet. 1999;8:323–330. doi: 10.1093/hmg/8.2.323. [DOI] [PubMed] [Google Scholar]
- Feng L, Novak EK, Hartnell LM, Bonifacino JS, Collinson LM, Swank RT. The Hermansky-Pudlak syndrome (HPS1) and HPS2 genes independently contribute to the production and function of platelet dense granules, melanosomes, and lysosomes. Blood. 2002;99:1651–1658. doi: 10.1182/blood.V99.11.4006. [DOI] [PubMed] [Google Scholar]
- Gu L, Zhu YJ, Yang X, Guo ZJ, Xu WB, Tian XL. Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol Sin. 2007;28(3):382–391. doi: 10.1111/j.1745-7254.2007.00468.x. [DOI] [PubMed] [Google Scholar]
- Guttentag SH, Akhtar A, Tao J, Atochina E, Rusiniak ME, Swank RT, Bates SR. Defective surfactant secretion in a mouse model of Hermansky-Pudlak syndrome. Am J Respir Cell Mol Biol. 2005;33:14–21. doi: 10.1165/rcmb.2004-0293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AM, Shifren A, Mazan MR, Gruntman AM, Lascola KM, Nolen-Walston RD, Kim CF, Tsai L, Pierce RA, Mecham RP, Ingenito EP. Matrix modulation of compensatory lung regrowth and progenitor cell proliferation in mice. Am J Physiol Lung Cell Mol Physiol. 2010;298(2):L158–L168. doi: 10.1152/ajplung.90594.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami M, Whitsett JA, Martis PC, Weaver TE. Reversibility of lung inflammation caused by SP-B deficiency. Am J Physiol Lung Cell Mol Physiol. 2005;289:L962–L970. doi: 10.1152/ajplung.00214.2005. [DOI] [PubMed] [Google Scholar]
- Intone WJ, Westbroek W, Golas GA and Adams D (2009) Chediak-Higashi syndrome. Gene reviews: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=chediak-higashi
- Kim JH, Jang YS, Eom KS, Hwang YI, Kang HR, Jang SH, Kim CH, Park YB, Lee MG, Hyun IG, Jung KS, Kim DG. Transforming growth factor beta1 induces epithelial-to-mesenchymal transition of A549 cells. J Korean Med Sci. 2007;22:898–904. doi: 10.3346/jkms.2007.22.5.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, Chapman HA. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Königshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, Rose F, Fink L, Seeger W, Schaefer L, Günther A, Eickelberg O. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119:772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo JS. Angiotensin converting enzyme activity in mice after subacute bleomycin administration. Toxicol Appl Pharmacol. 1981;59:395–404. doi: 10.1016/0041-008X(81)90211-8. [DOI] [PubMed] [Google Scholar]
- Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky-Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–628. doi: 10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.ASN.0000106015.29070.E7. [DOI] [PubMed] [Google Scholar]
- Lyerla TA, Rusiniak ME, Borchers M, Jahreis G, Tan J, Ohtake P, Novak EK, Swank RT. Aberrant lung structure, composition, and function in a murine model of Hermansky-Pudlak syndrome. Am J Physiol Lung Cell Mol Physiol. 2003;285:L643–L653. doi: 10.1152/ajplung.00024.2003. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Nakamura N, Sano J, Inayama Y, Kawano N, Yamanaka S, Miyagi Y, Nagashima Y, Ohbayashi C, Mizushima M, Manabe T, Kuroda M, Yokoi T, Matsubara O. Interstitial pneumonia in Hermansky-Pudlak syndrome: significance of florid foamy swelling/degeneration (giant lamellar body degeneration) of type-2 pneumocytes. Virchows Arch. 2000;437:304–313. doi: 10.1007/s004280000241. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Nishiike-Wada T, Wada Y, Miura Y, Otsuki T, Iguchi H. Long-lasting production of TGF-beta1 by alveolar macrophages exposed to low doses of asbestos without apoptosis. Int J Immunopathol Pharmacol. 2007;20:661–671. doi: 10.1177/039463200702000402. [DOI] [PubMed] [Google Scholar]
- Prueitt JL, Chi EY, Lagunoff D. Pulmonary surface-active materials in the Chediak-Higashi syndrome. J Lipid Res. 1978;19:410–415. [PubMed] [Google Scholar]
- Tang X, Yamanaka S, Miyagi Y, Nagashima Y, Nakatani Y. Lung pathology of pale ear mouse (model of Hermansky-Pudlak syndrome 1) and beige mouse (model of Chediak-Higashi syndrome): Severity of giant lamellar body degeneration of type II pneumocytes correlates with interstitial inflammation. Pathol Intern. 2005;55:137–143. doi: 10.1111/j.1440-1827.2005.01811.x. [DOI] [PubMed] [Google Scholar]
- Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Resp Dis. 1967;95:752–764. doi: 10.1164/arrd.1967.95.5.752. [DOI] [PubMed] [Google Scholar]
- Wei ML. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pig Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Yamada M, Kuwano K, Maeyama T, Hamada N, Yoshimi M, Nakanishi Y, Kasper M. Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol. 2008;129:453–462. doi: 10.1007/s00418-008-0388-9. [DOI] [PubMed] [Google Scholar]