

Abstract
Liver cytochrome P450s (P450s) play critical roles in drug metabolism, toxicology, and metabolic processes. Despite rapid progress in the understanding of these enzymes, a systematic investigation of the full spectrum of functionality of individual P450s, the interrelationship or networks connecting them, and the genetic control of each gene/enzyme is lacking. To this end, we genotyped, expression-profiled, and measured P450 activities of 466 human liver samples and applied a systems biology approach via the integration of genetics, gene expression, and enzyme activity measurements. We found that most P450s were positively correlated among themselves and were highly correlated with known regulators as well as thousands of other genes enriched for pathways relevant to the metabolism of drugs, fatty acids, amino acids, and steroids. Genome-wide association analyses between genetic polymorphisms and P450 expression or enzyme activities revealed sets of SNPs associated with P450 traits, and suggested the existence of both cis-regulation of P450 expression (especially for CYP2D6) and more complex trans-regulation of P450 activity. Several novel SNPs associated with CYP2D6 expression and enzyme activity were validated in an independent human cohort. By constructing a weighted coexpression network and a Bayesian regulatory network, we defined the human liver transcriptional network structure, uncovered subnetworks representative of the P450 regulatory system, and identified novel candidate regulatory genes, namely, EHHADH, SLC10A1, and AKR1D1. The P450 subnetworks were then validated using gene signatures responsive to ligands of known P450 regulators in mouse and rat. This systematic survey provides a comprehensive view of the functionality, genetic control, and interactions of P450s.
Cytochrome P450s (P450s) form a superfamily of monooxygenases critical for anabolic and catabolic metabolism in all organisms characterized so far (Nelson et al. 1996; Aguiar et al. 2005; Plant 2007). Specifically, P450 enzymes are involved in the metabolism of various endogenous and exogenous chemicals, including steroids, bile acids, fatty acids, eicosanoids, xenobiotics, environmental pollutants, and carcinogens (Ortiz De Montellano 2005). Mutations in P450 genes or deficiency in the enzymes result in a wide spectrum of human diseases such as glaucoma (CYP1B1) (Chavarria-Soley et al. 2008; Vasiliou and Gonzalez 2008), elevated cholesterol (CYP7A1) (Pullinger et al. 2002), congenital adrenal hyperplasia (CYP11B1) (Krone et al. 2006; Peters et al. 2007), and congenital hypoaldosteronism (CYP11B2) (Kuribayashi et al. 2003). In addition, modulation of P450s by environmental factors, for example, cigarette smoking and dietary flavonoids, can modify cancer susceptibility because some P450s are involved in carcinogen bioactivation by metabolism (Adonis et al. 2005; Moon et al. 2006; Huber et al. 2008). The diverse endogenous function and the essential role of P450s in drug metabolism and toxicology further underline the importance of these enzymes in human health (Ortiz De Montellano 2005).
In humans, there exist 18 P450 families, 44 subfamilies, and 57 putative functional enzymes (Nelson et al. 1996). Functionality, substrate specificity, and tissue specificity vary among P450 enzymes. For example, members of families CYP1, CYP2, and CYP3 —especially CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 in liver—metabolize about 95% of drugs in clinical use (Hodgson 2001; Williams et al. 2003); the CYP4 family members in liver are involved in fatty acid and eicosanoid metabolism (Masters and Marohnic 2006); members of the CYP11, CYP17, CYP19, and CYP21 families are steroidogenic enzymes in adrenal cortex, ovaries, and testes (Lewis and Lee-Robichaud 1998).
Because of the importance of P450s in human physiology and drug metabolism, the genetic variations and regulatory mechanisms of these enzymes have been extensively explored. So far, genetic polymorphisms have been identified in most P450 genes, and many of these variations contribute to interindividual differences in gene expression and/or enzyme activity and, consequently, underlie disease susceptibility or drug pharmacokinetics (Daly 2004; http://www.cypalleles.ki.se/index.htm). In fact, genetic polymorphisms, primarily single nucleotide polymorphisms (SNPs), of CYP1, CYP2, and CYP3 family members, represent the top candidate genes in pharmacogenetics, a field that investigates genetic factors that contribute to drug efficacy or side effects. For instance, polymorphisms in CYP2D6 and CYP2C19 have been linked to the response or adverse effects of various drugs used in the treatment of cancer, psychiatric disorders, and cardiovascular diseases (Anderson et al. 2003; van Schaik 2005; Vandel et al. 2007; Mega et al. 2009; Simon et al. 2009).
In addition to genetic polymorphisms within P450 genes, regulatory mechanisms at transcriptional, translational, and post-translational levels also contribute to the variability in P450 activities across individuals (Aguiar et al. 2005; Lim and Huang 2008). Nuclear receptors such as the constitutive androstane receptor (NR1I3, commonly known as CAR), pregnane X receptor (NR1I2, commonly known as PXR), peroxisome proliferator activated receptor alpha (PPARA), retinoid X receptor alpha (RXRA), liver X receptor alpha (NR1H3, commonly known as LXR), farnesol X receptor (NR1H4, commonly known as FXR), aryl hydrocarbon receptor (AHR), and glucocorticoid receptor (NR3C1) function as key regulators of P450 expression upon binding of foreign chemical inducers and endogenous hormones, growth factors, and cytokines (Waxman 1999; Cai et al. 2002; Gerbal-Chaloin et al. 2002; Plant 2007; Audet-Walsh and Anderson 2008; Gonzalez 2008; Lim and Huang 2008).
Despite the tremendous progress in the studies of P450s in the past few decades, gaps remain in the understanding of the full spectrum of functionality of individual P450 genes/enzymes, the networks connecting them, and the genetic control of each gene/enzyme. In this study, we focused on the liver since it represents the primary location of activity for the majority of P450s, and attempted to address these gaps via integration of genetics, gene expression, and enzyme activity measurements in a human cohort. In addition, we employed a weighted gene coexpression network approach that has been used extensively in yeast, mouse, and human to uncover biologically meaningful gene modules and explore novel P450 functions/pathways and coregulation structure among P450s (Zhang and Horvath 2005; Gargalovic et al. 2006; Ghazalpour et al. 2006; Horvath et al. 2006; Lum et al. 2006; Chen et al. 2008; Emilsson et al. 2008; Schadt et al. 2008; Zhu et al. 2008a,b). Furthermore, networks constructed based on Bayesian inference were used as an alternative approach to delineate potential regulatory mechanisms. In doing so, we aimed to gain a more comprehensive understanding of the functionality, genetic control, and interactions of P450s.
Results
The human liver cohort and overall data analysis scheme
The liver tissues of 466 Caucasian samples from a previously described human liver cohort (HLC) were used in the current study (Schadt et al. 2008). DNA, RNA, and microsomes were isolated from liver tissue samples for genotyping, gene expression profiling, and enzyme activity measurements, respectively. There were 213 females and 253 males, and the age ranged from 0–93, with an average of 50 and a standard deviation of 18.
As shown in Figure 1, we used the multilevel information collected from HLC and conducted (1) activity–activity correlation analysis between enzymatic activity measurements and (2) gene–gene correlation analysis between the expression levels to address the interrelationship between various P450s; (3) gene–activity correlation analysis between the genome-wide transcripts and activity measures to investigate the potential regulatory and functional relationships between genes and P450 enzymes; (4) gene coexpression network analysis to uncover gene modules relevant to P450 expression and activity as a means to explore novel P450 functions/pathways and coregulation structure among P450s; (5) expression SNP (eSNP) analysis to identify genetic variants that contribute to the interindividual variation in P450 expression; (6) enzyme activity SNP (aSNP) analysis to identify polymorphisms that may underlie the interindividual variation in P450 enzyme activity; (7) usage of eSNPs and aSNPs to infer causal relationships between the coexpression modules and P450s to identify potential novel regulators; and (8) Bayesian network (BN) analysis to further identify potential key regulators of the expression of P450s.
Figure 1.

Data sets collected and analysis scheme of the current study. The three data sets (P450 activity traits, gene expression, SNP genotyping) are shown in rectangles. The data analyses conducted are labeled above, below, or between the data sets. These include (1) activity–activity correlation among enzyme activity measures, (2) gene–gene correlation among gene expression levels, (3) activity–gene correlation between genes and activity levels, (4) coexpression network construction based on expression levels, (5) activity–module correlation between activity measurements and coexpression modules, (6) SNP–gene association (expression SNP or eSNP) between genotype and gene expression, (7) SNP–activity association (activity SNP or aSNP) between genotype and enzyme activity, and (8) Bayesian network construction based on both expression and genotype data.
P450 enzyme activity measurements in HLC
The activities of nine P450 enzymes (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) in isolated microsomes from 398 HLC liver samples were measured using probe substrate metabolism assays expressed as nmol/min/mg protein. Each enzyme was measured with a single substrate, with the exception of CYP3A4 activity, which was measured using two substrates, midazolam and testosterone, with the activity measures designated as CYP3A4M and CYP3A4T, respectively. The general statistics of the enzyme activity values are listed in Table S1. As each trait appeared to strongly deviate from normal distribution based on the Shapiro-Wilk test of normality (Supplemental Table S1) and QQ-plots (Supplemental Fig. S1A) with peak shifted toward lower activity (histograms in Supplemental Fig. S1B), we chose to use nonparametric methods to analyze these traits.
We assessed the effect of gender and age on P450 activities using the Wilcoxon rank sum test and Spearman correlation, respectively (Supplemental Table S1). CYP3A4T activity was significantly influenced by gender at P = 3.85 × 10−3, and even after Bonferroni correction for 10 assays, the association remained significant. Specifically, females showed higher CYP3A4T activity than did males. Age had a significant impact on the activities of CYP2C9 in both genders (P = 2.64 × 10−5 for all samples, 1.74 × 10−3 for males, and 4.24 × 10−3 for females), CYP1A2 in females only (P = 6.52 × 10−3), CYP2C8 in all samples (P = 9.12 × 10−3) and females (6.67 × 10−2), CYP2A6 in all samples (P = 1.02 × 10−2) and females (6.51 × 10−3), CYP3A4M and CYP3A4T in all samples (P = 2.94 × 10−2 and 2.30 × 10−2, respectively) and females (P = 4.18 × 10−3 and 1.05 × 10−2), and CYP2B6 in males (4.64 × 10−2). As shown in Supplemental Figure S2, the correlations observed are all in a positive direction; namely, P450 activities increase as age advances. After Bonferroni correction for 30 tests (10 assays × three gender categories), only the effect of age on CYP2C9 remained significant. We note that the positive correlations were mainly driven by low activity levels in very young (<20 yr) subjects. This is consistent with the observation that most P450s turn on after birth and mature over the first years of life (Hakkola et al. 1998; Koukouritaki et al. 2004). In order to address the relationship between age and P450 activity in adults only (more than 20 yr), we performed additional analyses and found that most of the correlations observed in the all-subject analysis disappeared, suggesting weaker age effect in adults. However, in the case of CYP2C9, the positive correlation remained in all adults (P = 3.50 × 10−4), adult males (P = 4.45 × 10−3), and adult females (1.70 × 10−2).
Interestingly, when both age and gender were considered simultaneously (Supplemental Fig. S3), gender demonstrated different effects at different age bands. For example, at younger ages (e.g., <10 yr), females appeared to have lower activity than males in most cases, whereas at older ages females tended to have higher activities than males. Similar trends were found for P450 expression levels (data not shown). Although the effect of age and gender was not very strong, our data suggest influence from both factors on P450 activities. Hence, we adjusted for age and gender for each P450 activity and expression trait in all subsequent analyses.
Relationship between P450s
Pair-wise Spearman correlations were employed to determine the relationships between the various enzyme activity measurements. The significance P-values and correlation values are listed in Supplemental Table S2. Without multiple-testing correction, the nine P450 enzymes analyzed using 10 assays were highly positively correlated with one another. After Bonferroni correction, most of the correlations still remained significant at a new significance threshold of P < 9.09 × 10−4, except for those between CYP2D6 and CYP2C19 and between CYP2D6 and CYP2A6.
As shown in Figure 2A, a hierarchical clustering of the activity traits provided a clearer view of the relationship among them: The two assays for CYP3A4 (3A4T and 3A4M) showed the highest similarity as expected; adjacent to CYP3A4 is the subcluster containing CYP1A2, CYP2C8, and CYP2C9; CYP2C19 and a clade comprised of CYP2E1, CYP2B6, and CYP2A6 are at the next level. Finally, CYP2D6 shows a more distant relationship with the other eight P450s.
Figure 2.

Dendrograms of the hierarchical clustering between P450s. (A) Dendrogram of the P450 enzyme activity traits. (B) Dendrogram of the P450 gene expression traits. The coding genes for the P450 enzymes shown in A are highlighted in red rectangles in B.
We applied similar analyses to the mRNA levels of the P450 genes and found that most of the P450 genes were highly correlated with one another, supporting the close relationship between the activities of the P450 enzymes described above. The significance P-values and correlation values are listed in Supplemental Table S3. A hierarchical clustering of the P450 gene expression values (Fig. 2B) revealed that a majority of the P450s, including the nine genes whose enzyme activities were measured as described above, were under a main clade. A comparison between panels A and B of Figure 2 indicated that there was a general agreement between the enzyme activity and the gene expression dendrograms. For instance, CYP2B6 and CYP2A6 were clustered closer to each other at both enzyme and expression levels. Similar kinship was found for CYP2C8, CYP2C9, CYP1A2, and CYP3A4. However, CYP2C19 and CYP2E1 were exceptions. They clustered more closely together but well apart from the other genes at the gene expression level, while they were more distant from each other at the activity level. The discordance was not a result of probe cross-hybridization at the mRNA level because the probes were reliable and specific.
Relationship between P450s and known regulators
In order to evaluate the role of known P450 regulators in modulating the expression and enzyme activity levels of different P450s in our cohort, we investigated the correlation of the expression of 48 key regulators reported in the literature with the P450 expression as well as the enzyme activity traits. These regulators investigated include NR1I2 (PXR), NR1I3 (CAR), RXRA, NR1H3 (LXR), NR1H4 (FXR), HNF4A, NR3C1 (GR), PGRMC1, PPARA, VDR, and many others (Supplemental Table S4; Waxman 1999; Cai et al. 2002; Gerbal-Chaloin et al. 2002; Tirona et al. 2003; Barbier et al. 2004; Fan et al. 2004; Gnerre et al. 2004, 2005; Seree et al. 2004; Debose-Boyd 2007; Hughes et al. 2007; Plant 2007; Audet-Walsh and Anderson 2008; Gonzalez 2008; Lee et al. 2008; Lim and Huang 2008; Wang et al. 2008). At an uncorrected significance threshold of P < 0.05, the expression levels of most of the known regulators are correlated with the enzyme activities of two or more P450s. In particular, expression of NR1I2, NR1I3, ARNT, CEBPG, ESR1, HNF4A, NCOR1, NCOR2, NFE2L2, NR1H3, NR5A2, THRB, and PGRMC1 was correlated with all measured P450 enzyme activity levels. Even after Bonferroni correction (P < 6.2 × 10−5), most of the correlations of HNF4A, ESR1, NR1I3, NCOR1, NCOR2, and PGRMC1 to P450s remained significant. Among all 48 known regulators tested, only the mRNA expression levels of NCOA1, ONECUT1, and PPARG were not correlated with any of the P450 enzyme activity measures. Due to the lack of reliable probes for FOXA3 and NR0B1 on the array, we were not able to determine their correlation with P450 activities.
When we looked at the relationship between the expression of the P450 genes and that of these key regulators, we also found significant correlations that were consistent with the results from the above analysis for the enzyme activities (Supplemental Table S4). Moreover, when the mRNA level of a key regulator is correlated to the activity of a P450, it is even more significantly correlated to the expression of the same P450. This result is not surprising given that many of these regulators are known to play regulatory roles at the transcriptional level (Waxman 1999; Cai et al. 2002; Gerbal-Chaloin et al. 2002; Tirona et al. 2003; Barbier et al. 2004; Fan et al. 2004; Gnerre et al. 2004, 2005; Seree et al. 2004; Debose-Boyd 2007; Hughes et al. 2007; Plant 2007; Audet-Walsh and Anderson 2008; Gonzalez 2008; Lee et al. 2008; Lim and Huang 2008; Wang et al. 2008).
P450 enzyme activity levels are highly correlated with their corresponding coding genes as well as many additional genes
In order to test whether the enzyme activity level is mainly regulated at the gene expression level and whether each of the enzymatic measures capture information from genes other than the corresponding P450 genes, we analyzed the correlation between a selected set of 5811 transcriptionally active transcripts (for details, see Methods) and the activity measurements of each P450. As summarized in Table 1 and detailed in Supplemental Table S5, thousands of genes are correlated with each of the P450 activity measurements at P < 0.01, with corresponding false discovery rates (FDRs) ranging from 0.34%–1.65% at this significance level. Notably, the activity levels of all P450 enzymes were significantly positively correlated with the expression of their corresponding coding genes (see representative plots for CYP1A2, CYP2C8, CYP2C9, CYP2D6, and CYP3A4 in Supplemental Fig. S4). Furthermore, the corresponding coding gene was among the top five correlated genes for each of the enzyme measurements of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2D6, CYP2E1, CYP3A4M, and CYP3A4T. Across various P450s, the correlated genes were highly enriched for similar functional pathways including drug and xenobiotic metabolism, amino acid and derivative metabolism, steroid metabolism, fatty acid metabolism, dehydrogenase, and P450 activities. However, the rank order of the enriched pathways for each of the P450-correlated gene sets was not the same, suggesting that each P450 enzyme has its own distinct functional footprint.
Table 1.
Summary of genes and functional pathways correlated with P450 activities

aThe genes in bold are those that code for the enzymes in the first column.
Gene coexpression network analysis
The fact that many genes were found to be correlated with each enzyme activity measurement suggests these genes are coregulated, and as such, the coexpression structure may provide insights into the regulation of P450s. We therefore reconstructed the coexpression network for a set of 5012 most varying genes identified in the HLC data using a previously described weighted coexpression network algorithm (Zhang and Horvath 2005).
A number of studies have previously demonstrated that coexpression networks are both scale-free and modular (Ghazalpour et al. 2006; Lum et al. 2006), thus highlighting functional components of the network that are often associated with specific biological processes. Therefore, to identify modules composed of highly interconnected expression traits within the coexpression network, we examined the topological overlap matrix (TOM) (Ravasz et al. 2002) associated with this network. The topological overlap between two genes not only reflects their more proximal interactions (e.g., two genes physically interacting or having correlated expression values) but also reflects the higher-order interactions that these two genes may have with other genes in the network. Figure 3 depicts a hierarchically clustered topological overlap map in which the most highly interconnected modules in the network are readily identified. Previous studies have shown that topological overlap leads to more cohesive and biologically meaningful modules (Ravasz et al. 2002; Zhang and Horvath 2005). To identify gene modules (subnetworks) formally from the topological overlap map, we employed a previously described dynamic cut-tree algorithm with near optimal performance on complicated dendrograms (for details, see Methods) (Langfelder et al. 2008).
Figure 3.

The HLC coexpression network and corresponding gene modules. (A) A topological overlap matrix of the gene coexpression network consisting of the top 12.5% most differentially expressed genes (5012 expression traits) identifies individual modules (boxed). Genes in the rows and columns are sorted by an agglomerative hierarchical clustering algorithm. The different shades of color signify the strength of the connections between the nodes (from white, signifying not significantly correlated, to red, signifying highly significantly correlated). The hierarchical clustering and the topological overlap matrix strongly indicate highly interconnected subsets of genes (modules). Modules identified are colored along both column and row and are boxed. (B) The corresponding graph of the HLC coexpression network. The colors of the nodes represent their module assignments as described in A. Pathway enrichment terms are assigned to individual modules.
From the HLC topological overlap map depicted in Figure 3, eight modules were identified and the membership of each module can be found in Supplemental Table S6. The eight modules contained 2998 genes with the remaining 2014 genes failing to fall into any module. We tested each of these modules for gene enrichment using Gene Ontology (GO) categories, KEGG, Panther, and GeneGo pathways. Table 2 lists the most significantly enriched category for each module and the complete lists of enriched categories can be found in Supplemental Table S7. Five modules were significantly enriched for at least one functional category or pathway, supporting that the HLC coexpression network is organized into functional units. For example, of the 54 P450 genes represented on our microarray, 52 are in the turquoise module of 1361 genes. The probability of this enrichment by chance is smaller than 1.97 × 10−73 by Fisher's exact test. The turquoise module is significantly (P-value < 1 × 10−16 by Fisher's exact test) enriched with genes in many known pathways involved in liver activities such as organic acid metabolism, lipid, fatty acid and steroid metabolism, carboxylic acid metabolism, generation of precursor metabolites and energy, oxidoreductase activity, nitrogen compound metabolism, amine metabolism, and amino acid and derivative metabolism.
Table 2.
Modules identified from the HLC gene coexpression network are enriched for GO categories

Gene module relevance to P450 enzyme activities and gene expression
To examine how each gene module is related to P450 enzyme activities or gene expression, we performed principal component analysis (PCA) for each module and then took as module relevance the correlation between the first principal component (module eigengene) and the P450 enzyme measures or expression levels. The module eigengenes explain 43%–61% of the variations in the expression of the corresponding module member genes. As shown in Figure 4, A (for activity) and B (for expression), and detailed in Supplemental Tables 8 and 9, among the eight modules the turquoise module is most significantly (P-value < 0.001) positively correlated with the expression of many of the P450 genes, and all P450 enzyme activity measurements except for CYP2E1. This is not surprising, as this module contains the majority of the P450 genes as well as the known P450 regulators NR1I2, NR1I3, ESR1, NR0B2 and PGRMC1. In addition, three other modules—namely, the red, pink, and brown modules—were also significantly correlated with both the enzyme activity and gene expression levels of many P450s (P-value < 0.05), although the specificity for P450s varied among these modules. The red module contains 75 genes and is positively correlated with the expression of 43 P450 genes as well as all P450 activity measures but not CYP2E1, CYP2A6, and CYP2B6. This module is not enriched for any particular functional pathways but notably contains genes involved in WNT/beta-catenin signaling such as TGFB3, SMO, FRAT1, and ACVR1B, as well as genes involved in the regulation of apoptosis including SOD2, IL4R, SKP2, IRS1, BAG3, GPER, and TLR2, confirming the literature observations of P450 regulation by beta-catenin-dependent signaling and the connection between apoptosis and P450s (Schrenk et al. 2004; Loeppen et al. 2005; Sekine et al. 2006; Braeuning 2009). On the other hand, both the pink and the brown modules are negatively correlated with P450 activities and gene expressions, with the former associating with CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP3A4M, and CYP3A4T enzyme activities and gene expression of 42 P450 genes, and the latter with 45 P450 genes and the activity measurements of all P450s except CYP2E1 and CYP2B6. The pink module has 70 gene members and is highly enriched for acute phase inflammatory genes (P = 2.20 × 10−9 after Bonferroni correction), supporting the link between inflammation and P450s (Aitken et al. 2006; Xie and Tian 2006; Zhou et al. 2006). The brown module contains 451 genes, of which 10 are involved in ribosome biogenesis and assembly.
Figure 4.

Module relevance to various P450 traits of interest. To examine how each gene module is related to P450 enzyme activity/gene expression level, we performed principal component analysis (PCA) for each module and then took as module relevance the value of the correlation between the first principal component (module eigengene) and the enzyme measure. (A) Clustering analysis on the module relevance to P450 enzyme activities; (B) Clustering analysis on the module relevance to P450 gene expression. Red color indicates positive correlation, and green represents negative correlation. The turquoise module is most significantly (P-value < 0.001) positively correlated with the expression of many of the P450 genes and all P450 enzyme activity measurements except for CYP2E1. Three other modules—namely, the red, pink, and brown modules—are also significantly correlated with both the enzyme activity and gene expression levels of many P450s (P-value < 0.05), although the specificity for P450s vary among these modules.
While it is interesting to uncover coexpression modules that are relevant to P450s, it is also important to identify the most critical genes within each module. A number of studies have shown that the gene intramodule connectivity is an effective metric for further identifying key genes in a module (Gargalovic et al. 2006; Ghazalpour et al. 2006; Horvath et al. 2006; Chen et al. 2008; Emilsson et al. 2008). Key genes with high connectivity measures are termed hub genes herein. For the HLC coexpression network, as shown in Supplemental Figure S5 as an example, the gene network connectivity measures such as within module connectivity k.in (for details, see Methods) are highly predictive of the relevance of a gene (the absolute value of correlation) to the trait CYP2C8 enzyme activity. Similar conclusions can be reached for all the other enzyme traits. The top hub genes for the P450-correlated turquoise, red, pink, and brown modules are listed in Table 2. These hubs include the antioxidative stress genes such as CAT and SOD2; the oxidoreductases HAO1 and EHHADH that are involved in carboxylic acid metabolism; acute phase inflammatory genes such as SAA1, SAA2, SAA3P, and CRP; as well as translation-related genes DKC1, EEF1E1, and EIF2S2. Given the central role of these genes in the P450-correlated modules, they may represent candidate regulatory factors for P450s.
Despite the usefulness of the coexpression network to identify modules, hub genes, and their relevance to P450s, this analysis cannot address whether the correlations observed are of causal, reactive, or independent nature. Because genetic variations can serve as causal anchors as previously described (Schadt et al. 2005), P450-correlated modules that are enriched for genes affected by genetic variations may harbor upstream regulators of P450s. To this end, we conducted the following analyses to systematically explore the genetic regulation of P450s.
Identification of SNPs associated with P450 gene expression (eSNPs)
In order to survey the contribution of genetic variations to P450 gene expression, we analyzed the association of the transcripts of the 54 P450 genes represented on the human 44k array with 782,476 unique SNPs that were represented, successfully genotyped, and passed quality control (for details, see Methods) in the HLC using both the Affymetrix 500K and Illumina 650Y panels (Schadt et al. 2008). At an FDR < 10% level, we identified 252 unique cis-eSNPs (defined by <1-Mb distance between SNP and gene location) that are associated with the expression of 19 of the 54 P450 genes represented on the array (summarized in Table 3, and fully listed in Supplemental Table S10). No trans-eSNPs were found at the same FDR cutoff, reflecting the weaker nature of trans-regulation. The P450 genes with cis-eSNPs are CYP1B1, CYP26C1, CYP2C18, CYP2C19, CYP2C9, CYP2D6, CYP2E1, CYP2F1, CYP2R1, CYP2S1, CYP2U1, CYP39A1, CYP3A5, CYP46A1, CYP4A11, CYP4F11, CYP4F12, CYP4V2, and CYP4X1. Among these, CYP3A5, CYP2D6, CYP4F12, and CYP2E1 each have more than 30 cis-eSNPs; thus, about one-third of the P450 genes appear to be under detectable cis-regulation.
Table 3.
Summary of SNPs associated with P450 expression (eSNPs) and those associated with protein/enzyme activity (aSNPs) identified for P450s

SNPs associated with P450 enzyme activities (aSNPs)
The identification of polymorphisms that associate strongly with P450 expression also leads us to investigate polymorphisms that associate strongly with P450 enzyme activities. In this article, we have named such SNPs as activity SNPs, or aSNPs. By analyzing the association between the 10 P450 activity measurements and the same set of 782,476 SNPs, as described above, we identified 54 aSNPs for nine of the enzyme activity measurements of eight P450s, namely, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4M, and CYP3A4T at FDR < 10% (summary in Table 3, and full list in Supplemental Table S11). Of the 54 aSNPs identified, 30 were associated with CYP2D6 activity, and they were all within 200-kb distance of the physical location of the CYP2D6 gene, demonstrating mainly cis genetic regulation of CYP2D6 enzyme activity. However, none of the remaining 24 aSNPs are located on the same chromosome as the genes encoding the corresponding P450 enzymes, and hence, they represent trans-regulatory elements. In fact, none of the known P450 candidate regulatory genes are within 1-Mb distance of these trans-aSNPs, and none of these aSNPs were associated with the expression of any adjacent genes; thus we are not able to apply any functional annotation to these aSNPs. The genes closest to the aSNPs are listed in Table 3, and these gene regions warrant further validation and investigation regarding their contribution to the regulation of P450 activity.
From this analysis, CYP2D6 is the only P450 whose eSNPs and aSNPs overlap at FDR < 10%. Specifically, 29 out of the 30 aSNPs for CYP2D6 activity were found to be also cis-eSNPs of the CYP2D6 gene, suggesting that this particular P450 is subject to genetic self-regulation at the transcription level, and such regulation is also reflected at the activity level. There was no overlap between activity and gene eSNPs for the rest of the P450s. The lack of correlation between aSNPs and eSNPs could be due to the stringent criteria used to detect the associations at FDR < 10%. To test this, we maintained the stringency for either the eSNP or the aSNP while relaxing the criteria for the other at P < 0.05 (irrespective of FDR) and repeated the comparison. As detailed in Supplemental Table S12, in addition to the 29 aSNPs of CYP2D6 activity mentioned above, six additional aSNPs were eSNPs at a loose cutoff of P < 0.05 while keeping the aSNP criteria fixed at FDR < 10%. On the other hand, when we fixed the eSNP criteria but relaxed the aSNP criteria, we found that more than half (137 out of 252) of the cis-eSNPs identified at FDR < 10% were also associated with P450 enzyme activity measurements at P < 0.05 (Supplemental Table S13). Among these, 52 out of the 54 cis-eSNPs of the CYP2D6 gene were associated with CYP2D6 activity, consistent with the findings on the eSNP–aSNP relationship discussed above for this P450. Interestingly, 42 out of the 56 CYP3A5 cis-eSNPs were found to be associated with CYP3A4M but not CYP3A4T activity, suggesting that CYP3A4 activity measured using midazolam reflects the activity levels of CYP3A5, which is consistent with the known role of CYP3A5 in midazolam metabolism and its limited contribution to testosterone metabolism (Williams et al. 2002). The P450 genes carrying the remaining 43 cis-eSNPs were not the corresponding coding genes for the P450 enzymes that were associated with the same SNPs, suggesting more complex interactions or crosstalk among P450s.
Validation of aSNPs and eSNPs
To test the validity of the aSNPs and eSNPs discovered above, we chose to genotype three aSNP/eSNPs of CYP2D6, namely, rs8138080, rs5751247, and rs17478227, in a previously described independent cohort that comprised 237 Caucasians (Gaedigk et al. 2008). These SNPs were found to be significantly correlated (P < 1 × 10−11) with CYP2D6 activity measured in vivo using the log10 urinary ratio of dextromethorphan (DM) to its metabolite dextrorphan (DX) (Supplemental Fig. S6). Furthermore, these SNPs were found to be in strong linkage disequilibrium (LD) with two previously known functional variants, CYP2D6*4 (1846G>A, 100C>T) and CYP2D6*10 (100C>T) (Supplemental Table S14). The 1846G>A polymorphism inactivates splicing at the acceptor site of exon 4 and activates a new cryptic site 1 nucleotide (nt) downstream of the natural splice, resulting in a frameshift and complete loss of functional activity. The nonsynonymous 100C>T results in a proline-to-serine amino acid change and a reduction of catalytic activity relative to the reference CYP2D6.1 protein. Therefore, the correlation of the three aSNP/eSNPs with CYP2D6 activity is most probably conferred by the *4 and *10 variants. However, the functional relationship between the SNPs defining the CYP2D6*4 and CYP2D6*10 alleles and variability in CYP2D6 RNA expression is less clear.
eSNP and aSNP enrichment in coexpression modules
As noted earlier, DNA variations can serve as anchors to infer causal relationship between genes or gene modules and traits. We therefore tested whether the P450-correlated modules identified from the coexpression network analysis are enriched for any genes whose expression or activity is associated with a SNP. For the 40,638 probes on the human 44k array (not limited to P450 genes), 6463 eSNPs were previously identified at FDR < 10% and 4264 of them were cis-acting (Schadt et al. 2008). Among the four gene modules that were correlated with P450 expression and activities, the turquoise and brown modules were significantly enriched for genes with eSNPs (P = 3.00 × 10−26 and 4.37 × 10−5, respectively) as well as for cis-eSNPs (P = 3.23 × 10−20 and 5.78 × 10−4, respectively). Moreover, the eSNPs and cis-eSNPs that were associated with the turquoise module genes were found to be significantly overrepresented among the aSNPs that were associated with P450 activities at P < 0.01 (enrichment P = 1.44 × 10−3 and 8.92 × 10−3 for turquoise module eSNP and cis-eSNPs, respectively). Similarly, among the P450 eSNPs at P < 0.01, we found overrepresentations of eSNPs (P = 1.10 × 10−5) and cis-eSNPs (P = 2.48 × 10−2) that were associated with the turquoise module genes, as well as enrichment of eSNPs associated with the brown module genes (P = 1.23 × 10−3). Thus, these two modules might harbor upstream regulators of P450s, as more member genes appear to be affected by DNA variations that also show evidence of association with P450 expression and activity levels. In fact, known P450 regulators such as NR0B2, PGRMC1, and NR1I3 had cis-eSNPs and were in the turquoise module. In addition, another known P450 regulator, HNF4A, was in the brown module, again supporting the importance of these two modules in the regulation of P450s.
Constructing a predictive BN from the HLC data
As an alternative approach to identifying the pathways and regulators for P450s, we used a Bayesian gene regulatory network reconstructed based on the HLC gene expression and genotyping data (Schadt et al. 2008). Unlike coexpression networks, which allow one to look at the overall gene–gene correlation structure at a high level, BNs are sparser but allow a more granular look at the relationships and directional predictions between genes (Zhu et al. 2004). Figure 5A (a high-resolution figure is available in Supplemental material) represents an overview of the P450 gene regulatory subnetwork composed of the P450 genes and the known P450 regulators, and the genes that are up to two edges away from them.
Figure 5.

P450 gene regulatory Bayesian subnetwork. (A) A P450 gene regulatory subnetwork composed of both the P450 genes and the genes that are two-edges away from any P450 gene. P450 genes and the known P450 regulators in the subnetwork are highlighted as red circles and blue squares, respectively. (B) The same subnetwork highlighted with liver genes responsive to ligands of known regulators of P450s, namely, AHR, CAR, or PXR (Slatter et al. 2006) in mouse (cyan nodes), rat (green nodes), and both species (red nodes). (C) A pruned P450 subnetwork composed of the key regulators identified in A and their downstream P450 targets. The known regulators and P450 genes are shown as blue rectangles and red ovals, respectively.
In order to test if the reconstructed network is predictive, we turned to data in the public domain. We obtained consensus compound signatures in mouse and rat livers that are responsive to 26 inducers of AHR, NR1I3 (CAR), and NR1I2 (PXR) (e.g., AHR ligand flutamide, CAR ligand androstenol, and PXR ligand hyperforin) (Slatter et al. 2006), and analyzed the overlap between these signatures and our network. Indeed, our P450 subnetwork is significantly enriched not only for mouse genes responsive to ligands of AHR, CAR, PXR, and all three receptors with enrichment P-values of 1.66 × 10−12, 1.08 × 10−14, 9.74 × 10−13, and 6.19 × 10−7, respectively, but also for rat genes responsive to ligands of AHR, CAR, PXR, and all three receptors with enrichment P-values of 9.82 × 10−15, 7.43 × 10−12, 2.45 × 10−18, and 3.64 × 10−9, respectively (Fisher's exact test; overlapping genes are shown in Supplemental Table S15 and highlighted in the subnetwork in Fig. 5B; high-resolution figure is available in Supplemental material).
As the reconstructed P450 subnetwork appeared to be predictive, we proceeded to identify causal regulators for the P450 genes. We used a previously described procedure (Schadt et al. 2008; Zhu et al. 2008b), which is briefly described here (see Methods). First we defined the signature for each gene as the set of genes in the subnetwork that could be reached by this gene following directed links throughout the entire subnetwork. The signature for each gene is composed of genes in the subnetwork that are predicted to be affected when that particular gene is perturbed in silico. We then ask if each of the 57 putative functional P450 genes as defined by Nelson et al. (1996) is significantly enriched among the predicted responsive signature by Fisher's exact test. If one is, then that particular gene perturbed in silico is inferred as a potential regulator of P450 genes. All genes in the subnetwork were tested. Based on the criteria that (1) Fisher's exact test P-value < 0.05 and (2) there are at least two P450 genes in the signature, we found 19 genes (CYP2C19, ACSM3, EHHADH, IL15RA, IPPK, FNDC3A, DMD, SEPP1, Corf114, AF147447, SLC27A5, AKR1D1, SLC10A1, SLCO2B1, CMBL, SUN1, HGD, SIRT5, and HMGCS1) to be putative regulators of the P450s. As shown in Figure 5C, three of these genes form a pathway starting with EHHADH and ending with BC019583. In this path, EHHADH and ACSM3 are upstream of the pathways and their regulation is mediated through CYP2C19, while the downstream P450 targets include CYP2C9, CYP3A7, CYP3A4, and CYP3A43. Another two putative P450 regulators, SLC10A1 and AKR1D1, form a separate branch and are upstream of eight P450 genes and seven known P450 regulators: PGRMC1, CEBPD, FOXA2, NR1I3, NR1I2, PPARG, and HNF4A. The subnetwork threaded by this path was highly enriched for the Ingenuity pathways “LPS IL-1-mediated inhibition of RXR function” and “RXR/FXR activation” (P = 4.81 × 10−6 and 2.15 × 10−4, respectively, after Bonferroni correction). As RXRA and NR1H4 (FXR) are also known regulators of P450s, the SLC10A1 → AKR1D1 path is interesting.
In addition to identifying genes that are predicted to be regulators of P450s, we also wanted to see which genes in the subnetwork are potential regulators of large numbers of downstream genes. These more global regulators represent genes that, when perturbed, could have a major impact on the network that is not limited to P450s. We identified SLC10A1, AKR1D1, GLYAT, ETNK2, LIME1, ZGPAT, BTD, ETNK2, KLKB1, FMO3, LIMK2, EHHADH, and ACSM3 to be the top global regulators, each with over 40 downstream nodes. Therefore, SLC10A1, AKR1D1, EHHADH, and ACSM3 were identified as both global regulators and P450 regulators. Furthermore, in the coexpression network discussed earlier, EHHADH, SLC10A1, and AKR1D1 were present in the same turquoise module, and their connectivity measures were ranked high (three, 13, and 14, respectively) among the 1361 genes within this module. Thus, there exists certain consistency between the putative regulators identified from the two different network approaches.
In conclusion, the reconstruction of a BN based on the HLC gene expression and genetic data enabled us to identify two primary putative regulatory pathways for P450 genes, with upstream paths SLC10A1 → AKR1D1 and EHHADH → ACSM3. Three of these genes—EHHADH, SLC10A1, and AKR1D1—were also found to be top hub genes in the P450-correlated turquoise module from the independent coexpression network. In the literature, EHHADH has been found to be responsive to ligands of AHR, NR1I3 (CAR), and NR1I2 (PXR); SLC10A1 is selectively responsive to AHR ligands; AKR1D1 is selectively responsive to PXR ligands (Slatter et al. 2006). Thus, these three upstream genes represent novel putative key regulators of P450 genes.
Discussion
In this study, we conducted a systematic investigation of liver P450 functionality, interconnectivity, and regulatory mechanisms via an integrated genetic and genomic approach. To our knowledge, this is the first comprehensive, large-scale study of P450s that involves the incorporation of genome-wide data from multiple dimensions, including genetic, gene expression, enzyme activity, and demographic factors, from a large HLC consisting of about 500 samples.
In an attempt to address the demographic confounding factors, we studied the impact of gender and age on P450 enzyme activity measurements. Previously it has been reported that CYP2E1, 3A4, 1A2, 2A6, and 2B6 are subject to gender influence in humans, but conflicting results have been obtained (Dhir et al. 2006; Anderson 2008; Scandlyn et al. 2008). Our results support that the activity of CYP3A4 (but not the others reported) is affected by gender, with females having higher activity measurement than males. However, the gender difference in CYP3A4 observed here was moderate and only reached significance threshold for the activity measurement using testosterone but not midazolam. One possible explanation for the lack of consistent observations of sexual dimorphism in human P450s compared with the extensive reports for rodents (Ahluwalia et al. 2004; Rinn and Snyder 2005; Yang et al. 2006) could be due to the genetic heterogeneity and more complex environmental influences present in human populations. Alternatively, signaling pathways that magnify sexual dimorphism are generally recognized to be overrepresented in rodents. An additional explanation is that gender effect varies by age as shown in Supplemental Figure S3. Thus, when different age groups are represented in different study populations or when all age groups are considered together, inconsistent conclusions could be reached. The role of gender in modifying P450 activity needs to be further explored when there is opportunity to obtain samples from more homogeneous populations to minimize confounding factors.
The effect of age on enzyme activity has been reported for several major P450s such as CYP2C19, CYP2D6, and CYP3A4 in humans (Ishizawa et al. 2005; Stevens 2006; Wauthier et al. 2007; Stevens et al. 2008). However, the lack of an age effect on these enzymes has also been indicated (Simon et al. 2001; Gorski et al. 2003; Parkinson et al. 2004). Here, we show evidence supporting the impact of age on CYP2C9 activity and a relatively weak gender-specific age effect for CYP1A2, CYP2A6, CYP2B6, CYP2C8, and CYP3A4. Considering a large cohort of about 500 individuals was used for this study, our results might be representative of the age influence on P450s in a general population. However, potential confounding factors such as unequal representation of ages in the cohort, gender, cause of death, and concomitant disease exist in the population under investigation.
It is interesting to observe positive correlations between age and various enzyme activity measurements in adults of age less than 20, especially in the case of CYP2C9, a main metabolizing enzyme for warfarin. Current warfarin dosing algorithms adjust for patient age, with lower doses associated with increasing age, which seemingly conflicts with the positive correlation between CYP2C9 activity and age. However, drug metabolism is a complex process that involves not only P450s but many other contributors. The current study only addresses oxidative drug biotransformation by liver microsomes in vitro, whereas warfarin dosage requirements in patients are a function of several processes in addition to oxidative biotransformation, such as absorption, distribution (changes in plasma protein binding), phase II (conjugation) reactions and excretion, as well as genetic variation in the target of warfarin action, vitamin K oxidoreductase complex 1 (VKORC1). In the elderly, this process can be further complicated by blood flow, diet, poly-pharmacy, obesity, and other chronic disorders. Furthermore, as shown in Supplemental Figure S3, the increase in CYP2C9 activity across the different age bands in adults is subtle, with the statistical significance mainly driven by the large population size involved in the current study. Therefore, the age-dependent adjustment for warfarin dose is likely due to factors other than CYP2C9 activity level and hence does not necessarily conflict with our finding.
When the relationships between P450s were studied at both the expression and activity levels, we found a general agreement between the two analyses. That is, most of the P450s are significantly correlated with one another. This is likely because P450s have arisen from a common ancestor gene through gene duplication and other relevant sequence rearrangement events and hence may be under similar regulatory control by a limited set of transcription factors (Gibbs et al. 2004). We noticed that the relationship among the P450s did not entirely concur with the sequence similarity based on phylogeny data (Nelson et al. 2004). For example, CYP2C8 and CYP2C9 clustered more closely with CYP2E1 and more remotely with CYP3A4 at the sequence level. However, at both the expression and enzyme activity levels, CYP3A4 showed greater similarity to CYP2C8 and CYP2C9 than CYP2E1 did. Thus, although P450 genes in each family or subfamily have higher sequence similarity, their expression and enzymatic activity levels are not necessarily more closely correlated.
We found that all of the P450 enzyme activity measurements analyzed were highly correlated with the expression of their corresponding coding genes, suggesting strong transcriptional regulation of P450s. We also observed significant correlations between P450s and many known key regulators, thus validating findings from the literature as well as the quality of our data. However, the presence of thousands of genes that correlate with each individual P450 was somewhat less expected. A possible explanation is that liver P450s may lie at the center of many endocrine and xenobiotic metabolic pathways that require crosstalk across numerous nuclear receptor networks. This is supported by the finding that P450-correlated gene sets are enriched for highly relevant metabolic pathways such as lipid, fatty acid, steroid, and amino acid metabolism.
The correlation analyses conducted above mainly focus on pair-wise relationships. In order to provide a comprehensive view of how genes are connected as a whole, we also analyzed the connectivity of the transcriptional networks of this cohort. This provided a basis for the identification of key functional modules within the networks that contribute to variability of traits of interest. We have previously described the characterization of transcriptional coexpression networks based on brain, adipose, and liver tissues in human and mouse (Gargalovic et al. 2006; Ghazalpour et al. 2006; Horvath et al. 2006; Lum et al. 2006; Chen et al. 2008; Emilsson et al. 2008). Building on this approach, we constructed a coexpression network based on the human liver tissue data to identify gene modules. We identified multiple modules demonstrating functional enrichment that correlated with both P450 expression and enzyme activity. The turquoise module harbored a majority of the P450 genes and showed the most significant positive correlation with P450 traits. Interestingly, three other modules that were highly correlated with P450s contained genes involved in oxidative stress and apoptosis such as superoxide dismutase 2 (SOD2), catalase (CAT), and caspase 4 (CASP4); acute phase response such as serum amyloid proteins SAA1, SAA2, and SAA3P and c-reactive protein (CRP); and translation such as dyskeratosis congenita 1 dyskerin (DKC1), eukaryotic translation initiation factor 2 subunit 2 (EIF2S2), and eukaryotic translation elongation factor 1 epsilon 1 (EEF1E1). The connections between oxidative stress genes as well as acute phase inflammatory genes with P450s have been noted before (Strolin-Benedetti et al. 1999; Saitoh et al. 2000; Barouki and Morel 2001), and the correlation of genes acting in translation with protein levels, and hence enzyme activity measurements, makes intuitive sense.
As the connectivity levels of the genes within these modules were found to be positively correlated with their module-to-trait relevance, we were also able to identify the hubs, or the key genes in each module. Among the top hub genes in the turquoise module, hydroxyacid oxidase 1 (HAO1) and enoyl coenzyme A hydratase 3-hydroxyacyl coenzyme A dehydrogenase (EHHADH) are oxidoreductases involved in fatty acid oxidation (Hardwick 2008; Tomaszewski et al. 2008), and catalase (CAT) plays a role in free radical scavenging and attenuates apoptosis. For the other modules that were correlated with P450 traits, the top hub genes were mostly acute phase response genes and those involved in translation as mentioned above. Although these top hub genes may represent novel regulators of P450s, coexpression modules cannot differentiate modules that are causally linked to P450s from those downstream of P450s. This can be partially resolved with the genetic information collected in the same cohort, as discussed below.
Genome-wide association studies between DNA variations and gene expression or enzyme activity can provide insights into the genetic regulation of P450s, and genetic information is a useful anchor to infer causality (Schadt et al. 2005). In the present setting, the term causality is used from the standpoint of statistical inference, where statistical associations between changes in DNA, changes in expression (or other molecular phenotypes), and changes in complex phenotypes like disease are examined for patterns of statistical dependency among these variables that support directionality among them, where the directionality then provides the source of causal information (highlighting putative regulatory control as opposed to physical interaction) for further experimental testing.
We identified cis-eSNPs that were associated with the expression of one-third of the P450s, suggesting that many P450s are regulated by cis-acting polymorphisms. We also identified aSNPs that were associated with the enzyme activity measurements for most of the P450s tested. These lines of evidence suggest that P450s are under strong genetic control. However, only the cis-eSNPs of the CYP2D6 gene were coincident with aSNPs of the corresponding enzyme activity, indicating that for many of the other P450s, the regulatory path from gene transcription to protein translation to activity is more complicated.
A comparison of the eSNPs and aSNPs with the putative functional polymorphisms previously reported in the literature (http://www.cypalleles.ki.se/index.htm) revealed only limited overlaps. For instance, only the known SNP rs776746 that affects CYP3A5 activity and Saquinavir pharmacokinetics (Katz et al. 2008) was among our cis-eSNPs. However, when LD was taken into consideration, 28 additional cis-eSNPs of CYP3A5 were in strong LD (R2 > 0.6) with rs776746 and thus could be considered as overlaps. There was no overlap between the aSNPs and the published functional SNPs. The limited overlap could result from the fact that the majority of the literature reports were based on single-gene analysis, whereas a genome-wide approach was used in our study, which involved aggressive multiple-testing correction. We indeed found more overlaps, including CYP2C9*2 (rs1799653, associated with lower enzyme activity and warfarin response) (Katz et al. 2008) as an aSNP for CYP2C9 at P = 9.63 × 10−4, CYP3A5*3 (rs776746) as an aSNP for CYP3A4M at P = 8.96 × 10−5, and CYP4F2*3 (rs2108622) as a cis-eSNP for CYP4F2 at P = 6.63 × 10−5 when a less stringent cutoff of P < 0.05 was considered. An additional explanation could be that many of the literature findings were derived from in vitro systems, whereas human tissues were used in our study (http://www.cypalleles.ki.se/index.htm). Moreover, the SNP coverage of the genotyping panels used in this study was less than optimal, as these platforms were not designed specifically for P450 studies but more for genome-wide coverage. Such design results in insufficient coverage of SNPs in the coding regions of P450s. For example, none of the known SNPs within the CYP2D6 gene boundary was represented in the Affymetrix and Ilumina panels, although many of these SNPs within genes were the focus of published literature studies. All these indicate that our study covers biological space different from the previous studies, and hence, any discoveries made in this study are potentially novel. The fact that we are able to recapitulate the associations between three CYP2D6 aSNPs and the enzyme activity levels in an independent cohort supports their true discovery nature, and the identification of the strong relationship between these aSNPs and known functional variants further highlights their relevance. Validation of the other newly identified aSNPs and eSNPs is necessary.
It is important to note that in the current manuscript, cis and trans were used in a more liberal sense, namely, we defined any aSNP/eSNPs that were within 1-Mb distance of P450 genes as cis-SNPs, whereas any SNPs out of this range were called trans-SNPs. These cis- and trans-calls are suggestive of cis- or trans-regulation but by no means directly demonstrate true cis- or trans-regulation.
By mapping genes with cis-eSNPs and aSNPs to the coexpression modules, we were able to differentiate the turquoise and brown modules as upstream causal modules from the red and pink modules. The top hub genes from the turquoise and brown modules were thus candidate regulatory genes for P450s. We also used a Bayesian inference-based approach and found a similar set of regulatory genes that included EHHADH, SLC10A1, and AKR1D1. The validity of the Bayesian P450 subnetwork was supported by (1) significant enrichment for the previously reported drug response genes to ligands of known P450 regulators AHR, NR1I3 (CAR), and NR1I2 (PXR) in two rodent species; and (2) the presence of the three putative regulators we identified among the drug signatures. EHHADH is enoyl coenzyme A hydratase 3-hydroxyacyl coenzyme A dehydrogenase and is known to be involved in fatty acid metabolism as well as PPAR signaling (Jia et al. 2003). SLC10A1 is solute carrier family 10 member 1 and functions as a sodium-dependent bile acid transporter. It is involved in pathways including LPS IL-1-induced inhibition of RXR, hepatic cholestasis, and FXR RXR activation, and therefore plays an important role in the coordinated regulation of bile acid homeostatis and drug metabolism (Eloranta and Kullak-Ublick 2005). AKR1D1 is aldo-keto reductase family 1 member D1 and plays a role in bile acid synthesis and steroid hormone metabolism (Lee et al. 2009). These genes are relevant to the functions and regulatory pathways of P450s, and this study implicates these genes as P450 regulators for the first time. Validation studies involving perturbation of these genes are underway.
Even though this study is the first comprehensive survey of human P450s in a large cohort, there are some limitations to the interpretation of the data. For example, medication information was not available for a majority of the individuals in the cohort and therefore not taken into consideration in the current study. Since many drugs can be inducers or inhibitors of P450s, noise could have been introduced to the analyses due to the inability to control for medication status. We also acknowledge that one of the limitations of BN approach is that feedback regulation cannot be accommodated. In addition, although we have demonstrated the utility of BNs in predicting how genetic and environmental perturbation signals propagate in biological systems in controlled study populations (Zhu et al. 2004, 2008b; Schadt et al. 2005; Chen et al. 2008; Yang et al. 2009), the accuracy to infer causal relationships between genes in a random or uncontrolled study design like the current liver cohort is lower (Wang et al. 2009). Furthermore, the directionality of the outcome of any perturbation of genes in a network such as whether the perturbation will raise or lower a phenotypic outcome depends on the genetic background and environmental conditions due to complex network feedbacks (Davey Smith and Ebrahim 2003; Chen et al. 2008). Last but not least, confounding effects from pleiotropy and LD can complicate causality inference, and the interpretation of causality results can be further complicated by factors such as morphogenic stability, developmental adaptation, and canalization (Davey Smith and Ebrahim 2003; Schadt et al. 2005; Fraser et al. 2010). Therefore, some of the conclusions should be interpreted with care, and more follow-up studies are needed to test the hypothesis put forward.
In summary, we conducted a genetic and genomic survey of the P450 system in a large HLC. We found that P450s were highly correlated among themselves, with known regulators, and with thousands of functionally relevant genes. Genetic association analysis revealed SNPs that are associated with P450 expression and/or activity measures, with evidence suggestive of both cis-regulation and more complex trans-regulation. A coexpression module that contained a majority of P450 genes and several known P450 regulators was found to be most significantly correlated with P450 expression and activity measures. The same module was also highly enriched for genes with regulatory SNPs and harbored several novel candidate P450 regulators that were also identified using a BN approach. The Bayesian subnetwork identified was highly enriched for gene signatures elicited by known ligands of P450 regulators.
This study provides broad insights into the regulation of P450 enzymes that underlie the interindividual pharmacokinetic and pharmacodynamic variability of P450s as well as the disease susceptibility, drug response, drug–drug interactions, and toxicity associated with P450s in the general population. The interrelationship between P450s, the regulatory mechanisms (e.g., novel regulators, eSNPs, aSNPs), and the networks uncovered may help define accessible peripheral biomarkers of toxicity or metabolism that are useful for pharmacokinetic and pharmacodynamic studies during drug development and for pharmacogenetic studies during post marketing to stratify populations, enhance drug efficacy, and reduce adverse effects.
Methods
Human liver cohort
As described previously (Schadt et al. 2008), a total of 780 liver samples (1–2 g) were obtained from Caucasian individuals from three independent liver collections at tissue resource centers at Vanderbilt University, the University of Pittsburgh, and Merck Research Laboratories (Slatter et al. 2006). DNA and RNA were isolated from all liver tissue samples. Of the 780 samples collected, high-quality RNA was successfully isolated and profiled on 466 Caucasian subjects. These 466 subjects included the 427 samples with successful genotype data described in the previous study (Schadt et al. 2008), 398 samples with enzyme activity measurements (25 of these subjects were not genotyped), and 14 subjects with gene expression data only. The sample size for each individual analysis varied and was a function of the data intersected in the particular analysis. For example, 466 samples were involved in gene–gene correlation analysis; 398 samples were used for trait–trait correlation and gene–trait correlation analyses; 427 samples were used for eSNP discovery. Age, gender, and ethnicity were confirmed or imputed in cases of missing data as described previously (Schadt et al. 2008).
Human liver microsome preparation
Each of the liver samples was processed into cytosol and microsomes at CellzDirect using a standard differential centrifugation method. Frozen liver tissues were thawed on ice in homogenization buffer (50 mM Tris at pH 7.4, 150 mM KCl, 2 mM EDTA), homogenized with a Potter-Elvehjem type tissue grinder, and centrifuged at low speed (∼13,000gmax) for 20 min at 4°C to remove plasma membranes, mitochondria, nuclei, and lysosomes. The resulting supernatant was centrifuged at high speed (∼100,000gmax) for 60 min at 4°C to yield a cytosolic fraction and microsomal pellet. The pellet was resuspended in homogenization buffer, recentrifuged, and resuspended in 0.25 M sucrose with a Potter-Elvehjem tissue grinder fitted with a Teflon pestle. A small aliquot of the microsomal preparation was retained for protein determination, and the remainder was stored at −70°C. The concentration of microsomal protein was determined with a bicinchoninic acid kit (BCA protein assay, Pierce Chemical) based on instructions provided by the manufacturer. The concentration of microsomal protein per gram of liver tissue was determined. Microsomes were diluted to an equivalent concentration before evaluation.
P450 enzyme activity measurements
The activities of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 were measured in human liver microsomal preparations at CellzDirect. Two substrates were used for the CYP3A4 activity measurement: testosterone and midazolam, with the corresponding enzyme activity designated as CYP3A4T and CYP3A4M, respectively. Microsomal incubations were performed in triplicate in a final incubation volume of 150 μL. The substrate, in 0.1 M phosphate buffer (pH 7.4), was prewarmed to 37°C with a total organic solvent content of ≤1%. Microsomal protein was added to a final concentration of ∼0.1 mg/mL (±0.05 mg/mL). Reactions were initiated by addition of NADPH (1 mM final concentration) and incubated for the indicated times with gentle agitation. The rate of substrate turnover was linear under these reaction conditions during method validations. Control incubations included pooled human liver microsomes (CellzDirect; pooled from 15 male and female individuals) as positive control samples as well as samples without NADPH. The reactions were terminated by the addition of organic solvent, and internal standards were added, mixed, and centrifuged at ∼3000 rpm for 5 min. The organic layer was transferred to a clean tube and evaporated to dryness at ∼40°C under nitrogen. The sample was reconstituted and analyzed by LC-MS/MS.
Metabolite standard curves with eight concentrations were included with each analytical run. In addition, bioanalytical metabolite quality control samples (QC) at three concentrations with four replicates spaced throughout the analytical run were measured. All analytical runs were evaluated based on predefined acceptance criteria as follows: having at least two-thirds of the quality control samples, with 50% at each level, and within ±20% of the nominal values.
Mass spectrometry data were acquired, integrated, regressed, and quantified with MassLynx software, version 3.4 (Micromass). Data were graphed and calculated using Microsoft Excel 97. Reaction velocities (v) were calculated using the following equation:
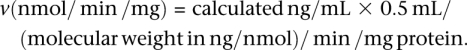 |
All enzyme activity data have been deposited to the Sage Commons Repository and are freely available at http://sage.fhcrc.org/downloads/.
RNA profiling
The microarray design, RNA sample preparation, amplification, hybridization, and expression analysis were previously described in detail (Schadt et al. 2008). Briefly, liver samples of the 466 Caucasian individuals were homogenized and total RNA extracted using TRIzol reagent. Each RNA sample was profiled on a custom Agilent 44,000 feature microarray composed of 39,280 oligonucleotide probes targeting transcripts representing 34,266 known and predicted genes, including high-confidence, noncoding RNA sequences. Arrays were quantified on the basis of spot intensity relative to background, adjusted for experimental variation between arrays using average intensity over multiple channels, and fitted to an error model to determine significance (type I error) (He et al. 2003). Gene expression is reported as the mean-log ratio relative to the pool derived from 192 liver samples selected for balance from the Vanderbilt and Pittsburgh samples as the RNA from the Merck samples had been amplified at an earlier date.
Thorough quality control was conducted to assess parameters such as study site, RNA quality, batch effects, age, and gender. The most significant covariate was study site, which was confounded with amplification driven mostly by (but not solely) the processing of the Merck samples separately and lack of representation in the reference pool. In addition to study site, gender and age were also significant covariates. These covariates were all controlled for in the analysis. Other relevant factors that may have direct effects on the liver, such as drug use, steatosis, alcohol use, and/or smoking were also evaluated. However, due to the nature of the cohort and Institutional Review Board (IRB) restrictions, information on these factors was very sparse, and thus was not incorporated directly in this analysis. All microarray data associated with the HLC have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE9588.
DNA processing and genotyping
DNA isolation and genotyping were described in detail previously (Schadt et al. 2008). Briefly, DNeasy tissue kits from Qiagen Inc. were used to carry out all DNA extractions from 20–30 mg of each liver sample. Genotyping of 548 and 397 Caucasian DNA samples was performed with two platforms, respectively: the commercial release of the Affymetrix 500K genotyping array at the Perlegen genotyping facility in Mountain View, CA and the Illumina Sentrix humanHap650Y genotyping beadchip at the Illumina genotyping facility in La Jolla, CA. In total, 517 and 384 DNA samples were successfully genotyped on the Affymetrix 500K SNP and Illumina 650K SNP arrays, respectively. The Affymetrix 500K array consisted of 500,568 SNPs in total, and the Illumina array consisted of 655,352 tag SNP markers. Analysis was restricted to those SNPs that had a genotyping call rate >90% and a minor allele frequency >4% and that did not deviate significantly from Hardy-Weinberg equilibrium in the HLC. In total, 310,744 and 557,240 SNPs met these criteria from the Affymetrix and Illumina sets, respectively, and these represented 782,476 unique SNPs. Genotype data for 228 individuals that satisfy privacy policy have been deposited to dbGaP under accession number phs000253.v1.p1.
Correlation and hierarchical clustering of P450 enzyme activity traits
Pairwise Spearman correlation among the residuals of the P450 activity traits after adjusting for covariates (age, gender, and study site) was calculated using the cor.test function in R. The hclust function in R was used to perform hierarchical cluster analysis using the “complete linkage” method to find similar clusters. The iterative algorithm joins the most similar traits at each stage based on the distances computed by the Lance–Williams dissimilarity update formula.
Correlation between gene expression and P450 enzyme activity
We selected 5855 transcriptionally variable genes with significance level P < 0.05 (as determined by error model; for details on the error-model, see Weng et al. 2006) in at least 50 samples. These genes represent those whose expression levels vary across samples and, thus, are more biologically relevant. Partial Spearman correlation was calculated between the selected transcripts and the activity measurements of the nine P450s by adjusting both the transcripts and enzyme activity traits for covariates including age, gender, and study site. Transcripts correlated with each P450 activity trait at P < 0.01 were reported. The FDR levels at this significance cutoff were calculated using the Q-value approach (Storey and Tibshirani 2003).
Constructing the HLC coexpression network
The 5012 (the top 12.5%) most differentially expressed genes were selected for constructing a weighted gene coexpression network (Zhang and Horvath 2005). Different from the traditional unweighted gene coexpression network, where two genes (nodes) are either connected or disconnected, the weighted gene coexpression network analysis assigns a connection weight to each gene pair using soft-thresholding and thus is robust to parameter selection. The weighted network analysis begins with a matrix of the Pearson correlations between all gene pairs and then converts the correlation matrix into an adjacency matrix using a power function f(x) = xβ. The parameter β of the power function is determined in such a way that the resulting adjacency matrix, namely, the weighted coexpression network, is approximately scale-free. To measure how well a network satisfies a scale-free topology, we use the fitting index proposed by Zhang and Horvath (2005), that is, the model fitting index R2 of the linear model that regresses log[p(k)] on log(k), where k is connectivity and p(k) is the frequency distribution of connectivity. The fitting index of a perfect scale-free network is 1. For this data set, we select the smallest β (=5.5) that leads to an approximately scale-free network with the truncated scale-free fitting index R2 > 0.8. The distribution p(k) of the resulting network approximates a power law: p(k) ∼ k−1.75.
To explore the modular structures of the coexpression network, the adjacency matrix is further transformed into a TOM (Ravasz et al. 2002). As the topological overlap between two genes reflects not only their direct interaction but also their indirect interactions through all the other genes in the network, previous studies (Ravasz et al. 2002; Zhang and Horvath 2005) have shown that topological overlap leads to more cohesive and biologically more meaningful modules. To identify modules of highly coregulated genes, we used average linkage hierarchical clustering to group genes based on the topological overlap of their connectivity, followed by a dynamic cut-tree algorithm to dynamically cut clustering dendrogram branches into gene modules (Langfelder et al. 2008). Eight modules are identified, and the module size, in number of genes, varies from 55–1361.
To distinguish between modules, each module was assigned a unique color identifier, with the remaining, poorly connected genes colored gray. Figure 3A shows the hierarchical clustering over the TOM and the identified modules. In this type of map, the rows and the columns represent genes in a symmetric fashion, and the color intensity represents the interaction strength between genes. This connectivity map highlights that genes in the liver transcriptional network fall into distinct network modules, where genes within a given module are more interconnected with each other (blocks along the diagonal of the matrix) than with genes in other modules. There are a couple of network connectivity measures, but one particularly important one is the within module connectivity (k.in). k.in of a gene was determined by taking the sum of its connection strengths (coexpression similarity) with all other genes in the module that the gene belongs to.
Identification of aSNPs and eSNPs
DNA isolation, genotyping with Affymetrix 500K genotyping array and Illumina 650Y panel, and quality control procedures were described in detail previously (Schadt et al. 2008). Briefly, a total of 85,508 SNPs were represented in both genotyping platforms, and 782,476 unique SNPs were successfully genotyped in the HLC at a call rate > 90%, MAF > 5%, and nonsignificant deviation from the Hardy-Weinberg equilibrium at the 0.0001 significance level.
The Kruskal-Wallis test was used to determine association between age and gender-adjusted expression or enzyme activity traits and genotypes of the approximately 800 k SNPs. We chose this nonparametric method because it is robust to underlying genetic model and trait distribution. P-values were computed using nag_mann_whitney (for loci with two observed genotypes) and nag_kruskal_wallis_test (for loci with three observed genotypes) routines from the NAG C library (http://www.nag.co.uk). We employed FDR for multiple-test correction. FDR was estimated as the ratio of aSNPs or eSNPs found in the original data set to the average number of aSNPs or eSNPs found in data sets with randomized sample labels. Since the number of tests was large, we found the empirical null distribution was very stable, and five permutation runs were sufficient for convergence to estimate FDR. For eSNPs, FDR computation was performed separately for cis (<1 Mb probe-to-SNP distance) and trans associations resulting in a nominal P-value cutoff of P < 5 × 10−4 for cis to reach FDR < 10%. No P-value cutoff can be found for trans eSNPs at FDR < 10%. For aSNPs, the P-value cutoff was 3.5 × 10−6 at FDR < 10%.
Validation of CYP2D6 aSNP/eSNPs
The validation cohort comprised 237 Caucasian subjects previously described in detail (Gaedigk et al. 2008). Briefly, subjects were phenotyped with the CYP2D6 probe drug DM and the urinary ratio of DM to its metabolite DX determined (DM/DX). This ratio serves as a measure of CYP2D6 activity in vivo (DM/DX > 0.3 indicates poor metabolism). As the ratio of DM/DX follows a nonnormal distribution, we used the log10 transformation log10_DMDX. CYP2D6 genotype analysis included three long-range aSNP/eSNPs of CYP2D6 identified from the current study (rs8138080, rs5751247, and rs17478227) and gene deletion, duplication, and conversion events in addition to 24 common and rare allelic variants, including 1584C>G, 31G>A (*35), 100C>T (*4, *10), 124G>A (*12), 138Tins (*15), 843T>G (many), 883G>C (*11), 1023C>T (*17), 1659G>A (*29), 1707Tdel (*6), 1716G>A (*45/46), 1758G>T (*8), 1846G>A (*4), 2549Adel (*3), 2613AGAdel (*9), 2850C>T (many), 2935A>C (*7), 2988G>A (*41), 3183G>A (*29), 3201C>T (*56), 3259GTins (*42), 3790C>T (many), 4125_9bp ins (*18), and 2D7 conv (E9 conv/*36). The aSNP/eSNPs rs8138080, rs5751247, and rs17478227 were genotyped using made to order TaqMan SNP Genotyping Assays (Applied Biosystems) on a 7900 real-time PCR system. Correlation between the aSNP/eSNPs and log10_DMDX was assessed using linear regression, and the LD among the genotyped CYP2D6 SNPs was analyzed using the R Bioconductor package snpMatrix.
Reconstruction of HLC Bayesian gene regulatory network
A total of 8188 genes were selected for inclusion in the network reconstruction process based on criteria: (1) variance of the gene expression is among the top 20% of gene expression variance; or (2) the LOD scores of genes' eQTL are above genome-wide significance level. The 8188 genes were input into a BN reconstruction software program based on a previously described algorithm (Zhu et al. 2007; 2008b). Genetics information was used as priors as follows: genes with cis-eQTLs are allowed to be parent nodes of genes with coincident trans eQTLs, p(cis → trans) = 1, but genes with trans eQTLs are not allowed to be parents of genes with cis eQTLs, p(trans → cis) = 0. Bayesian information criteria (BIC) was used in the reconstruction process. One thousand BNs were reconstructed using different random seeds to start the reconstruction process. From the resulting set of 1000 networks generated by this process, edges that appeared in >30% of the networks were used to define a consensus network. Edges in this consensus network were removed if (1) the edge was involved in a loop and (2) the edge was the most weakly supported of all edges making up the loop.
Identification of P450 regulators from HLC BN
In order to determine whether a subnetwork is enriched for P450s, the following procedure was used. For a given subnetwork, the procedure first computes the signature of each node that could be reached by the node following directed links throughout the entire subnetwork. Then, the signature of each node was intersected with the set of P450 genes. If the overlap was significant as determined by an enrichment test P-value < 0.05 and if there were at least two P450 targets in signature, we declared the node (gene) a regulator of the P450 genes.
Acknowledgments
We thank Jonathan Derry for valuable discussions on the manuscript. This work was funded in part by United States Public Health Service grants R37 CA090426 and P30 ES000267 (F.P.G.), GM60346 and ES017499-0109 (E.S.), and NIH contract no. N01-DK-9-2310 for the Liver Tissue Procurement and Distribution System (S.S.).
Author contributions: J.G.S., R.G.U., F.P.G., E.S., S.C.S., T.H.R., J.S.L., E.E.S., A.K., and P.Y.L. conceived and designed the experiments. F.P.G., S.C.S., E.S., T.H.R., R.G.U., and J.G.S. contributed liver biopsy samples. X.Y., B.Z., and P.Y.L. designed the data analysis scheme. X.Y., B.Z., C.M., E.C., E.E.S., A.G., K.H., J.Z., and H.Z. analyzed the data. A.G. and J.S.L. performed validation experiments. X.Y., B.Z., J.Z., and A.G. wrote the paper with revisions from J.G.S., E.S., R.G.U., F.P.G., J.S.L., E.E.S., A.K., and P.Y.L. C.S. and A.K. coordinated the study.
Footnotes
[Supplemental material is available online at http://www.genome.org. Genotype data for 228 individuals who satisfy privacy policy have been submitted to the NCBI dbGaP (http://www.ncbi.nlm.nih.gov/gap/) under accession no. phs000253.v1.p1.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.103341.109.
References
- Adonis M, Martinez V, Marin P, Berrios D, Gil L 2005. Smoking habit and genetic factors associated with lung cancer in a population highly exposed to arsenic. Toxicol Lett 159: 32–37 [DOI] [PubMed] [Google Scholar]
- Aguiar M, Masse R, Gibbs BF 2005. Regulation of cytochrome P450 by posttranslational modification. Drug Metab Rev 37: 379–404 [DOI] [PubMed] [Google Scholar]
- Ahluwalia A, Clodfelter KH, Waxman DJ 2004. Sexual dimorphism of rat liver gene expression: Regulatory role of growth hormone revealed by deoxyribonucleic acid microarray analysis. Mol Endocrinol 18: 747–760 [DOI] [PubMed] [Google Scholar]
- Aitken AE, Richardson TA, Morgan ET 2006. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol 46: 123–149 [DOI] [PubMed] [Google Scholar]
- Anderson GD 2008. Gender differences in pharmacological response. Int Rev Neurobiol 83: 1–10 [DOI] [PubMed] [Google Scholar]
- Anderson JL, Carlquist JF, Horne BD, Muhlestein JB 2003. Cardiovascular pharmacogenomics: Current status, future prospects. J Cardiovasc Pharmacol Ther 8: 71–83 [DOI] [PubMed] [Google Scholar]
- Audet-Walsh E, Anderson A 2008. Dexamethasone induction of murine CYP2B genes requires the glucocorticoid receptor. Drug Metab Dispos 37: 580–588 [DOI] [PubMed] [Google Scholar]
- Barbier O, Fontaine C, Fruchart JC, Staels B 2004. Genomic and non-genomic interactions of PPARalpha with xenobiotic-metabolizing enzymes. Trends Endocrinol Metab 15: 324–330 [DOI] [PubMed] [Google Scholar]
- Barouki R, Morel Y 2001. Repression of cytochrome P450 1A1 gene expression by oxidative stress: Mechanisms and biological implications. Biochem Pharmacol 61: 511–516 [DOI] [PubMed] [Google Scholar]
- Braeuning A 2009. Regulation of cytochrome p450 expression by Ras- and β-catenin–dependent signaling. Curr Drug Metab 10: 138–158 [DOI] [PubMed] [Google Scholar]
- Cai Y, Konishi T, Han G, Campwala KH, French SW, Wan YJ 2002. The role of hepatocyte RXRα in xenobiotic-sensing nuclear receptor-mediated pathways. Eur J Pharm Sci 15: 89–96 [DOI] [PubMed] [Google Scholar]
- Chavarria-Soley G, Sticht H, Aklillu E, Ingelman-Sundberg M, Pasutto F, Reis A, Rautenstrauss B 2008. Mutations in CYP1B1 cause primary congenital glaucoma by reduction of either activity or abundance of the enzyme. Hum Mutat 29: 1147–1153 [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, et al. 2008. Variations in DNA elucidate molecular networks that cause disease. Nature 452: 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly AK 2004. Pharmacogenetics of the cytochromes P450. Curr Top Med Chem 4: 1733–1744 [DOI] [PubMed] [Google Scholar]
- Davey Smith G, Ebrahim S 2003. ”Mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 32: 1–22 [DOI] [PubMed] [Google Scholar]
- Debose-Boyd RA 2007. A helping hand for cytochrome p450 enzymes. Cell Metab 5: 81–83 [DOI] [PubMed] [Google Scholar]
- Dhir RN, Dworakowski W, Thangavel C, Shapiro BH 2006. Sexually dimorphic regulation of hepatic isoforms of human cytochrome p450 by growth hormone. J Pharmacol Exp Ther 316: 87–94 [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Kullak-Ublick GA 2005. Coordinate transcriptional regulation of bile acid homeostasis and drug metabolism. Arch Biochem Biophys 433: 397–412 [DOI] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428 [DOI] [PubMed] [Google Scholar]
- Fan LQ, You L, Brown-Borg H, Brown S, Edwards RJ, Corton JC 2004. Regulation of phase I and phase II steroid metabolism enzymes by PPARα activators. Toxicology 204: 109–121 [DOI] [PubMed] [Google Scholar]
- Fraser HB, Moses AM, Schadt EE 2010. Evidence for widespread adaptive evolution of gene expression in budding yeast. Proc Natl Acad Sci 107: 2977–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS 2008. The CYP2D6 activity score: Translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther 83: 234–242 [DOI] [PubMed] [Google Scholar]
- Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, et al. 2006. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci 103: 12741–12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbal-Chaloin S, Daujat M, Pascussi JM, Pichard-Garcia L, Vilarem MJ, Maurel P 2002. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J Biol Chem 277: 209–217 [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, Brozell A, Schadt EE, Drake TA, Lusis AJ, et al. 2006. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2: e130 doi: 10.1371/journal.pgen.0020130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521 [DOI] [PubMed] [Google Scholar]
- Gnerre C, Blattler S, Kaufmann MR, Looser R, Meyer UA 2004. Regulation of CYP3A4 by the bile acid receptor FXR: Evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 14: 635–645 [DOI] [PubMed] [Google Scholar]
- Gnerre C, Schuster GU, Roth A, Handschin C, Johansson L, Looser R, Parini P, Podvinec M, Robertsson K, Gustafsson JA, et al. 2005. LXR deficiency and cholesterol feeding affect the expression and phenobarbital-mediated induction of cytochromes P450 in mouse liver. J Lipid Res 46: 1633–1642 [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ 2008. Regulation of hepatocyte nuclear factor 4α-mediated transcription. Drug Metab Pharmacokinet 23: 2–7 [DOI] [PubMed] [Google Scholar]
- Gorski JC, Vannaprasaht S, Hamman MA, Ambrosius WT, Bruce MA, Haehner-Daniels B, Hall SD 2003. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther 74: 275–287 [DOI] [PubMed] [Google Scholar]
- Hakkola J, Tanaka E, Pelkonen O 1998. Developmental expression of cytochrome P450 enzymes in human liver. Pharmacol Toxicol 82: 209–217 [DOI] [PubMed] [Google Scholar]
- Hardwick JP 2008. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem Pharmacol 75: 2263–2275 [DOI] [PubMed] [Google Scholar]
- He YD, Dai H, Schadt EE, Cavet G, Edwards SW, Stepaniants SB, Duenwald S, Kleinhanz R, Jones AR, Shoemaker DD, et al. 2003. Microarray standard data set and figures of merit for comparing data processing methods and experiment designs. Bioinformatics 19: 956–965 [DOI] [PubMed] [Google Scholar]
- Hodgson J 2001. ADMET: Turning chemicals into drugs. Nat Biotechnol 19: 722–726 [DOI] [PubMed] [Google Scholar]
- Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, Laurance MF, Zhao W, Qi S, Chen Z, et al. 2006. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci 103: 17402–17407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber WW, Rossmanith W, Grusch M, Haslinger E, Prustomersky S, Peter-Vorosmarty B, Parzefall W, Scharf G, Schulte-Hermann R 2008. Effects of coffee and its chemopreventive components kahweol and cafestol on cytochrome P450 and sulfotransferase in rat liver. Food Chem Toxicol 46: 1230–1238 [DOI] [PubMed] [Google Scholar]
- Hughes AL, Powell DW, Bard M, Eckstein J, Barbuch R, Link AJ, Espenshade PJ 2007. Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metab 5: 143–149 [DOI] [PubMed] [Google Scholar]
- Ishizawa Y, Yasui-Furukori N, Takahata T, Sasaki M, Tateishi T 2005. The effect of aging on the relationship between the cytochrome P450 2C19 genotype and omeprazole pharmacokinetics. Clin Pharmacokinet 44: 1179–1189 [DOI] [PubMed] [Google Scholar]
- Jia Y, Qi C, Zhang Z, Hashimoto T, Rao MS, Huyghe S, Suzuki Y, Van Veldhoven PP, Baes M, Reddy JK 2003. Overexpression of peroxisome proliferator-activated receptor-α (PPARα)–regulated genes in liver in the absence of peroxisome proliferation in mice deficient in both L- and D-forms of enoyl-CoA hydratase/dehydrogenase enzymes of peroxisomal β-oxidation system. J Biol Chem 278: 47232–47239 [DOI] [PubMed] [Google Scholar]
- Katz DA, Murray B, Bhathena A, Sahelijo L 2008. Defining drug disposition determinants: A pharmacogenetic-pharmacokinetic strategy. Nat Rev Drug Discov 7: 293–305 [DOI] [PubMed] [Google Scholar]
- Koukouritaki SB, Manro JR, Marsh SA, Stevens JC, Rettie AE, McCarver DG, Hines RN 2004. Developmental expression of human hepatic CYP2C9 and CYP2C19. J Pharmacol Exp Ther 308: 965–974 [DOI] [PubMed] [Google Scholar]
- Krone N, Grischuk Y, Muller M, Volk RE, Grotzinger J, Holterhus PM, Sippell WG, Riepe FG 2006. Analyzing the functional and structural consequences of two point mutations (P94L and A368D) in the CYP11B1 gene causing congenital adrenal hyperplasia resulting from 11-hydroxylase deficiency. J Clin Endocrinol Metab 91: 2682–2688 [DOI] [PubMed] [Google Scholar]
- Kuribayashi I, Kuge H, Santa RJ, Mutlaq AZ, Yamasaki N, Furuno T, Takahashi A, Chida S, Nakamura T, Endo F, et al. 2003. A missense mutation (GGC[435Gly]→AGC[Ser]) in exon 8 of the CYP11B2 gene inherited in Japanese patients with congenital hypoaldosteronism. Horm Res 60: 255–260 [DOI] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S 2008. Defining clusters from a hierarchical cluster tree: The Dynamic Tree Cut package for R. Bioinformatics 24: 719–720 [DOI] [PubMed] [Google Scholar]
- Lee SS, Cha EY, Jung HJ, Shon JH, Kim EY, Yeo CW, Shin JG 2008. Genetic polymorphism of hepatocyte nuclear factor-4α influences human cytochrome P450 2D6 activity. Hepatology 48: 635–645 [DOI] [PubMed] [Google Scholar]
- Lee WH, Lukacik P, Guo K, Ugochukwu E, Kavanagh KL, Marsden B, Oppermann U 2009. Structure-activity relationships of human AKR-type oxidoreductases involved in bile acid synthesis: AKR1D1 and AKR1C4. Mol Cell Endocrinol 301: 199–204 [DOI] [PubMed] [Google Scholar]
- Lewis DF, Lee-Robichaud P 1998. Molecular modelling of steroidogenic cytochromes P450 from families CYP11, CYP17, CYP19 and CYP21 based on the CYP102 crystal structure. J Steroid Biochem Mol Biol 66: 217–233 [DOI] [PubMed] [Google Scholar]
- Lim YP, Huang JD 2008. Interplay of pregnane X receptor with other nuclear receptors on gene regulation. Drug Metab Pharmacokinet 23: 14–21 [DOI] [PubMed] [Google Scholar]
- Loeppen S, Koehle C, Buchmann A, Schwarz M 2005. A β-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis 26: 239–248 [DOI] [PubMed] [Google Scholar]
- Lum PY, Chen Y, Zhu J, Lamb J, Melmed S, Wang S, Drake TA, Lusis AJ, Schadt EE 2006. Elucidating the murine brain transcriptional network in a segregating mouse population to identify core functional modules for obesity and diabetes. J Neurochem 97: 50–62 [DOI] [PubMed] [Google Scholar]
- Masters B, Marohnic CC 2006. Cytochromes P450: A family of proteins and scientists-understanding their relationships. Drug Metab Rev 38: 209–225 [DOI] [PubMed] [Google Scholar]
- Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, Walker JR, Antman EM, Macias W, Braunwald E, et al. 2009. Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 360: 354–362 [DOI] [PubMed] [Google Scholar]
- Moon YJ, Wang X, Morris ME 2006. Dietary flavonoids: Effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro 20: 187–210 [DOI] [PubMed] [Google Scholar]
- Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, et al. 1996. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 6: 1–42 [DOI] [PubMed] [Google Scholar]
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW 2004. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14: 1–18 [DOI] [PubMed] [Google Scholar]
- Ortiz De Montellano PR 2005. Cytochrome P450: Structure, mechanism, and biochemistry Springer, New York [Google Scholar]
- Parkinson A, Mudra DR, Johnson C, Dwyer A, Carroll KM 2004. The effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme activity in human liver microsomes and inducibility in cultured human hepatocytes. Toxicol Appl Pharmacol 199: 193–209 [DOI] [PubMed] [Google Scholar]
- Peters CJ, Nugent T, Perry LA, Davies K, Morel Y, Drake WM, Savage MO, Johnston LB 2007. Cosegregation of a novel homozygous CYP11B1 mutation with the phenotype of non-classical congenital adrenal hyperplasia in a consanguineous family. Horm Res 67: 189–193 [DOI] [PubMed] [Google Scholar]
- Plant N 2007The human cytochrome P450 sub-family: Transcriptional regulation, inter-individual variation and interaction networks. Biochim Biophys Acta 1770: 478–488 [DOI] [PubMed] [Google Scholar]
- Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, Verhagen A, Rivera CR, Mulvihill SJ, Malloy MJ, et al. 2002. Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest 110: 109–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi A-L 2002. Hierarchical organization of modularity in metabolic networks. Science 297: 1551–1555 [DOI] [PubMed] [Google Scholar]
- Rinn JL, Snyder M 2005. Sexual dimorphism in mammalian gene expression. Trends Genet 21: 298–305 [DOI] [PubMed] [Google Scholar]
- Saitoh T, Kokue E, Shimoda M 2000. The impact of acute phase response on the plasma clearance of antipyrine, theophylline, phenytoin and nifedipine in rabbits. J Vet Pharmacol Ther 23: 153–158 [DOI] [PubMed] [Google Scholar]
- Scandlyn MJ, Stuart EC, Rosengren RJ 2008. Sex-specific differences in CYP450 isoforms in humans. Expert Opin Drug Metab Toxicol 4: 413–424 [DOI] [PubMed] [Google Scholar]
- Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, Sieberts SK, Monks S, Reitman M, Zhang C, et al. 2005. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet 37: 710–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. 2008. Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6: e107 doi: 10.1371/journal.pbio.0060107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrenk D, Schmitz HJ, Bohnenberger S, Wagner B, Worner W 2004. Tumor promoters as inhibitors of apoptosis in rat hepatocytes. Toxicol Lett 149: 43–50 [DOI] [PubMed] [Google Scholar]
- Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M 2006. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43: 817–825 [DOI] [PubMed] [Google Scholar]
- Seree E, Villard PH, Pascussi JM, Pineau T, Maurel P, Nguyen QB, Fallone F, Martin PM, Champion S, Lacarelle B, et al. 2004. Evidence for a new human CYP1A1 regulation pathway involving PPAR-α and 2 PPRE sites. Gastroenterology 127: 1436–1445 [DOI] [PubMed] [Google Scholar]
- Simon T, Becquemont L, Hamon B, Nouyrigat E, Chodjania Y, Poirier JM, Funck-Brentano C, Jaillon P 2001. Variability of cytochrome P450 1A2 activity over time in young and elderly healthy volunteers. Br J Clin Pharmacol 52: 601–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon T, Verstuyft C, Mary-Krause M, Quteineh L, Drouet E, Meneveau N, Steg PG, Ferrieres J, Danchin N, Becquemont L 2009. Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 360: 363–375 [DOI] [PubMed] [Google Scholar]
- Slatter JG, Templeton IE, Castle JC, Kulkarni A, Rushmore TH, Richards K, He Y, Dai X, Cheng OJ, Caguyong M, et al. 2006. Compendium of gene expression profiles comprising a baseline model of the human liver drug metabolism transcriptome. Xenobiotica 36: 938–962 [DOI] [PubMed] [Google Scholar]
- Stevens JC 2006. New perspectives on the impact of cytochrome P450 3A expression for pediatric pharmacology. Drug Discov Today 11: 440–445 [DOI] [PubMed] [Google Scholar]
- Stevens JC, Marsh SA, Zaya MJ, Regina KJ, Divakaran K, Le M, Hines RN 2008. Developmental changes in human liver CYP2D6 expression. Drug Metab Dispos 36: 1587–1593 [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strolin-Benedetti M, Brogin G, Bani M, Oesch F, Hengstler JG 1999. Association of cytochrome P450 induction with oxidative stress in vivo as evidenced by 3-hydroxylation of salicylate. Xenobiotica 29: 1171–1180 [DOI] [PubMed] [Google Scholar]
- Tirona RG, Lee W, Leake BF, Lan LB, Cline CB, Lamba V, Parviz F, Duncan SA, Inoue Y, Gonzalez FJ, et al. 2003. The orphan nuclear receptor HNF4α determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med 9: 220–224 [DOI] [PubMed] [Google Scholar]
- Tomaszewski P, Kubiak-Tomaszewska G, Pachecka J 2008. Cytochrome P450 polymorphism–molecular, metabolic, and pharmacogenetic aspects. II. Participation of CYP isoenzymes in the metabolism of endogenous substances and drugs. Acta Pol Pharm 65: 307–318 [PubMed] [Google Scholar]
- Vandel P, Talon JM, Haffen E, Sechter D 2007. Pharmacogenetics and drug therapy in psychiatry: The role of the CYP2D6 polymorphism. Curr Pharm Des 13: 241–250 [DOI] [PubMed] [Google Scholar]
- van Schaik RH 2005. Cancer treatment and pharmacogenetics of cytochrome P450 enzymes. Invest New Drugs 23: 513–522 [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Gonzalez FJ 2008. Role of CYP1B1 in glaucoma. Annu Rev Pharmacol Toxicol 48: 333–358 [DOI] [PubMed] [Google Scholar]
- Wang K, Chen S, Xie W, Wan YJ 2008. Retinoids induce cytochrome P450 3A4 through RXR/VDR-mediated pathway. Biochem Pharmacol 75: 2204–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Narayanan M, Zhong H, Tompa M, Schadt EE, Zhu J 2009. Meta-analysis of inter-species liver co-expression networks elucidates traits associated with common human diseases. PLoS Comput Biol 5: e1000616 doi: 10.1371/journal.pcbi.1000616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauthier V, Verbeeck RK, Calderon PB 2007. The effect of ageing on cytochrome p450 enzymes: Consequences for drug biotransformation in the elderly. Curr Med Chem 14: 745–757 [DOI] [PubMed] [Google Scholar]
- Waxman DJ 1999. P450 gene induction by structurally diverse xenochemicals: Central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys 369: 11–23 [DOI] [PubMed] [Google Scholar]
- Weng L, Dai H, Zhan Y, He Y, Stepaniants SB, Bassett DE 2006. Rosetta error model for gene expression analysis. Bioinformatics 22: 1111–1121 [DOI] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Hamman MA, Hall SD, Wrighton SA 2002. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30: 883–891 [DOI] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Ward A, Angove HC, Matak Vinkovic D, Jhoti H 2003. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 424: 464–468 [DOI] [PubMed] [Google Scholar]
- Xie W, Tian Y 2006. Xenobiotic receptor meets NF-κB, a collision in the small bowel. Cell Metab 4: 177–178 [DOI] [PubMed] [Google Scholar]
- Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ 2006. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res 16: 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Deignan JL, Qi H, Zhu J, Qian S, Zhong J, Torosyan G, Majid S, Falkard B, Kleinhanz RR, et al. 2009. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet 41: 415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S 2005. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17. doi: 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A, Lin M, Mallick S, Forman BM, Thummel KE, et al. 2006. Mutual repression between steroid and xenobiotic receptor and NF-κB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest 116: 2280–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Lum PY, Lamb J, GuhaThakurta D, Edwards SW, Thieringer R, Berger JP, Wu MS, Thompson J, Sachs AB, et al. 2004. An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet Genome Res 105: 363–374 [DOI] [PubMed] [Google Scholar]
- Zhu J, Wiener MC, Zhang C, Fridman A, Minch E, Lum PY, Sachs JR, Schadt EE 2007. Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput Biol 3: e69 doi: 10.1371/journal.pcbi.0030069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Schadt EE 2008a. A systems biology approach to drug discovery. Adv Genet 60: 603–635 [DOI] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE 2008b. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet 40: 854–861 [DOI] [PMC free article] [PubMed] [Google Scholar]