

Abstract
MicroRNAs (miRNAs) and small interfering RNAs (siRNAs), bound to Argonaute proteins (RISC), destabilize mRNAs through base-pairing with the mRNA. However, the gene expression changes after perturbations of these small RNAs are only partially explained by predicted miRNA/siRNA targeting. Targeting may be modulated by other mRNA sequence elements such as binding sites for the hundreds of RNA binding proteins (RNA-BPs) expressed in any cell, and this aspect has not been systematically explored. Across a panel of published experiments, we systematically investigated to what extent sequence motifs in 3′ untranslated regions (UTRs) correlate with expression changes following transfection of small RNAs. The most significantly overrepresented motifs in down-regulated mRNAs are two novel U-rich motifs (URMs), UUUUAAA and UUUGUUU, recently discovered as binding sites for the ELAVL4 (also known as HuD) RNA-BP. Surprisingly, the most significantly overrepresented motif in up-regulated mRNAs is the heptanucleotide AU-rich element (ARE), UAUUUAU, which is known to affect mRNA stability via at least 20 different RNA-BPs. We show that destabilization mediated by the transfected miRNA is generally attenuated by ARE motifs and augmented by URM motifs. These ARE and URM signatures were confirmed in different types of published experiments covering eight different cell lines. Finally, we show that both ARE and URM motifs couple to presumed endogenous miRNA binding sites in mRNAs bound by Argonaute proteins. This is the first systematic investigation of 3′ UTR motifs that globally couple to regulation by miRNAs and may potentially antagonize or cooperate with miRNA/siRNA regulation. Our results suggest that binding sites of miRNAs and RNA-BPs should be considered in combination when interpreting and predicting miRNA regulation in vivo.
MicroRNAs (miRNAs) are small endogenous RNA molecules that regulate gene expression posttranscriptionally (Chekulaeva and Filipowicz 2009). Hundreds of mRNAs are down-regulated following miRNA transfection (Lim et al. 2005; Wang and Wang 2006; Grimson et al. 2007) and up-regulated following miRNA inhibition (Krützfeldt et al. 2005; Frankel et al. 2007), presumably mediated by miRNA targeting. However, a substantial fraction of mRNA expression changes is not accounted for by direct miRNA targeting. A large fraction of genes that contain a predicted miRNA site do not display detectable down-regulation after transfection of the miRNA (Grimson et al. 2007; Baek et al. 2008; Hammell et al. 2008; Selbach et al. 2008), and some reports claim that only 30%–40% of mRNAs, which are up-regulated after inhibition, contain predicted miRNA sites (Krützfeldt et al. 2005; Frankel et al. 2007). Other expression changes seen after transfections may be accounted for by impaired endogenous miRNA targeting through saturation of RISC (Khan et al. 2009) and, of course, by secondary effects. We reasoned that there may be other, as yet undiscovered, sequence signals that modulate miRNA targeting. Such signals would clearly be important for understanding basic miRNA biology, design of small interfering RNAs (siRNAs), and development of small RNA therapeutics.
Of the hundreds of RNA binding proteins (RNA-BPs) in the human genome (Finn et al. 2008), some are known to regulate mRNA turnover and protein translation often mediated by sequence elements in the 3′ untranslated region (UTR). The CPE binding motif (Piqué et al. 2008), FMR binding motifs (Darnell et al. 2001; Schaeffer et al. 2001), AU-rich elements (AREs) (Barreau et al. 2005), and newly discovered ELAVL4 (also known as HuD) binding motifs (Bolognani et al. 2009) are but four of the most well studied. AREs recruit at least 20 different binding proteins (ARE-BPs) that signal rapid degradation or increased stability of mRNAs in response to stress or developmental cues (Barreau et al. 2005). Multiple copies of the core ARE motif (the heptanucleotide UAUUUAU) in a 3′ UTR increases mRNA decay potency (Lagnado et al. 1994; Zubiaga et al. 1995), and it is the most frequent conserved motif in 3′ UTRs after miRNA sites and the polyA signal (Xie et al. 2005).
The connection between ARE and miRNA-mediated regulation remains an open question. A few cases of cooperative (Jing et al. 2005; Kim et al. 2009; Sun et al. 2009) and competitive (Bhattacharyya et al. 2006) binding have been described, and ARE associated proteins such as PAIP1, FXR1, and KSRP have been associated with miRNA regulation (Caudy et al. 2002; Jin et al. 2004; Trabucchi et al. 2009). In addition, an ARE motif together with the binding of a miRNA loaded RISC in the TNFalpha 3′ UTR induces up-regulation rather than down-regulation under certain cellular conditions (Vasudevan et al. 2007), and another computational study mentioned that UAUUUA hexamers correlated with mRNA expression changes after both miR-1 and miR-124 transfection in HeLa cells (Sood et al. 2006).
We hypothesized that miRNA regulation may be modulated systematically and genome wide by the presence or absence of sequence elements such as binding sites for RNA-BPs. To investigate this with a nonbiased approach, we searched for recurring sequence signatures correlating with gene expression changes after miRNA perturbations. We analyzed a panel of 11 different miRNA transfection experiments in HeLa cells (Lim et al. 2005; Grimson et al. 2007) and used a nonparametric correlation statistic to systematically evaluate recurrent overrepresentation of all oligonucleotides (length 5–7) in 3′ UTRs of up- or down-regulated genes.
We discover both known and novel sequence motifs that recurrently correlate with changes in gene expression after miRNA and siRNA transfections. Strikingly, we identified the ARE stability motif, UAUUUAU, as the most significantly overrepresented motif in up-regulated genes (false discovery rate [FDR] < 1 × 10−4, permutation test), and two U-rich motifs (URMs), UUUUAAA and UUUGUUU, as the most significantly overrepresented motifs in down-regulated genes (FDR < 1 × 10−4). We strengthen this finding by showing that two of these top-scoring motifs can be used as features of 3′ UTRs to significantly improve the prediction of effective target sites after miRNA transfections. Furthermore, the endogenous relevance of the motifs are supported by analysis showing significant enrichment of the top scoring motifs in 3′ UTRs bound to Argonaute in HEK293 cells.
Results and Discussion
Discovery of novel coregulatory sequence motifs after miRNA transfections
We analyzed the expression profiles of 11 different miRNA transfections in HeLa cells (Lim et al. 2005; Grimson et al. 2007). For each word of length 5–7 (21,504 words in total), we used a nonparametric correlation statistic to measure overrepresentation in 3′ UTRs of up and down-regulated genes respectively (see Methods). Confirming many previous analyses (Lim et al. 2005; Sood et al. 2006; Grimson et al. 2007; Nielsen et al. 2007), the most overrepresented word in 3′ UTRs of the down-regulated mRNAs in each of the 11 experiments was the 6- or 7-mer seed site of the transfected miRNA (data not shown). We subsequently quantified the extent that a given word correlated with up- or down-regulation across all the experiments, independently of the transfected miRNA mimics. This approach revealed 91 and 993 words significantly enriched (FDR < 1 × 10−4, permutation test; see Methods) in up- and down-regulated genes, respectively (see Tables 1, 2; Supplemental Tables S1, S2). Because many of the most significant words have very similar sequences, we clustered the words and obtained nine independent sequence motifs enriched in up-regulated genes and eight in down-regulated genes (see Methods; Supplemental Tables S3, S4); two motifs were enriched in both up- and down-regulated genes. The ARE stability motif, UAUUUAU, was the most significantly overrepresented motif in up-regulated genes. URMs similar to the recently discovered HuD binding motifs (Bolognani et al. 2009), UUUUAAA and UUUGUUU, were the most significantly overrepresented motifs in down-regulated genes. These two URMs are subsequently named URM1 (UUUUAAA) and URM2 (UUUGUUU).
Table 1.
Top 10 words recurrently correlated with up-regulation following miRNA transfection

Word correlations were calculated for mRNA expression profiles of 11 different miRNA transfections in HeLa cells. Recurrent word correlation in up-regulated genes across all experiments was measured by the mean word correlation rank across all transfection experiments (see Methods). All words in the table have a highly significant recurrent word correlation (FDR < 1 × 10−4, permutation test). Word correlations in individual experiments are listed in the last column in the following order: miR-181a, miR-9, miR-128, miR-132, miR-133a, miR-142, miR-148b, miR-122, miR-7, miR-124, and miR-1.
Table 2.
Top 10 words recurrently correlated with down-regulation following miRNA transfection

Word correlations were calculated for mRNA expression profiles of 11 different miRNA transfections in HeLa cells. Recurrent word correlation in down-regulated genes across all experiments was measured by the mean word correlation rank across all transfection experiments (see Methods). All words in the table have a highly significant recurrent word correlation (FDR < 1 × 10−4, permutation test). Word correlations in individual experiments are listed in the last column in the following order: miR-181a, miR-9, miR-128, miR-132, miR-133a, miR-142, miR-148b, miR-122, miR-7, miR-124, and miR-1.
Genes up-regulated after miRNA transfections are enriched for the ARE stability motif UAUUUAU
The words most overrepresented in up-regulated genes, measured across all experiments, were substrings or shifted variants of the ARE motif UAUUUAU (Table 1). Importantly, UAUUUAU was significantly correlated with up-regulation in every one of the individual experiments. Figure 1A shows the word-expression correlation for the transfection with strongest (miR-128, FDR < 1 × 10−7) and weakest (miR-133a, P < 0.03, FDR < 0.11) UAUUUAU correlation in up-regulated genes. Our correlation statistic corrects for mononucleotide sequence composition bias in 3′ UTRs, and we found that the UAUUUAU correlations were comparable or even stronger when correcting for di- and tri-nucleotide composition bias (see Supplemental material). Our analysis also suggests that ARE motif correlations were not due to a longer overrepresented motif (see Supplemental material).
Figure 1.

The ARE stability motif is overrepresented in genes up-regulated after miRNA transfection. (A) UAUUUAU overrepresentation in up-regulated 3′ UTRs after transfection of two different miRNAs in HeLa cells. The plot shows the UAUUUAU running sum (left to right) of word overrepresentation scores in the ranked list of 3′ UTRs (black line) and running sums from 500 random permutations of the UAUUUAU word scores (red lines, see Methods). miR-128 is included as it has the strongest UAUUUAU correlation (FDR < 1 × 10−7) among the 11 different miRNA transfections in HeLa cells, while miR-133a transfection has the weakest correlation (P < 0.03, FDR < 0.11). (B) The gene set defined by UAUUUAU overrepresentation at the 0.05 level comprised 448 genes expressed in HeLa cells, and this gene set was significantly up-regulated in the 11 HeLa experiments (P < 2.5 × 10−60, Kolmogorov-Smirnov [KS] one-tailed test, standardized expression changes pooled for all 11 experiments). (C) For each of the seven time points following miR-124 transfection in HepG2 cells, the plot shows the significance level (−log10 P-value, Kolmogorov-Smirnov one-tailed test) for down-regulation of miR-124 targets relative to all expressed genes (blue line) and the UAUUUAU word correlation Z-score in up-regulated genes (red line, numbers correspond to the rank of the correlation score, N = 21 504 words).
We went on to test the significance of this finding using a different statistical test. We defined the set of genes having statistically significant overrepresentation of the ARE stability motif UAUUUAU (448 genes expressed in HeLa cells at P < 0.05; see Supplemental Table S5; Supplemental material). These 448 genes had on average about two ARE sites and an average 3′ UTR length of 1400 nucleotides (nt). We compared the change in expression of these ARE motif containing genes in all the experiments to a background set (defined by all genes expressed in HeLa cells minus the ARE genes), and found that they were significantly up-regulated (P < 2.5 × 10−60, Kolmogorov-Smirnov one-tailed test; Fig. 1B). Our analysis further suggested that the consistent up-regulation of ARE genes could not be explained directly be expression changes of known ARE-BPs after the miRNA transfections (see Supplemental material).
We also examined the ARE stability motif in five siRNA experiments targeting MAPK14, PIK3CB, PRKCE, and two against VHL (Jackson et al. 2006a,b). In 4/5 of the siRNA experiments, we observed a significant (P < 0.01, FDR < 0.19) enrichment of the ARE stability motif in the 3′ UTR of genes that were up-regulated after the siRNA transfection. Specific reports of the effects of scrambled siRNAs support our more general finding: for instance, a scrambled control siRNA caused a dose-dependent up-regulation of a gene, SREBF1, in three different cell types (Vankoningsloo et al. 2008) and our analysis shows that the 3′ UTR of this gene has significant overrepresentation of the ARE stability motif (P < 0.02; Supplemental Table S5).
We analyzed a set of experiments designed to more directly probe miRNA targeting using immunoprecipitation of epitope-tagged Argonaute2 (AGO2, also known as EIF2C2) followed by expression analysis of the mRNAs bound by AGO. This set of experiments quantitatively profiled mRNA association with AGO following transfection of miR-1 and miR-124 in HEK293T cells (Hendrickson et al. 2008). Consistent with our other findings, we found six words, including the ARE stability motif, containing the sequence AUUUA among the top-500 words most negatively correlated with AGO2 association across both transfection experiments (Supplemental Table S7), whereas no AUUUA words were found in the top-500 words positively correlating with AGO2 association (Supplemental Table S8). As in the HeLa transfections, our results suggest that transfected miRNA target genes respond oppositely, whereas endogenous miRNA targets respond correspondingly (Khan et al. 2009), to changes in expression of genes with the ARE stability motif.
Finally, we found that the expression changes of genes with the ARE stability motif is temporally coupled to the response of predicted direct targets. Figure 1D shows the highly correlated enrichment of ARE motifs in up-regulated genes and predicted miRNA targets in down-regulated genes at successive time points after transfection with miR-124 mimic into HepG2 cells (data from Wang and Wang 2006). Additionally supporting that the observed correlation is not an artifact of HeLa cells, we analyzed an additional data set measuring mRNA expression changes after miR-34a transfection in five different cancer cell lines (He et al. 2007) and found significant enrichment of ARE motifs in up-regulated genes in all five cell lines (FDR < 0.05 in each experiment).
Two novel URMs associated with miRNA regulation
Two URMs, UUUUAAA (URM1) and UUUGUUU (URM2), had strongest significant recurrent correlation with down-regulation after miRNA (Table 2; Supplemental Table S4) and siRNA transfections. In single experiments, the correlation for URM1 motifs was sometimes of a magnitude almost equivalent to the seed site of the transfected miRNA. Indeed, even the weakest single experiment correlation was still highly significant (FDR < 1 × 10−7; see Supplemental material). This was further supported by a different statistical test, which found that genes with overrepresentation (P < 0.05) of URM1 were significantly down-regulated compared with all genes without URM1 overrepresentation (P < 2.9 × 10−69, Kolmogorov-Smirnov one-tailed test, pooled data). We also found that these two motifs were somewhat correlated with up-regulation after miRNA transfections (Supplemental Tables S1, S3), and this pattern was also supported by the siRNA transfection data (P < 0.01 in each experiment). Analysis of an additional data set measuring mRNA expression changes after miR-34a transfection in five different cancer cell lines (He et al. 2007) found consistent significant enrichment of URM motifs in both down- and up-regulated genes in all five cell lines (FDR < 0.05 in each experiment).
These two motifs were recently identified as high affinity binding sites for HuD (Bolognani et al. 2009), which is almost exclusively expressed in neurons where it is involved in memory and learning processes (Akamatsu et al. 2005). Further supporting the association of these motifs with RNA protein binding, a set of intronic URMs strikingly similar to the URM2 motif was recently found to regulate alternative splicing via the TIA1/TIAL proteins (Aznarez et al. 2008). It is unlikely that the HuD protein is mediating regulation via these motifs in HeLa cells since HuD mRNA expression cannot be detected in HeLa cells. The most likely explanation is that other, more highly expressed RNA-BPs are binding the same motifs in these cells, now obviously a crucial area for future experimental investigation.
ARE stability motifs attenuate destabilization mediated by transfected miRNAs
Because genes enriched in ARE motifs responded oppositely compared to direct targets of the transfected miRNA, we analyzed the consequence of having both UAUUUAU overrepresentation (P < 0.05) and target sites of a transfected miRNA. In the 11 HeLa transfection experiments, we found that genes with miRNA target sites and ARE overrepresentation were significantly up-regulated compared with those without ARE overrepresentation (P < 1.3 × 10−8, Kolmogorov-Smirnov one tailed test, pooled data; Fig. 2A), and the same result was obtained when only considering conserved miRNA target sites (P < 4.1 × 10−5). This result suggests that ARE stability motifs may to some extent explain why a significant proportion of predicted miRNA target genes are not measurably destabilized after miRNA transfection (see Fig. 3A).
Figure 2.

ARE and URM motifs affect miRNA mediated repression. (A) Expression changes for the 11 HeLA miRNA transfection experiments were standardized and pooled. ARE genes are defined as genes with UAUUUAU overrepresentation at the P < 0.05 level. miRNA target genes are genes with a target site of the transfected miRNA in the given experiments. The plot shows that miRNA target genes with ARE stability motifs were significantly up-regulated compared with miRNA targets without ARE motifs (P < 1.3 × 10−8, Kolmogorov-Smirnov one-tailed test, pooled data). (B) The same analysis was carried out for endogenous miRNA target genes. Endogenous miRNA target genes are defined as genes with predicted conserved target sites for an endogenous miRNA and no target sites for the transfected miRNA mimic in the individual experiment. Endogenous miRNA target genes with ARE stability motifs were significantly up-regulated compared with endogenous miRNA targets without ARE motifs (P < 9.1 × 10−13, Kolmogorov-Smirnov one-tailed test). (C) Similar to A, but using genes with URM1 motif overrepresentation. miRNA target genes with URM1 overrepresentation were significantly down-regulated compared with genes with miRNA target sites alone (P < 2.5 × 10−9, Kolmogorov-Smirnov one-tailed test).
Figure 3.

Schematic of the effects of ARE and URM motifs in genes after miRNA transfections. (A) Effect on extent of down-regulation with transfected miRNA target site alone, the attenuation of down-regulation with ARE motifs in addition to the transfected target site, and the enhancement of down-regulation with URM motifs in addition to transfected target sites. Down-regulation is multiplicative, suggesting a synergistic mechanism of URM1 and transfected target sites. (B) Endogenous miRNA targets are more up-regulated after miRNA transfection if they also have ARE stability motifs. Up-regulation mediated by endogenous target sites and ARE motifs is multiplicative when they co-occur in a 3′ UTR, suggesting a synergistic mechanism.
URM motifs augment destabilization mediated by transfected miRNAs
We analyzed the expression changes of the set of genes that have both URM1 motif overrepresentation and sites for the transfected miRNA. We found that genes that had both URM1 overrepresentation and predicted miRNA target sites were significantly more down-regulated than genes with predicted miRNA sites alone (P < 2.5 × 10−9, Kolmogorov-Smirnov one-tailed test, pooled data; Fig. 2C). The same analysis considering only conserved sites showed a similar but weaker trend (P < 0.09). Although the two signals did not co-occur more than expected in 3′ UTRs of top-300 down-regulated mRNAs in each experiment (Fisher's exact test), a different more sensitive test revealed some evidence for cooperativity. We tested whether the effects of the two signals are nonadditive (or noncooperative) in genes where they do co-occur, using a previously published test for cooperativity of closely spaced miRNA target sites (Grimson et al. 2007). We simulated expression changes of genes having additive (noncooperative) occurrences of URM1 overrepresentation and transfected miRNA target sites (see Methods), and found that true genes having both signals were more down-regulated than simulated genes (P < 1.2 × 10−6, Kolmogorov-Smirnov one-tailed test, pooled data; see Supplemental material). This indicates that miRNA target sites and URM1 motifs work cooperatively inside 3′ UTRs when they do co-occur (Fig. 3A). Overall, these results suggest that URM1 motifs augment miRNA-mediated destabilization after small RNA transfection and that this motif could be used to improve computational miRNA and siRNA target prediction.
URM and ARE stability motifs are significantly enriched in 3′ UTRs bound by Argonaute in HEK293 cells
We then tested whether ARE and URM motifs are enriched in genes that are bound to Argonaute in HEK293 cells, using data produced using the PAR-CLIP method (Hafner et al. 2010). The PAR-CLIP method is a new technology that involves crosslinking and immunoprecipitation of epitope tagged Argonaute 1–4 (AGO) (also known as EIF2C1–EIF2C4) to directly sequence mRNA regions bound endogenously by AGO and plausibly under miRNA regulation. We first investigated enrichment of all 7-mer words in the corresponding full-length sequence of 3′ UTRs bound by AGO (see Methods). The words most significantly enriched in bound relative to nonbound 3′ UTRs were almost all variants of the two URM motifs (Z-score > 40, P = 0, two-tailed, Bonferroni correction; Table 3). We also found significant enrichment of the ARE motif at a level comparable to target sites of individual miRNAs expressed at highest levels in HEK293 cells (P < 4.8 × 10−114; Table 3).
Table 3.
Words enriched in 3′ UTRs bound by Argonaute in HEK293 cells

Enrichment of words (7-mers) in 3′ UTRs of the 2679 genes bound by Argonaute in HEK293 cells (PAR-CLIP data from Hafner et al. 2010). Expected word occurrences were estimated from nonbound 3′ UTRs having length and nucleotide composition matching the bound 3′ UTRs (reported values are mean ± SD; see Methods). Two-tailed Bonferroni corrected P-values are estimated from the Z-scores. Many URM motifs are found among the top enriched words and significantly enriched words outside top-20 include the ARE motif and 7-mer seed sites of the miRNAs expressed at highest levels in HEK293 cells (four last rows).
We investigated motif enrichment in the cross-linked regions of the 3′ UTRs bound by AGO. Not surprisingly the words most significantly enriched in the cross-linked AGO binding regions all corresponded to predicted seed target sites of highly expressed miRNAs (P = 0, dinucleotide background model). However, we did also observe a more modest enrichment of URM1 and ARE motifs (P < 3.1 × 10−13 and P < 2.6 × 10−15, respectively; Supplemental material). In the 2679 genes bound by AGO, there was also significant co-occurrence of genes with target sites of the top three expressed endogenous miRNAs (miR-15/16, miR-17/93, miR-19) and the URM and ARE motifs (P < 2.35 × 10−22 for each motif, Fisher's exact, two-tailed). Eighty-four percent of the AGO bound predicted endogenous miRNA target genes also have URM motifs, and 54% of the genes have both URM1 and URM2 motifs, compared with 62% and 31%, respectively, for the nonbound predicted target genes. We analyzed if there were any local spatial biases between predicted miRNA target sites and either of the URM and ARE motifs in the AGO bound 3′ UTRs, and found a significant enrichment of URM1 motifs ∼50 nt upstream of endogenous miRNA target sites (Supplemental material). In summary, we found strong enrichment of both ARE and URM motifs in the 3′ UTRs of AGO bound mRNAs and additionally some enrichment in the actual sequences cross-linked to Argonaute. Overall, these results raise the possibility of cooperative binding of RNA-BP with AGO in these cells.
Endogenous miRNA target genes are more up-regulated if they contain ARE stability motifs
Transfection of small RNAs impairs the normal regulation by endogenous miRNAs in a cell (Khan et al. 2009). Not surprisingly, we find that many 7-mer words overrepresented in 3′ UTRs of up-regulated genes after miRNA transfection correspond to seed sites of miRNAs highly expressed in HeLa cells. Words corresponding to the top-10 expressed miRNA seed sequences in HeLa cells (aggregated from expression of individual miRNAs) rank significantly higher than all other miRNA seed sites (P < 4.0 × 10−7, Wilcoxon rank-sum one-tailed test). For example, we found the seed sites of the highly expressed miR-17/93 (GCACUUU), miR-30 (UGUUUAC), miR-15/16 (UGCUGCU), and let-7 (CUACUUC) ranked third, 31st, 34th, and 88th among all 7-mer words (UAUUUAU ranking first).
Since genes enriched in ARE motifs and endogenous miRNA targets responded in the same direction, we wanted to investigate the relationship between the two signals. We found genes that contained both a predicted endogenous miRNA target site (from the top-10 expressed HeLa miRNA seed sites) and ARE motif overrepresentation, were significantly up-regulated compared with changes in expression of endogenous miRNA targets without the ARE motif overrepresentation (P < 1.3 × 10−8, Kolmogorov-Smirnov one-tailed test, pooled data; Fig. 2B). This suggests that ARE motifs have a stabilizing effect on endogenous miRNA targets after small RNA transfection.
We analyzed co-occurrence of genes with ARE motif overrepresentation and endogenous miRNA target genes (one or more conserved sites) in HeLa cells, but found no significant overlap of the two gene sets in the top 300 up-regulated genes (Fisher's exact two-tailed test). We then tested whether the effects of the two signals are synergistic (nonadditive) in genes where they do co-occur. We simulated expression changes of genes having additive (noncooperative) occurrences of ARE overrepresentation and endogenous miRNA target sites (see Methods), and found that genes having both ARE overrepresentation and endogenous miRNA target sites were more up-regulated than simulated genes (P < 1.1 × 10−3, Kolmogorov-Smirnov one-tailed test, pooled data; see Supplemental material). Intriguingly, this result indicates that endogenous miRNA target sites and ARE motifs work cooperatively inside 3′ UTRs when they do co-occur (Fig. 3B).
The functional consequences of ARE and URM motifs after miRNA transfections
Consistent with literature (Barreau et al. 2005; Stoecklin and Anderson 2006), gene set enrichment analysis (GSEA) found that genes with ARE motif overrepresentation are enriched for inflammatory pathways (see Fig. 4A; see Methods; Supplemental Table S12). However, over and above that we found that both up-regulated genes and genes with ARE motif overrepresentation were significantly enriched for genes involved in cytokine-cytokine receptor interaction (see Fig. 4B; see Methods; Supplemental Table S15). This gene set is manually curated and includes genes such as interleukin 15 receptor alpha (IL15RA), ACVR2B, ACVR1B, and lymphotoxin beta receptor (LTBR), which all have one to three occurrences of the ARE stability motif in their 3′ UTRs. Notably, over 70% of these genes contain predicted target sites for miRNAs highly expressed in HeLa cells where predicted target sites are defined by conserved miRanda sites, 11/ 14 genes (John et al. 2004), or a conserved seed site, 10/14 (Friedman et al. 2008).This result indicates that the cytokine–cytokine receptor interaction pathway is enriched for functional ARE motifs and that up-regulation of these genes may have profound consequences for the cell after small RNA transfection. Since short double-stranded RNAs can cause an immune response (Robbins et al. 2008, 2009), we cannot rule out that the perturbed ARE motif is a secondary effect of a general immune response after transfection of miRNA mimics or antagonists (as discussed in the next section). However, we found no mRNAs consistently differentially regulated across most of the experiments, like one might expect from a general immune response (Supplemental material).
Figure 4.

Functional consequences of ARE and URM motifs after miRNA transfections in HeLa cells, (A) Gene set enrichment analysis (GSEA) showing that genes with ARE motif overrepresentation are enriched for inflammatory pathways (INFLAMPATHWAY). (B) Genes up-regulated after miRNA transfections were enriched for cytokine-cytokine receptor interaction (HSA04060_CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION). (C) Genes with overrepresentation of the URM1 motif were enriched for a manually curated gene set (CHESLER_HIGHEST_FOLD_RANGE_GENES) of neurologically relevant transcripts with high fold range between mouse inbred strains. (D) Genes down-regulated after miRNA transfections were also enriched for this gene set.
We used the same approach to identify gene sets enriched for genes with overrepresentation of the URM1 motif (Supplemental Table S13). The gene set with strongest enrichment was a manually curated gene set consisting of 50 neurologically relevant transcripts with high fold range among different mouse inbred strains (Fig. 4C; Chesler et al. 2005). This result is in accordance with the fact that HuD is an important in neuronal regulator (Akamatsu et al. 2005). This gene set was also significantly enriched for genes down-regulated after miRNA transfections in HeLa cells (Supplemental Table S15) and include genes such as lin-7 homolog C (LIN7C), protein phosphatase 1 catalytic subunit (PP1CB), and cysteine and glycine-rich proteins (CSRP1/CSRP2) (Fig. 4D).
Inhibition of endogenous miRNAs perturbs genes with ARE and URM motifs
We also tested whether genes with ARE or URM motifs change in gene expression after miRNA inhibition: inhibition of miR-16 and miR-106b in HeLa cells (Linsley et al. 2007) and inhibition of miR-21 in MCF7 cells (Frankel et al. 2007). Our results show a strong enrichment of the ARE stability motif (UAUUUAU rank fifth; Supplemental Table S9) in genes that are up-regulated following inhibition of an endogenous miRNA in three experiments. Variants of both URM motifs were also found among the top motifs correlated with up-regulation after miRNA inhibition (Supplemental Table S9), and no enrichment was found in down-regulated genes. After inhibition of miR-21, URM1 had the second strongest enrichment in up-regulated genes (FDR < 1 × 10−7) after the 7-mer miR-21 seed site.
Inhibition of miR-34a in senescent TIG3 fibroblast cells (Christoffersen et al. 2009) had a notably different correlation of the ARE stability motif: It is the motif most correlated with down-regulation (FDR < 1 × 10−7; Supplemental Table S11). Recent work by Vasudevan et al. (2007) has shown that miRNA transfections in growth-arrested cells can cause ARE-mediated up-regulation of the direct transfected miRNA targets. We speculate that AREs have a general stabilizing role in this cellular state that is perturbed after miRNA inhibition. Neither of the HuD motifs was among the words most correlated with up-regulation after inhibition of miR-34a, potentially due to an unusual modest inhibitory effect of miR-34a in this experiment (7-mer seed site has rank 153 in up-regulated mRNAs).
Overall, we found based on a limited number of miRNA inhibition experiments that genes up-regulated after both miRNA transfections and inhibitions (three out of four experiments) have strong enrichment of ARE stability motifs, and that this association may be reversed when cells are growth-arrested. Conversely, URM motifs were consistently enriched together with the directly perturbed miRNA targets: enriched in down-regulated genes after transfection and up-regulated genes after inhibition.
Summary
There are many hundreds, if not thousands, of RNA-BPs for which the precise regulatory functions are unknown, and it is plausible that many of these bind mRNAs via sequence motifs to cooperate with or antagonize miRNA/siRNA regulation. We tested the possibility that there are sequence motifs that are universally associated with small RNA regulation by systematically searching for significant recurrent sequence signatures across several small RNA perturbation experiments. As expected, our unbiased search revealed significant motifs corresponding to seed sites of the small transfected miRNA in each individual experiment, and in addition, our results confirm the finding in a recent study showing impaired endogenous miRNA targeting allegedly mediated through saturation of RISC (Khan et al. 2009). However, more surprisingly, we find other sequence motifs in 3′ UTRs that recurrently correlate with expression changes induced by different miRNA transfections. First, we find that the well-studied ARE motif is highly overrepresented in up-regulated genes after small RNA perturbations in HeLa cells and impacts the effectiveness of RNAi in a range of other experiments covering eight different cell types: Argonaute immunoprecipitation (Hendrickson et al. 2008), selected siRNA perturbations (Jackson et al. 2006a,b), temporal studies of small RNA overexpression (Wang and Wang 2006), miR-34a transfection in five different cell lines (He et al. 2007), and miRNA inhibition (Frankel et al. 2007; Linsley et al. 2007; Christoffersen et al. 2009).
Readers may be surprised that AREs are most strongly associated with up-regulated genes after miRNA overexpression. One possible model for this observation could be simply that AREs are associated with endogenous miRNA sites, and we know that genes controlled by endogenous miRNAs are derepressed after small RNA overexpression potentially through competition for RISC (Khan et al. 2009). Supporting this model, we find that AREs are significantly enriched in 3′ UTRs bound by AGO in HEK293 cells and show significant synergism with endogenous miRNA sites in up-regulation of mRNAs after transfections (Fig. 3B). Furthermore, the absence of ARE motif enrichment in down-regulated mRNAs after miRNA transfections suggests that bona fide endogenous miRNA targets may have features that are different from targets of a transfected miRNA mimic. This contrast has implications for experimental approaches to target prediction; it suggests that miRNA targeting rules obtained from overexpression may be biased and that targets learned from miRNA inhibition or Argonaute immunoprecipitation may have more predictive value. Overall, our result supports that ARE and miRNA mediated regulation are interlinked to regulate the mRNA degradation rate, and is consistent with recent work that found significant overrepresentation of ARE genes in up-regulated mRNAs following Ago1 knockout in Drosophila S2 cells (Hong et al. 2009).
The second major result we present is the discovery of two URMs, recently discovered as binding sites for the neuronal expressed RNA-BP HuD, which are coupled to miRNA regulation. These motifs are highly enriched in 3′ UTRs of both down- and up-regulated mRNAs after miRNA and siRNA transfections. Furthermore, the two motifs are the top significantly enriched words, above individual endogenous miRNA seed sites, in 3′ UTRs bound by AGO in HEK293 cells. These results suggest that both target sites of transfected miRNA mimics and endogenous miRNAs are more efficient when they occur in 3′ UTRs with signatures of URM motifs, potentially a very important practical finding for quantitative mi/siRNA target prediction.
In summary, this study provides a resource for the growing field of mRNA regulation by discovering a set of highly significant sequence signatures recurrently associated with expression changes mediated by experimental small RNA perturbations. More importantly, the study shows several lines of independent evidence that there may be binding signatures of RNA-BPs that work both synergistically and antagonistically to externally driven RNAi. In addition, the direct measurements of AGO bound mRNAs suggest that this is true also for physiological targeting by cellular miRNAs. Our results should motivate focused experiments and have profound consequences for miRNA target prediction, siRNA design, and development of therapeutic small RNAs.
Methods
Experimental and sequence data
Microarray expression data sets were obtained from the NCBI Gene Expression Omnibus: 11 different miRNA transfections in HeLa cells measured 24 h after transfection (accession nos. GSE2075 and GSE8501) (Lim et al. 2005; Grimson et al. 2007), miR-124 transfection time-series in HepG2 cells (accession no. GSE6207) (Wang and Wang 2006), and miR-16/106b inhibition in HeLa cells (accession no. GSE6838) (Linsley et al. 2007). Data from miR-21 inhibition in MCF7 cells were obtained from the corresponding investigators (Frankel et al. 2007). Data for the Argonaute 2 association analysis were obtained from the supplementary material of the original article (Hendrickson et al. 2008). Data for the analysis of crosslinked Argonaute bound (AGO1–4) regions in HEK293 cells were obtained from the supplementary material of the original article (Hafner et al. 2010). Microarray data from miR-34a inhibition in senescent TIG3 fibroblast cells (Christoffersen et al. 2009) were obtained from ArrayExpress (accession no. E-MEXP-2241). We used the log-transformed mRNA expression ratios and P-values calculated by the groups behind the original data sets. Only transcripts expressed above the median expression level in the given experiment were included in the analysis. When experiments were pooled, we transformed the expression changes to Z-scores of mean 0 and standard deviation 1.
3′ UTR sequences were obtained from Ensembl (version 49). When genes were annotated with multiple transcription units and UTR sequences, we used the longest UTR sequence given for each gene. Mature miRNA sequences were obtained from miRBase (version 11.0) (Griffiths-Jones et al. 2007). miRNA expression information was obtained from the mammalian miRNA expression atlas (Landgraf et al. 2007). The following cell line identifiers were used for HeLa, HepG2, and HEK293 cells, respectively: hsa_Cervix-HeLa-adh, hsa_Hepatoma-HepG2, and hsa_Kidney-embryo-HEK293.
Prediction of miRNA target sites
miRNA target sites were defined as 7-mers complementary to bases 2–8 of the miRNA or complementary to bases 2–7 of the miRNA followed by an adenosine. Conserved miRNA target sites were obtained from TargetScan (http://www.targetscan.org, release 5.1).
Finding overrepresented words in a ranked list of sequences
We used a nonparametric statistical framework for scoring and ranking words based on their overrepresentation in a ranked list of sequences. While the overall method is new, the different parts are inspired by previous work (Farh et al. 2005; Subramanian et al. 2005; Sood et al. 2006; Cheng and Li 2008; van Dongen et al. 2008). By ordering the 3′ UTR sequences by mRNA expression change as observed in a miRNA transfection experiment, the method statistically quantifies the extent that a word is imbalanced and more frequently overrepresented in the extremity of the list. The procedure does not assume a general form (i.e., linear) for the correlation between word occurrences and mRNA expression change. Length and AU-content of 3′ UTR sequences are properties that relate to word occurrences. Because these two properties are generally also dependent on mRNA expression changes in a miRNA transfection experiment (please refer to section on 3′ UTR length in Supplemental material and a discussion of AU-content by Elkon and Agami 2008), we quantify word occurrences in a particular sequence relative to shuffled sequences. Our correlation measure should therefore not be affected by a systematic bias in AU-content or 3′ UTR length of up or down regulated genes.
For a given sequence and word, we estimate a P-value for the null hypothesis that the number of observed word occurrences k can be explained by the length and nucleotide composition of the sequence. The null distribution of word occurrences P(k) is estimated from 5000 mononucleotide permutations of the original sequence (it is straight-forward to also correct for di- or tri-nucleotide composition) (see Supplemental material). The P-value, converted into a log-odds score, can be directly estimated by s = −log10[N(k) + 1]/[5000 + 1], where N(k) is the number of shuffles where the word occurred m times or more. We add 1 as a crude correction for sampling uncertainty and to avoid log(0).
The following part of the statistical assessment is almost identical to the method described by Cheng and Li (2008). The two key differences are that we choose not to weigh the word overrepresentation scores by log expression changes and that we ask if the word overrepresentation correlates with expression change separately in up- and down-regulated genes.
Given a ranked list of sequences, {x1…xn}, and the corresponding overrepresentation scores {sj,1…sj,n} for a word, wj, we can determine if the scores are distributed nonuniformly according to the ranking {x1…xn} using a running sum statistic represented by the sequence of mean-adjusted cumulated scores rj,m:
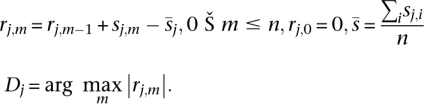 |
rj,m is a sequence starting and ending in zero, and overrepresentation of high scores near the ends of the list {sj,1…Sj,n} can be quantified by the element Dj in the list with greatest absolute value. This approach resembles the Kolmogorov-Smirnov test statistic measuring the largest horizontal distance between two cumulative distribution functions. We cannot directly compare D-values obtained for different words because the score distributions for each word can be quite different. By estimating the Dj null distribution for a given word, {PDj,1… PDj,500}, from 500 permutations of {sj,1…Sj,n}, we calculate a normalized word overrepresentation correlations score, WSj, that allows us to compare correlation scores for different words:
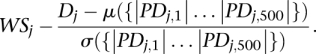 |
Ranking by absolute expression change, we can now score and rank words based on their overrepresentation in 3′ UTRs of down- or up-regulated genes by analyzing the two ranked list of genes with expression change less than zero or greater than zero, respectively. There are two reasons why we query the positive and negative expression changes separately instead of using the full list of genes: (1) for each word, we obtain a score for both up- and down-regulation (words could have bimodal overrepresentation, in both down- and up-regulated genes); and (2) using the full list of genes, some words produce high WS scores even though they are mostly enriched in nonregulated genes (taking up the middle of the list)—we minimize this problem by isolating nonregulated genes in the ends of the two separate lists. In relation to the second reason, other methods deal with this problem by using observed expression change as weights for the scores in the list (Subramanian et al. 2005; Cheng and Li 2008), but we find that our direct approach performs well.
Assessing recurrent word overrepresentation across experiments
The overrepresentation score WS and corresponding rank (adjusted for ties) of a given word in a given experiment is computed for all words (5-, 6-, and 7-mers, 21,504 words in total) in all experiments. To identify words that are recurrently correlated with up- or down-regulation across a set of experiments, we construct a word rank matrix with a column for each experiment. We use the mean word rank across the experiments as a measure for recurrent overrepresentation and evaluate the false discovery rate of a given mean word rank using 10,000 permutations of the rank matrix (using the R RankProd package) (Breitling et al. 2004; Hong et al. 2006).
Clustering of words into motifs
Motifs were computed separately from words overrepresented in up- or down-regulated genes following the 11 HeLa miRNA transfections. We conservatively estimate motifs by assuming that the most important motifs generate at least one word occurrence in the top-25 list of overrepresented words, and at least three occurrences in top-200 (all words in top-200 have a FDR < 0.001). Our clustering procedure is as follows: words in top-25 are single-linkage clustered, grouping word X with word Y, if (1) their overlap is at least 4 nt and contains at least two different nucleotides and (2) no other words in top-25 have a greater overlap with X. These initial clusters are extended by aligning the remaining words in top-200 to each cluster, requiring an overlap of at least 5 nt containing two different nucleotides. The resulting motifs are now defined as all clusters of size three or more.
Evaluating word overrepresentation in 3′ UTRs bound by Argonaute in HEK293 cells
We analyzed the data from a recent study using immunopurification of Flag-HA tagged Argonaute 1–4 (AGO) to sequence cross-linked bound RNA fragments in HEK293 cells (Hafner et al. 2010). We used the 2679 AGO bound 3′ UTRs (having at least one reported cross-linked AGO bound region) and evaluated overrepresentation of all 7-mers in these sequences relative to nonbound 3′ UTRs. Overrepresentation was measured using a Z-score statistic comparing observed occurrences in bound 3′ UTRs to the distribution of expected word occurrences estimated by randomly sampling 1000 cohorts of nonbound 3′ UTRs having length and nucleotide composition matching the bound 3′ UTRs.
Functional analysis of motifs using GSEA
In one analysis, the 4621 genes with occurrences of UAUUUAU in the 3′ UTR were sorted by their UAUUUAU overrepresentation P-value. This sorted gene list was evaluated for gene set enrichment using GSEA (parameters: 1000 permutations, MSigDB gene sets c2 [curated gene sets] and c5 [GO gene sets], classical nonweighted scoring scheme) (Subramanian et al. 2005). The same analysis was carried out for the URM1 motif. In another analysis, genes were sorted by their median expression change across all eleven HeLa miRNA transfections, and GSEA was used to identify gene sets correlating with expression change (parameters: 1000 permutations, MSigDB gene sets c2 [curated gene sets] and c5 [GO gene sets], weighted scoring scheme).
Assessing cooperativity of motifs and miRNA target sites
We simulated expression changes of genes having noncooperative occurrences of ARE-motif overrepresentation (P < 0.05) and endogenous miRNA target sites (a similar approach was used to simulate expression changes of noncooperative occurrences of URM1 and transfected miRNA target sites). The test closely follows a previously published test for cooperativity of closely spaced miRNA target sites (Grimson et al. 2007). We defined three disjoint gene sets: Set1, genes with ARE-motif overrepresentation but without endogenous miRNA target sites (one or more conserved site); Set2, genes with endogenous miRNA target sites but without ARE-motif overrepresentation; and Set3, genes with endogenous miRNA target sites and with ARE-motif overrepresentation. We sampled expression changes from Set1 and Set2 and simulated noncooperative co-occurrences by summing their log expression changes, and this procedure was repeated 100 times for each gene in Set3. To accommodate for the greater variance of the simulated compounded distribution (due to microarray measurement noise), the expression change of each gene in Set3 was summed with the expression change from a randomly selected gene.
Acknowledgments
We thank Erik Larsson, Christina Leslie, Chris Sander, Mads Hedegaard, and Jeppe Vinther for helpful discussions. A.J., J.W., and A.K. were supported by a grant from the Novo Nordisk Foundation.
Footnotes
[Supplemental material is available online at http://www.genome.org and at http://people.binf.ku.dk/andersbj/utrwords/.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.103259.109.
References
- Akamatsu W, Fujihara H, Mitsuhashi T, Yano M, Shibata S, Hayakawa Y, Okano HJ, Sakakibara S, Takano H, Takano T, et al. 2005. The RNA-binding protein HuD regulates neuronal cell identity and maturation. Proc Natl Acad Sci 102: 4625–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznarez I, Barash Y, Shai O, He D, Zielenski J, Tsui LC, Parkinson J, Frey BJ, Rommens JM, Blencowe BJ 2008. A systematic analysis of intronic sequences downstream of 5′ splice sites reveals a widespread role for U-rich motifs and TIA1/TIAL1 proteins in alternative splicing regulation. Genome Res 18: 1247–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB 2005. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res 33: 7138–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124 [DOI] [PubMed] [Google Scholar]
- Bolognani F, Contente-Cuomo T, Perrone-Bizzozero NI 2009. Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res 38: 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling R, Armengaud P, Amtmann A, Herzyk P 2004. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett 573: 83–92 [DOI] [PubMed] [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM 2002. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev 16: 2491–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekulaeva M, Filipowicz W 2009. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol 21: 452–460 [DOI] [PubMed] [Google Scholar]
- Cheng C, Li LM 2008. Inferring microRNA activities by combining gene expression with microRNA target prediction. PLoS One 3: e1989 doi: 10.1371/journal.pone.0001989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Lu L, Shou S, Qu Y, Gu J, Wang J, Hsu HC, Mountz JD, Baldwin NE, Langston MA, et al. 2005. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet 37: 233–242 [DOI] [PubMed] [Google Scholar]
- Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH 2009. p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ 17: 236–245 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB 2001. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107: 489–499 [DOI] [PubMed] [Google Scholar]
- Elkon R, Agami R 2008. Removal of AU bias from microarray mRNA expression data enhances computational identification of active microRNAs. PLoS Comput Biol 4: e1000189 doi: 10.1371/journal.pcbi.1000189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP 2005. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310: 1817–1821 [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. 2008. The Pfam protein families database. Nucleic Acids Res 36: D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH 2007. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem 283: 1026–1033 [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel D 2008. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, Dongen SV, Enright AJ 2007. miRBase: Tools for microRNA genomics. Nucleic Acids Res 36: D154–D158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP 2007. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell 27: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammell M, Long D, Zhang L, Lee A, Carmack CS, Han M, Ding Y, Ambros V 2008. mirWIP: MicroRNA target prediction based on microRNA-containing ribonucleoprotein-enriched transcripts. Nat Meth 5: 813–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. 2007. A microRNA component of the p53 tumour suppressor network. Nature 447: 1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, Herschlag D, Ferrell JE, Brown PO 2008. Systematic identification of mRNAs recruited to Argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS One 3: e2126 doi: 10.1371/journal.pone.0002126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J 2006. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 22: 2825–2827 [DOI] [PubMed] [Google Scholar]
- Hong X, Hammell M, Ambros V, Cohen SM 2009. Immunopurification of Ago1 miRNPs selects for a distinct class of microRNA targets. Proc Natl Acad Sci 106: 15085–15090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Leake D, Reynolds A, Schelter J, Guo J, Johnson JM, Lim L, Karpilow J, Nichols K, et al. 2006a. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12: 1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS 2006b. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 12: 1179–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K, Warren ST 2004. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci 7: 113–117 [DOI] [PubMed] [Google Scholar]
- Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J 2005. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120: 623–634 [DOI] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS 2004. Human microRNA targets. PLoS Biol 2: e363 doi: 10.1371/journal.pbio.0020363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Betel D, Miller ML, Sander C, Leslie CS, Marks DS 2009. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotech 27: 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M 2009. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 23: 1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M 2005. Silencing of microRNAs in vivo with “antagomirs.” Nature 438: 685–689 [DOI] [PubMed] [Google Scholar]
- Lagnado CA, Brown CY, Goodall GJ 1994. AUUUA is not sufficient to promote poly(A) shortening and degradation of an mRNA: The functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A). Mol Cell Biol 14: 7984–7995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433: 769–773 [DOI] [PubMed] [Google Scholar]
- Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, et al. 2007. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol 27: 2240–2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CB, Shomron N, Sandberg R, Hornstein E, Kitzman J, Burge CB 2007. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA 13: 1894–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqué M, López JM, Foissac S, Guigó R, Méndez R 2008. A combinatorial code for CPE-mediated translational control. Cell 132: 434–448 [DOI] [PubMed] [Google Scholar]
- Robbins M, Judge A, Ambegia E, Choi C, Yaworski E, Palmer L, McClintock K, MacLachlan I 2008. Misinterpreting the therapeutic effects of small interfering RNA caused by immune stimulation. Hum Gene Ther 19: 991–999 [DOI] [PubMed] [Google Scholar]
- Robbins M, Judge A, MacLachlan I 2009. siRNA and innate immunity. Oligonucleotides 19: 89–102 [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H 2001. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J 20: 4803–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63 [DOI] [PubMed] [Google Scholar]
- Sood P, Krek A, Zavolan M, Macino G, Rajewsky N 2006. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci 103: 2746–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Anderson P 2006. Posttranscriptional mechanisms regulating the inflammatory response. Adv Immunol 89: 1–37 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, Li H, Rossi JJ 2009. Sequence context outside the target region influences the effectiveness of miR-223 target sites in the RhoB 3′ UTR. Nucleic Acids Res 38: 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG 2009. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459: 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S, Abreu-Goodger C, Enright AJ 2008. Detecting microRNA binding and siRNA off-target effects from expression data. Nat Methods 5: 1023–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankoningsloo S, de Longueville F, Evrard S, Rahier P, Houbion A, Fattaccioli A, Gastellier M, Remacle J, Raes M, Renard P, et al. 2008. Gene expression silencing with “specific” small interfering RNA goes beyond specificity: A study of key parameters to take into account in the onset of small interfering RNA off-target effects. FEBS J 275: 2738–2753 [DOI] [PubMed] [Google Scholar]
- Vasudevan S, Tong Y, Steitz JA 2007. Switching from repression to activation: microRNAs can up-regulate translation. Science 318: 1931–1934 [DOI] [PubMed] [Google Scholar]
- Wang X, Wang X 2006. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res 34: 1646–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M 2005. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubiaga AM, Belasco JG, Greenberg ME 1995. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol 15: 2219–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]