

Abstract
The reaction pathways for 5′-hydroxylation and N-demethylation of nicotine catalyzed by cytochrome P450 were investigated by performing a series of first-principle electronic structure calculations on a catalytic reaction model system. The computational results indicate that 5′-hydroxylation of nicotine occurs through a two-state stepwise process, i.e. an initial hydrogen atom transfer from nicotine to Cpd I (i.e. the HAT step) followed by a recombination of the nicotine moiety with the iron-bound hydroxyl group (i.e. the rebound step) on both the high-spin (HS) quartet and low-spin (LS) doublet states. The HAT step is the rate-determining one. This finding represents the first case that exhibits genuine rebound transition state species on both the HS and the LS states for Cα-H hydroxylation of amines. N-demethylation of nicotine involves a N-methylhydroxylation to form N-(hydroxymethyl)nornicotine, followed by N-(hydroxymethyl)nornicotine decomposition to nornicotine and formaldehyde. The N-methylhydroxylation step is similar to 5′-hydroxylation, namely that a rate-determining HAT step followed by a rebound step. The decomposition process occurs on the deprotonated state of N-(hydroxymethyl)nornicotine assisted by a water molecule and the energy barrier is significantly lower than that of the N-methylhydroxylation process. Comparison of the rate-determining free energy barriers for the two reaction pathways predicts a preponderance of 5′-hydroxylation over the N-demethylation by roughly a factor of 18:1, which is in excellent agreement with the factor of 19:1 derived from available experimental data.
Introduction
Cigarette smoking is currently the most single preventable cause of mortality in the world and leads to ∼5.4 million deaths annually. Unless urgent action is taken, tobacco could kill up to one billion people during this century.1 Despite the widespread information about the enormous negative health consequences of cigarette smoking, millions of people continue to smoke and the use of tobacco is still rising globally.1,2 The vast majority of smokers attempt to quit each year, but only a few percents of them quit successfully.3,4 Although there are some pharmacotherapies (e.g. nicotine replacement therapies, dopamine reuptake inhibitor, antidepressant, and antianxiety agent) existing for smoking cessation, relapse rates continue to be high and side effects are common in these treatments.3,5
Nicotine, an alkaloid found in tobacco leaves, is the primary addictive component responsible for dependence on cigarette smoking.6,7 The primary pharmacologic function of tobacco smoking is to deliver nicotine to the brain.8,9 Nicotine-dependent individuals regulate their smoking to maintain nicotine concentration in their blood and brain.10,11 Conversion of nicotine to inactive cotinine and other minor detoxification products by hepatic enzyme cytochrome P450 2A6 (CYP2A6) is the principal nicotine-metabolizing pathway by which biologically active nicotine is removed from circulation.12-16 Individuals lacking CYP2A6 activity in nicotine metabolism are at lower risk to become smokers or at least smoke less.17-20 Hence, inhibition of CYP2A6 could be a valuable strategy for smoking cessation and tobacco exposure reduction.21 Some recently reported studies have focused on CYP2A6 inhibition as a means of smoking cessation.22-24 A number of compounds, such as tranylcypromine and methoxsalen, have been identified as inhibitors of CYP2A6.25,26 However, these compounds generally lack selectivity for targeting CYP2A6 and may also inhibit other cytochrome P450 enzymes and, therefore, could result in unexpected side effects.26-29 To date, no CYP2A6 inhibitor has been used actually for the treatment of tobacco dependence.3,30,31 It is highly desirable to design novel, potent and selective CYP2A6 inhibitors which can selectively bind to CYP2A6 and inhibit the metabolism of nicotine.
For rational design of novel inhibitors of CYP2A6, it is important to understand the detailed mechanism concerning how the enzyme catalyzes the nicotine metabolism, especially the transition state structure of the rate-determining reaction step. A detailed understanding of the mechanism of CYP2A6-catalyzed nicotine metabolism could provide a valuable mechanistic base for rational design of possible stable analogues of the rate-determining transition-state as a novel type of CYP2A6 inhibitors.
Nicotine metabolism has attracted considerable attention for several decades.4,32-39 In human hepatic microsomes, the primary metabolite of nicotine is cotinine (Scheme 1). This primary nicotine-metabolizing pathway is initiated by the CYP2A6-catalyzed hydroxylation at the 5′-position.33,40 The product 5′-hydroxynicotine, which exists in equilibrium with △1′(5′)-iminium ion, is further oxidized to cotinine by a cytosolic aldehyde oxidase with a very low Km (Michaelis-Menten constant) value.41-43 Nicotine can also be oxidized on the methyl carbon to form N-(hydroxymethyl)nornicotine that exists in equilibrium with the N-methylene-iminium ion.34 N-(hydroxymethyl)nornicotine then undergoes spontaneous breakdown to nornicotine and formaldehyde in smokers.4,36 Another minor metabolic pathway for nicotine is through 2′-hydroxylation.39 Since the amount of the 2′-hydroxylation product is so small and cannot even be detected,16 our computational studies will only focus on the 5′-hydroxylation and the N-demethylation pathways of nicotine metabolism.
Scheme 1.

CYP2A6-catalyzed metabolic pathways of nicotine
So far, we have not seen a report about reaction coordinate calculations on nicotine metabolism. Nevertheless, reaction mechanisms for alkane hydroxylation and N-demethylation of other amines have been studied computationally by many researchers.44-49 Earlier studies on the hydroxylation mechanism of alkanes revealed a two-state reactivity (TSR) scenario, originating from the high-spin (HS) quartet and low-spin (LS) doublet states of Cpd I, i.e., reactivity patterns and product distribution were determined by the interplay of the two states. The reaction mechanism involved two phases: a hydrogen atom transfer (HAT) phase and a rebound phase. The rate-determining step was the HAT phase. On the HS state the reaction was a stepwise one and there formed a radical that had a significant barrier for rebound, whereas on the LS state the reaction was effectively concerted and the rebound was barrierless.44-46 Later, reports on amines revealed that Cα-H hydroxylation of amines proceeded in a spin-selective manner (SSM), whereby the reaction proceeded mostly via a single spin state. The rate-determining step was also the HAT step. The reaction on both spin states were effectively concerted, i.e. the rebound process was barrier-free.47-49 However, a recent work by Molinie et al. on the heavy-atom isotope effects for the N-demethylation of nicotine indicated that the isotopically sensitive step was not associated with the initial HAT step and that the principal kinetic limiting step was the hydroxyl transfer from iron-hydroxo species to the N-methylene-iminium ion species.50
In light of the above-mentioned background, possible reaction pathways for nicotine metabolism could be various and the mechanisms pose some intriguing puzzles. First, is nicotine Cα-H hydroxylation a concerted process like those of other amines or a stepwise one? Second, if the reaction is a stepwise process, which step (the HAT one or the rebound one) is rate-determining? In addition, what kind of mechanism (TSR or SSM) does nicotine Cα-H hydroxylation follow? We herein performed reaction coordinate calculations using first-principle electronic structure method to examine the possible reaction pathways for 5′-hydroxylation and N-demethylation of nicotine catalyzed by Cpd I. On the basis of the reaction coordinate calculations, we determined the rate-determining steps and structures of the transition state. We also compared the free energy barriers for the two reaction pathways, and tried to understand the predominance of 5′-hydroxylation over N-demethylation which was extensively observed in experimental studies.16,35,51-54
Computational Methods
(S)-nicotine can have various stable molecular species associated with different protonation states in aqueous solution.55 The dominant molecular species under physiologic conditions (pH 7.4) is a cationic structure, denoted by SRH, with the pyrrolidine nitrogen protonated. Hence, the SRH species of nicotine was used in all reaction coordinate calculations reported here. The active species of CYP2A6, Cpd I was modeled as an iron-oxo-porphine complex without the side chains, and the proximal thiolate ligand was truncated to SH. This was established as a sufficient model in previous computational studies on other reaction systems catalyzed by cytochrome P450 enzymes.44,45,56-61
Unless stated explicitly otherwise, all calculations in the present study were carried out with the hybrid density functional method B3LYP62-65 using the Gaussian03 program.66 Geometries of all molecular species were fully optimized without geometric constraints. For geometry optimization, we used double-ζ LACVP basis set67,68 on iron and 6-31G* basis set on the remaining atoms. The mixed basis set was denoted by B1 for convenience. Vibrational frequency calculations were carried out to ensure that the optimized geometries are indeed associated with local minima or saddle points on the potential energy surfaces, and to determine the zero-point vibration energies and thermal corrections to the Gibbs free energies. Intrinsic reaction coordinate (IRC)69,70 calculations were performed at the same B3LYP/B1 level to verify the expected connection of the first-order saddle point with the two local minima found on the potential energy surface. The geometries optimized at the B3LYP/B1 level were used to carry out single-point energy calculations subsequently using the B3LYP functional with larger basis sets, i.e. triple-ζ LACV3P+* basis set67,68 on iron and 6-311+G* basis set on all the other atoms (denoted by B2). The weak polarization effect of the protein environment and the strong polarization effect of the nonenzymatic environment were modeled using the PCM solvation method with dielectric constants of ε = 5.62 (chlorobenzene) and ε = 78.39 (water) separately. The effect of hydrogen bond between the protein backbone and the sulfur ligand was mimicked by adding two ammonia molecules to the system which point toward the sulfur of the proximal ligand at fixed distances of rNH⋯S = 2.660 Å.47-49,56-59,71
We note that B3LYP was considerd to be dubitable in general on its reliability for transition metal complexes.72-74 It was suggested74,75 that the safest way to approach a problem requiring the identification of the ground spin state of a species was to calculate this property with more than one functional, preferably a GGA-type functional and a hybrid functional. In case of large differences between different sets of results, caution was required, and in the absence of other evidence, the most reliable way to make the prediction would be to use a functional such as B3LYP* with 15% Hartree-Fock exchange.76,77 Nevertheless, there were extensive exceptions, B3LYP functional performed well for hexacoordinate Fe complexes,78,79 especially for heme derivatives.59,80-84 The Cpd I model, computational methods and basis sets used in this study had been extensively used and proved to be sufficient to obtain reliable results for the reactions catalyzed by cytochrome P450 enzymes.59,82-84 Hence, in the present study, all calculations were carried out by using the B3LYP method. In order to further examine the computational results, we also used some other density functional, including BP86, B3LYP*, and B3PW91, to evaluate the energy barriers for the rate-determining steps.
Results and Discussion
Figures 1 and 2 show the calculated energy profiles for the formation of 5′-hydroxynicotine and N-(hydroxymethyl)nornicotine, whereas the geometries of the corresponding 4/2TSH and 4/2TSreb species are depicted in Figures 3 and 4. Here, we note that the superscript 4/2 represents the quartet (HS) or doublet (LS) spin state of the system. As usual for P450 reactions, the mechanisms involve two spin states nascent from the degenerate state of Cpd I. On both spin states, the reaction involves two steps, namely an HAT step followed by a rebound step.
Figure 1.
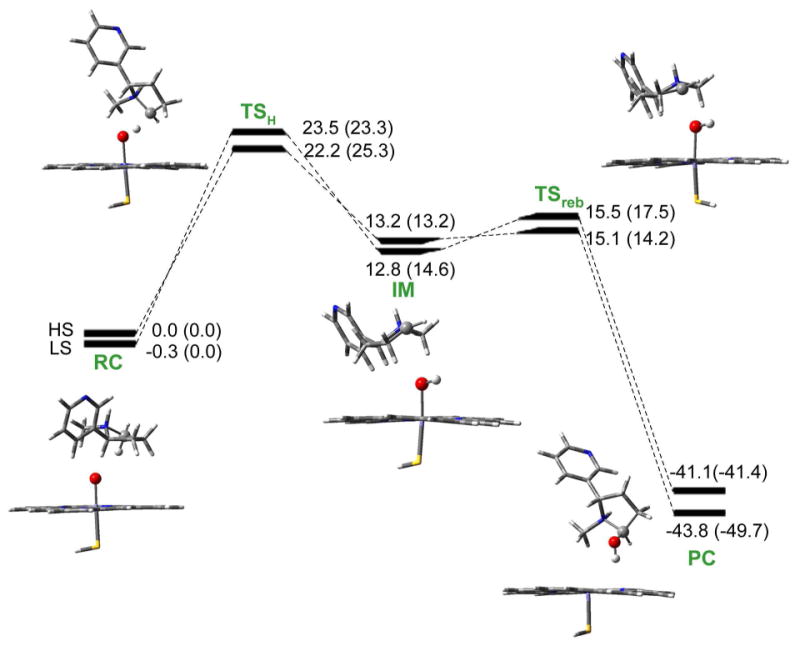
Energy profiles with zero-point corrections for 5′-hydroxylation of nicotine by Cpd I. Values outside the parentheses are relative energies in the gas phase, while the values in parentheses involve effects of bulk polarity and NH⋯S hydrogen bond. RC: reactant complex. TS: transition state. IM: intermediate. PC: product complex. All energetic values are in kcal/mol relative to 4RC (quartet spin state of RC).
Figure 2.
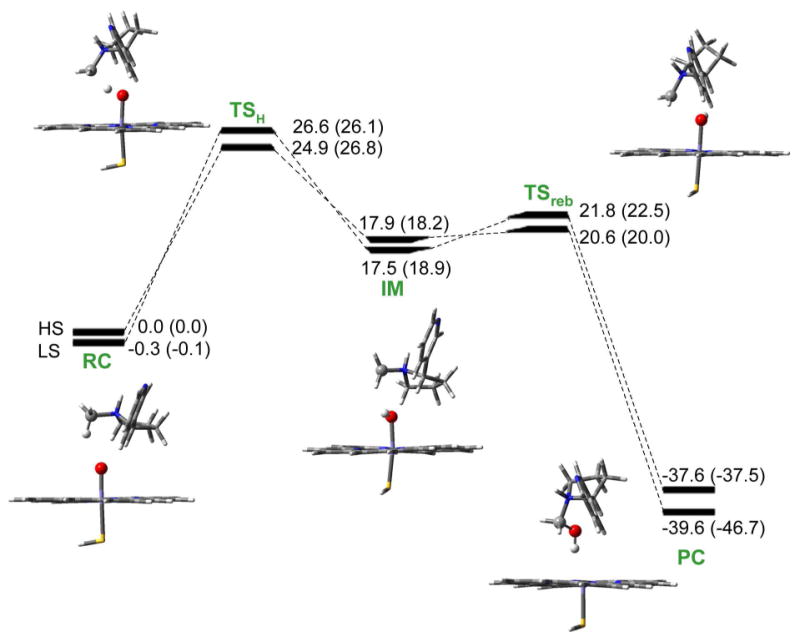
Energy profiles with zero-point corrections for N-methylhydroxylation of nicotine by Cpd I. Values outside the parentheses are relative energies in the gas phase, while the values in parentheses involve effects of bulk polarity and NH⋯S hydrogen bond. RC: reactant complex. TS: transition state. IM: intermediate. PC: product complex. All values are in kcal/mol relative to 4RC.
Figure 3.
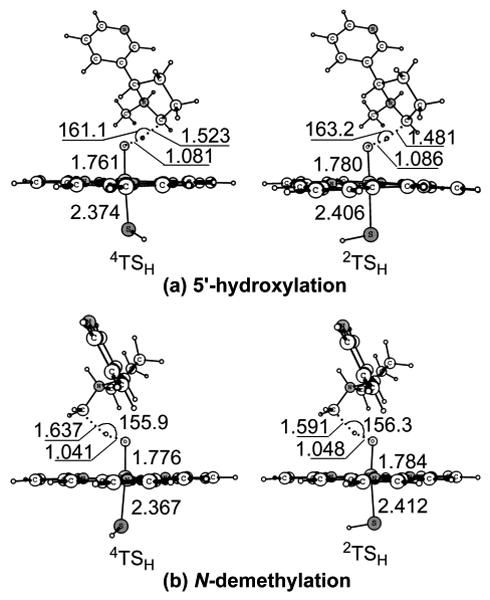
Key geometric features of the transition states optimized at the B3LYP/B1 level for the hydrogen atom transfer step. Bond lengths are in angstrom and bond angles in degrees.
Figure 4.
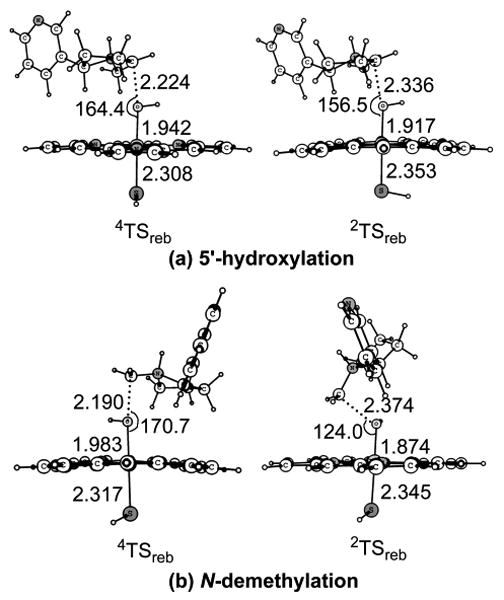
Key geometric features of the transition states optimized at the B3LYP/B1 level for the rebound step. Bond lengths are in angstrom and bond angles in degrees.
Hydrogen transfer from nicotine to Cpd I
The first step of nicotine Cα-H hydroxylation is an HAT step. In 5′-hydroxylation pathway, as seen in Figure 1, the two reactive states of Cpd I initially form two closely lying reactant complexes with nicotine, 4/2RC. This is followed by a hydrogen transfer from 5′-carbon of nicotine to the oxygen of Cpd I via a pair of transition states, 4/2TSH. The hydrogen transfer transition states 4/2TSH lead to the corresponding intermediates, 4/2IM, where the nicotine moiety is weakly coordinated to the hydroxyl group of the iron-hydroxo complex. In the gas phase, the calculated HS and LS energy barriers are 22.2 and 23.5 kcal/mol, respectively. When the corrections of the bulk polarity and the NH⋯S hydrogen bond capability of the protein pocket are taken into account, the calculated corresponding energy barriers are 25.3 and 23.3 kcal/mol. The barrier difference between the two states is 2.0 kcal/mol. With this barrier difference, the reaction proceeds mostly through the LS state.
For N-methylhydroxylation, as seen in Figure 2, the first step is also a hydrogen transfer process forming the intermediates. The calculated energy barriers are 24.9/26.6 kcal/mol for the HS/LS state in the gas phase, and become 26.8/26.1 kcal/mol when the environmental effects are added. The reaction proceeds in a TSR mechanism, and LS state is favored by 0.7 kcal/mol.
The key geometric features of the optimized HAT transition states for the two reaction pathways are depicted in Figure 3. The transition states are hydrogen-transfer species with partially broken C-H bond and partially formed O-H bond. In the optimized geometries of 4/2TSH, the C-H-O angles are 161.1°/163.2° for the 5′-hydroxylation pathway and 155.9°/156.3° for the N-demethylation pathway. The large spin densities (ρnic = 0.59/0.61 in 4/2TSH for 5′-hydroxylation and ρnic = 0.66/0.67 in 4/2TSH for N-methylhydroxylation, see Table S6) on the nicotine moiety indicate that the transition states involve hydrogen-transfer of the radical type. The hydrogen atom transfer reaction can be further evidenced from the even larger spin densities (ρnic = 1.00/1.00 in 4/2IM for 5′-hydroxylation and ρnic = 1.00/1.00 in 4/2IM for N-methylhydroxylation, see Table S6) of the nicotine moiety in the subsequently formed intermediates. For 5′-hydroxylation, the optimized distance from the 5′-carbon to the migrating hydrogen is 1.523/1.481 Å, while the optimized distance from the migrating hydrogen to the oxygen of Cpd I is 1.081/1.086 A. For N-methylhydroxylation, the optimized distance from the N-methyl-carbon to the migrating hydrogen is 1.637/1.591 Å, while the optimized distance from the migrating hydrogen to the oxygen of Cpd I is 1.041/1.048 Å.
Recombination of the nicotine moiety with the iron-bound hydroxyl group
The second step of nicotine Cα-H hydroxylation is a rebound step. It is seen from Figures 1 and 2 that in the intermediate clusters 4/2IM, the nicotine moiety is weekly connected to the hydroxyl group of the iron-hydroxo complex. Then the intermediate clusters undergo a rebound process via transition states 4/2TSreb and produce the iron-carbinolamine product complexes. In the gas phase, the calculated rebound barriers are 1.9/2.7 kcal/mol associated with the HS/LS states for 5′-hydroxylation and are 2.7/4.3 kcal/mol for N-methylhydroxylation. When the environmental effects are taken into account, the calculated rebound barriers are 1.0/2.9 kcal/mol and 1.8/3.6 kcal/mol for 5′-hydroxylation and N-methylhydroxylation, respectively. With these low rebound barriers, the intermediates can easily go through the transition states and down to the product complexes.
Figure 4 shows the geometric information about the transition states involved in the rebound step for the two reaction pathways. In TSreb, a C-O bond gradually forms between the nicotine moiety and the hydroxyl group of the iron-hydroxo complex, while the O-Fe bond gradually breaks. In the 5′-hydroxylation pathway, the distances from the 5′-carbon to the hydroxyl group of the iron-hydroxo complex are 2.224/2.336 Å on the HS/LS states, and the corresponding angles of the Fe-O-C portion are 164.4°/156.6°. In the N-demethylation pathway, the C-O distances are 2.190/2.374 Å and the Fe-O-C angles are 170.7°/124.0° on the HS/LS states.
Based on the above discussion, Cα-H hydroxylation of nicotine by Cpd I consists of two steps as seen in Figures 1 and 2: a bond-activation hydrogen transfer step, responsible for the C-H cleavage by Cpd I, and a rebound step, whereby the nicotine moiety makes a C-O bond and generates the iron-carbinolamine product complexes. The reaction is stepwise and, after the hydrogen transfer phase, the intermediate IM undergoes a rebound process through a distinct transition state TSreb with a low energy barrier. This finding represents the first case that exhibits rebound energy barriers and genuine rebound transition state species on both the HS and LS states for Cα-H hydroxylation of amines, although the rebound barriers are quite low compared to the corresponding energy barriers for the hydrogen transfer step. The existence of the rebound energy barriers for P450-catalyzed Cα-H hydroxylation of nicotine might be attributed to the fact that nicotine is a protonated amine (with the pyrrolidine nitrogen protonated), whereas the other amines examined in previously reported studies of P450-catalyzed Cα-H hydroxylation are all in the free base form (deprotonated form). It is currently unclear whether the rebound energy barriers exist in all protonated amines for their P450-catalyzed Cα-H hydroxylation.
Since the TSH species are higher in energy than all the other successive species, and the bond-activation hydrogen transfer step has higher energy barriers than the subsequent rebound step, the rate-determining step will be the hydrogen transfer one. We shall focus on the calculated hydrogen transfer energy barriers and consider the C-H bond-activation energy barriers as the determinants of the competition of the two reaction pathways.
Summarized in Table 1 are the energy barriers for the hydrogen transfer step accounting for the effect of polarity and NH⋯S hydrogen bond to the sulfur ligand of Cpd I. For the 5′-hydroxylation, the calculated HS and LS barriers in the gas phase for the hydrogen transfer are 22.2 and 23.5 kcal/mol, respectively. The calculated energy barrier for the HS transition state is lower than that for the corresponding LS species by 1.3 kcal/mol. With this barrier difference, the reaction will proceed mostly through the HS state. The bulk polarity of the hydrophobic heme pocket does not dramatically affect the HS barrier, but the hydrogen bonding-like interaction with the sulfur ligand of Cpd I raises the HS barrier by 2.7 kcal/mol. Together, the hydrogen bonding and the bulk polarity increase the HS barrier by 3.1 kcal/mol. The hydrogen bonding and bulk polarity exert opposing effects on the LS state, so that the combination of both factors leave the LS barrier almost unchanged, only 0.2 kcal/mol lower than that in the gas phase. It follows that the bulk polarity and the NH⋯S hydrogen bond capability of the protein pocket can exert a remarkable effect on the energy barriers for the C-H activation step and invert the barrier order of the two states, and the LS state is now favored by 2.0 kcal/mol.
Table 1.
Energy barriers (in kcal/mol) with zero-point corrections calculated using B3LYP functional for the first step of nicotine Cα-H hydroxylation at different levels of theory. B3//B1 refers to the B3LYP/B2//B3LYP/B1 level.
| B2//B1 | B2//B1+solv. | B2//B1+2NH⋯S | B2//B1+2NH⋯S+solv. | |
|---|---|---|---|---|
| (a) 5′-hydroxylation | ||||
| HS | 22.2 | 22.0 | 24.9 | 25.3 |
| LS | 23.5 | 22.0 | 24.1 | 23.3 |
| (b) N-methylhydroxylation | ||||
| HS | 24.9 | 25.2 | 26.3 | 26.8 |
| LS | 26.6 | 25.9 | 26.7 | 26.1 |
For N-methylhydroxylation, the HS and LS barriers are 24.9 and 26.6 kcal/mol, respectively, in the gas phase, and the HS is preferred by 1.7 kcal/mol. As is in the 5′-hydroxylation pathway, when the environmental effects are taken into account, the HS barrier becomes higher and the LS becomes lower. The combined effects of the bulk polarity and the NH⋯S hydrogen bonding increase the HS barrier by 1.9 kcal/mol, while decreasing the LS barrier by 0.5 kcal/mol. The environmental effects bring the energy barriers of the two states closer, with a negligible difference of 0.7 kcal/mol.
The energy barriers calculated in the present study are higher than the previously found barriers for amine Cα-H hydroxylation. This is because the amines used in the previous studies are deprotonated, whereas the nicotine used in this study is a protonated one, with a proton on the pyrrolidine nitrogen. In the deprotonated amines, the lone pair of the N atom forms a πC–N orbital with the Cα atom. The πC–N orbital then conjugates with the p orbital on the aromatic group and forms a conjugated πPh-πC–N orbital.48,49 This πPh-πC–N interaction delocalizes the electron distribution of the N-C-H reaction center and, thus, stabilizes the transition state. However, the protonated nicotine in this study has a proton on the nitrogen atom of the pyrrolidine ring such that no lone pair is available on the N atom. The lack of the delocalization effect is likely the primary reason for the significantly higher energy barriers.
In order to further theoretically examine the calculated energy barriers, we also evaulated the energy barrier for the rate-determining hydrogen transfer step by using several other functional in the final DFT energy calculations. The calculated energies and barriers are collected in Table S3 and Table 2. The reactant complex 4RC is 1.0 kcal/mol higher than 2RC when the BP86 and BLYP functional are used. On the other hand, the DFT calculations using the B3LYP*, B3LYP, and B3PW91 functionals predict a degeneration of 4RC and 2RC (see Table S3), and the degeneration of HS and LS reactant complex is commonly observed in calculated cytochrome P450 reactions.82,83 Thus, the GGA functionals (BP86 and BLYP) overestimated the stability of the HS state, whereas the hybrid functionals B3LYP*, B3LYP, and B3PW91 led to similar stabilities of the reactant complexes on HS and LS states. It is seen from Table 2 that the energy barriers calculated using the BP86 and BLYP functionals are systematically lower compared with the hybrid functionals. Energy barriers calculated using the B3LYP*, B3LYP, and B3PW91 functionals are of the similar values. By using different density functional, the calculated differences between the energy barriers for the 5′-hydroxylation and N-methylhydroxylation rang from 2.7 to 3.5 kcal/mol, as one can see in Table 2. Thus, no matter what functional is used, the DFT calculations can consistently predict that 5′-hydroxylation is significantly favored than N-methylhydroxylation in the metabolism of nicotine.
Table 2.
Energy barriers (in kcal/mol) in the gas phase with zero-point corrections calculated for the first step of nicotine Cα-H hydroxylation using different functional with B2 basis set.
| (a) 5′-hydroxylation | (b) N-methylhydroxylation | Barrier differencea | |||
|---|---|---|---|---|---|
| HS | LS | HS | LS | ||
| BP86 | 15.4 | 17.0 | 18.9 | 21.0 | 3.5 |
| BLYP | 18.8 | 20.2 | 21.6 | 23.3 | 2.8 |
| B3LYP* | 20.9 | 23.5 | 23.6 | 26.5 | 2.7 |
| B3LYP | 22.2 | 23.5 | 24.9 | 26.6 | 2.7 |
| B3PW91 | 20.0 | 22.6 | 23.2 | 26.1 | 3.2 |
The difference between the lowest barriers calculated for the two reaction pathways.
The predominance of 5′-hydroxylation over N-methylhydroxylation also exists when entropic contributions are taken into account. Table 3 shows the free energy barriers with thermal corrections calculated for the hydrogen-transfer step of nicotine Cα-H hydroxylation. The calculated 5′-hydroxylation barrier (25.8 kcal/mol) is lower than the N-methylhydroxylation barrier (27.5 kcal/mol) by 1.7 kcal/mol.
Table 3.
Free energy barriers (in kcal/mol) calculated at the B3LYP/B2//B3LYP/B1+2NH⋯S+solv level (with thermal corrections) for the first step of nicotine Cα-H hydroxylation.
| (a) 5′-hydroxylation | (b) N-methylhydroxylation | ||
|---|---|---|---|
| HS | 26.7 | HS | 27.6 |
| LS | 25.8 | LS | 27.5 |
Based on the calculated free energy barrier, the corresponding reaction rate constants can be estimated by using the conventional transition-state theory (CTST),85 i.e.,
| (1) |
where kB is the Boltzmann constant, T is the absolute temperature, h is Plack's constant, and ΔGav is the free energy barrier. Thus the reaction rate ratio of the two reaction pathways can be estimated as
| (2) |
If we assume that the product abundance of the 5′-hydroxylation and N-methylhydroxylation are determined by the relative free energy barriers for the rate-determining hydrogen transfer step (25.8 kcal/mol vs. 27.5 kcal/mol), then the computational data discussed here predict a preponderance of 5′-hydroxynicotine over the N-(hydroxymethyl)nornicotine by roughly a factor of 18:1. In experimental studies, 5′-hydroxynicotine and N-(hydroxymethyl)nornicotine were not stable. 5′-hydroxynicotine existed in equilibrium with △1′(5′)-iminium ion, and N-(hydroxymethyl)nornicotine existed in equilibrium with the N-methylene-iminium ion. The iminium ion species were both trapped as the corresponding 5′-cyano and N-cyanomethyl adducts by the inclusion of cyanide ion in the reaction mixture.33-35,40,86,87 For both in vivo and in vitro studies, 5′-hydroxynicotine was further oxidized to cotinine by a cytosolic aldehyde oxidase, and this step was much quicker than the hydroxylation one.41-43 On the other hand, N-(hydroxymethyl)nornicotine was believed to undergo spontaneous breakdown to nornicotine and formaldehyde in smokers.4,36 Thus, we might expect the 18:1 ratio for the rate-determining step predicted in our calculations to be conserved and continue onward to the cotinine and nornicotine products of nicotine metabolism.
Experimental studies on nicotine oxidation by CYP2A6 had been performed by many researchers using different sources of enzyme. The experimentally derived free energy barrier (using Eq 1) for nicotine oxidation is 19∼23 kcal/mol.13,15,16,54 Our calculated free energy barriers, i.e. 25.8 kcal/mol for 5′-hydroxylation and 27.5 kcal/mol for N-methylhydroxylation, are reasonably close to the up limit (∼23 kcal/mol) of the experimental values. The calculated free energy barriers being higher than the experimental ones is probably due to the fact that the calculations did not account for all of the protein environment in the actual enzymatic reaction system, although the bulk polarity and NH⋯S hydrogen bonding were taken into account. The experimental studies showed in common that 5′-hydroxylation should be the primary pathway of nicotine metabolism by the P450 enzyme.16,35,51-54 The most recent experimental data on CYP 2A6-catalyzed nicotine metabolism showed that in the presence of human liver cytosol the ratio of the cotinine to nornicotine products is about 19:1.16 Our predicted product ratio, 18:1, is very close to the experimental ratio.
Decomposition of N-(hydroxymethyl)nornicotine
In the N-demethylation pathway of nicotine metabolism, N-(hydroxymethyl)nornicotine is formed at the end of N-methylhydroxylation, and the next step is N-(hydroxymethyl)nornicotine decomposition to yield nornicotine and formaldehyde. The conventional concept about this process is a spontaneous reaction in a nonenzymatic environment. Thus, we examined this reaction step without any part of the enzyme. According to the reaction coordinate calculations, decomposition of N-(hydroxymethyl)nornicotine involves a proton transfer from the hydroxyl oxygen to the pyrrolidine nitrogen, while the C-N bond between the hydroxymethyl carbon and pyrrolidine nitrogen gradually breaks.
The N-(hydroxymethyl)nornicotine formed at the end of N-methylhydroxylation is protonated on the pyrrolidine nitrogen. Since the decomposition of N-(hydroxymethyl)nornicotine requires a proton transfer from the hydroxyl oxygen to the pyrrolidine nitrogen, this migrating proton would be hindered by the proton which already exists on the nitrogen atom and have to attack the nitrogen atom through other sides (see TS structures in Figure 5). In aqueous solution, protonated N-(hydroxymethyl)nornicotine can interchange with the deprotonated one and is able to reach a thermodynamic equilibrium. Thus, we also considered the decomposition of deprotonated N-(hydroxymethyl)nornicotine. We examined two different processes for N-(hydroxymethyl)nornicotine decomposition: one associated with a direct proton transfer, and the other associated with a water-assisted proton transfer.
Figure 5.
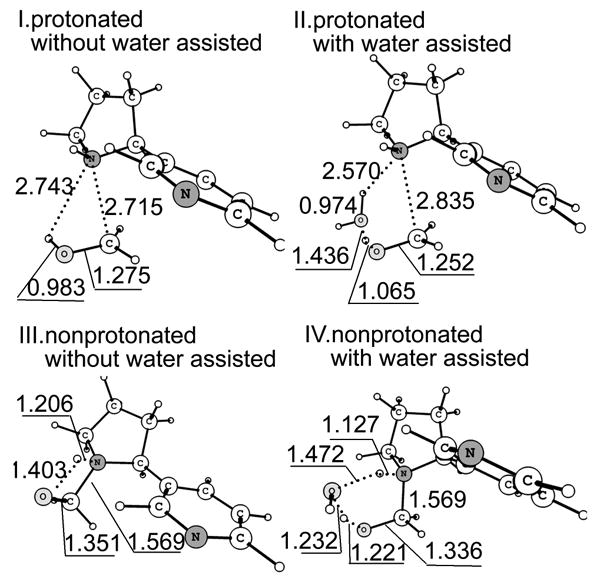
Optimized geometries of the transition states for protonated/deprotonated N-(hydroxymethyl)nornicotine decomposition with/without water assisted. Bond lengths are in angstrom and bond angles in degrees.
Collected in Table 4 are the energetic effects brought by the water molecule on the decomposition of protonated and deprotonated N-(hydroxymethyl)nornicotine, whereas depicted in Figure 5 are the structures of the corresponding TS species. It should be pointed out that bulk solvent effects on ionic solutes are usually much stronger than those on the neutral solutes with similar molecular sizes. However, the data summarized in Table 4 indicate that the bulk solvent effect on the energy barrier for the decomposition of deprotonated N-(hydroxymethyl)nornicotine is much stronger than that on the corresponding energy barrier for the decomposition of protonated N-(hydroxymethyl)nornicotine. This is because the bulk solvent stabilizes RC and TS very similarly for the decomposition of protonated N-(hydroxymethyl)nornicotine (-71.6 and -71.2 kcal/mol for RC and TS, respectively, without water assisted, -71.4 and -70.1 kcal/mol for RC and TS, respectively, with water assisted). In contrast, the bulk solvent stabilizes RC and TS rather differently for the decomposition of deprotonated N-(hydroxymethyl)nornicotine (-14.8 and -25.2 kcal/mol for RC and TS, respectively, without water assisted, -19.5 and -27.9 kcal/mol for RC and TS, respectively, with water assisted).
Table 4.
Energy barriers (in kcal/mol) with zero-point corrections calculated for decomposition of protonated/deprotonated N-(hydroxymethyl)nornicotine with/without water assisted.
| Gas phase | Solution | |
|---|---|---|
| Protonated N-(hydroxymethyl)nornicotine | ||
|
| ||
| without water | 44.3 | 44.7 |
| with water | 36.1 | 37.5 |
|
| ||
| Deprotonated N-(hydroxymethyl)nornicotine | ||
|
| ||
| without water | 39.7 | 29.2 |
| with water | 18.2 | 9.8 |
As seen in Table 4, for protonated N-(hydroxymethyl)nornicotine, the activation energy in the direct proton-transfer process is 44.7 kcal/mol when the solvation effect is take into account, whereas in the water-assisted reaction pathway the energy barrier decreases to 37.5 kcal/mol. For deprotonated N-(hydroxymethyl)nornicotine, the direct proton-transfer barrier is 29.2 kcal/mol and the water-assisted proton-transfer barrier is only 9.8 kcal/mol. These energetic results suggest that for both protonated and deprotonated N-(hydroxymethyl)nornicotine the water molecule significantly decreases the energy barrier. Further, the energy barrier for the protonated N-(hydroxymethyl)nornicotine is always much higher than that for the deprotonated one. Thus, the computational data indicate that the decomposition of N-(hydroxymethyl)nornicotine will occur through the deprotonated species and be assisted by a water molecule.
The significant decrease of the energy barrier caused by the water molecule can also be derived from the structures of the transition states shown in Figure 5. When a water molecule is added to the reaction system, the structure of the transition state changes from a strained quadrangle to a nearly strainless hexagon. The energy barrier for the deprotonated N-(hydroxymethyl)nornicotine decomposition is much lower than the former steps. With this low energy barrier, N-(hydroxymethyl)nornicotine can easily decompose to nornicotine and formaldehyde without any enzyme.
Conclusion
We performed a series of first-principle electronic structure calculations to examine the fundamental reaction pathways for 5′-hydroxylation and N-demethylation of nicotine catalyzed by the active species of P450 enzyme, Cpd I. Our calculations indicate that on both the high-spin (HS) quartet and low-spin (LS) doublet states 5′-hydroxylation of nicotine occurs through a stepwise process, i.e. a bond-activation hydrogen transfer step, responsible for the C-H cleavage by Cpd I, and a rebound step, whereby the nicotine moiety makes a C-O bond and generates the iron-carbinolamine product complexes. The reaction follows a two-state reactivity (TSR) mechanism. The hydrogen transfer step is the rate-determining one, since it has higher energy barriers than the subsequent rebound step. This is the first case that exhibits rebound barriers and genuine rebound transition state species on both the HS and LS states for Cα-H hydroxylation of amines, although the rebound barriers are quite low compared to those for the hydrogen transfer step. N-demethylation of nicotine involves a N-methylhydroxylation followed by the decomposition of N-(hydroxymethyl)nornicotine. The N-methylhydroxylation process is similar to the 5′-hydroxylation, namely that a rate-determining hydrogen transfer step in a TSR mechanism followed by the rebound step. Comparison of the free energy barriers for the rate-determining hydrogen transfer step reveals that 5′-hydroxylation is significantly more favored than N-demethylation, by a ratio of 18:1. This is in accord with the experimental observation that product formation occurs much faster at the 5′-methylene group than at the N-methyl group of nicotine. In the N-demethylation reaction pathway, after the N-methylhydroxylation process, N-(hydroxymethyl)nornicotine decomposes to nornicotine and formaldehyde with a very low barrier. This decomposition process occurs on the deprontonated N-(hydroxymethyl)nornicotine species and is assisted by a water molecule.
Supplementary Material
Acknowledgments
This work was supported in part by NIH (grants R01DA013930 and R01DA025100 to C.-G. Zhan), NKBRSF (grant 2007CB815202 to K. Han), and NSFC (grants 20721004 and 20833008 to K Han). D. Li worked in C.-G. Zhan's laboratory at the University of Kentucky as an exchange graduate student from Dalian Institute of Chemical Physics, Chinese Academy of Sciences and she is in the Ph.D. grogram of the Graduate School of the Chinese Academy of Sciences. The authors also acknowledge the Center for Computational Sciences (CCS) at University of Kentucky for supercomputing time on IBM X-series Cluster with 340 nodes or 1,360 processors.
Footnotes
Supporting Information Available: Ten tables showing the energies, free energies, Mulliken spin densities, imaginary frequencies, and Cartesian coordinates in the two reaction pathways. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.WHO Report on the Global Tobacco Epidemic, 2008. World Health Organization (WHO); 2008. [Google Scholar]
- 2.Hatsukami DK, Stead LF, Gupta PC. Lancet. 2008;371:2027. doi: 10.1016/S0140-6736(08)60871-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reus VL, Smith BJ. Int J Clin Pract. 2008;62:1753. doi: 10.1111/j.1742-1241.2008.01885.x. [DOI] [PubMed] [Google Scholar]
- 4.Hukkanen J, Jacob P, Benowitz NL. Pharmacol Rev. 2005;57:79. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- 5.Glynn DA, Cryan JF, Kent P, Flynn RA, Kennedy MP. Adv Ther. 2009;26:369. doi: 10.1007/s12325-009-0022-9. [DOI] [PubMed] [Google Scholar]
- 6.Scherer G. Psychopharmacology (Berl) 1999;145:1. doi: 10.1007/s002130051027. [DOI] [PubMed] [Google Scholar]
- 7.Hogg RC, Bertrand D. Science. 2004;306:983. doi: 10.1126/science.1106030. [DOI] [PubMed] [Google Scholar]
- 8.Henningfield JE, Miyasato K, Jasinski DR. J Pharmacol Exp Ther. 1985;234:1. [PubMed] [Google Scholar]
- 9.Zins BJ, Sandborn WJ, Mays DC, Lawson GM, McKinney JA, Tremaine WJ, Mahoney DW, Zinsmeister AR, Hurt RD, Offord KP, Lipsky JJ. J Clin Pharmacol. 1997;37:426. doi: 10.1002/j.1552-4604.1997.tb04320.x. [DOI] [PubMed] [Google Scholar]
- 10.Russell MAH, Feyerabend C. Drug Metab Rev. 1978;8:29. doi: 10.3109/03602537808993776. [DOI] [PubMed] [Google Scholar]
- 11.Henningfield JE, Goldberg SR. Pharmacol Biochem Behav. 1988;30:221. doi: 10.1016/0091-3057(88)90449-2. [DOI] [PubMed] [Google Scholar]
- 12.Cashman JR, Park SB, Yang ZC, Wrighton SA, Jacob P, Benowitz NL. Chem Res Toxicol. 1992;5:639. doi: 10.1021/tx00029a008. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, Funae Y, Shimada N, Kamataki T, Kuroiwa Y. Drug Metab Dispos. 1996;24:1212. [PubMed] [Google Scholar]
- 14.Messina ES, Tyndale RF, Sellers EM. J Pharmacol Exp Ther. 1997;282:1608. [PubMed] [Google Scholar]
- 15.Yamazaki H, Inoue K, Hashimoto M, Shimada T. Arch Toxicol. 1999;73:65. doi: 10.1007/s002040050588. [DOI] [PubMed] [Google Scholar]
- 16.Murphy SE, Raulinaitis V, Brown KM. Drug Metab Dispos. 2005;33:1166. doi: 10.1124/dmd.105.004549. [DOI] [PubMed] [Google Scholar]
- 17.Tyndale RF, Sellers EM. Drug Metab Dispos. 2001;29:548. [PubMed] [Google Scholar]
- 18.Pianezza ML, Sellers EM, Tyndale RF. Nature. 1998;393:750. doi: 10.1038/31623. [DOI] [PubMed] [Google Scholar]
- 19.Oscarson M. Drug Metab Dispos. 2001;29:91. [PubMed] [Google Scholar]
- 20.Malaiyandi V, Sellers EM, Tyndale RF. Clin Pharmacol Ther. 2005;77:145. doi: 10.1016/j.clpt.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Sellers EM, Kaplan HL, Tyndale RF. Clin Pharmacol Ther. 2000;68:35. doi: 10.1067/mcp.2000.107651. [DOI] [PubMed] [Google Scholar]
- 22.Yano JK, Denton TT, Cerny MA, Zhang XD, Johnson EF, Cashman JR. J Med Chem. 2006;49:6987. doi: 10.1021/jm060519r. [DOI] [PubMed] [Google Scholar]
- 23.Denton TT, Zhang X, Cashman JR. J Med Chem. 2005;48:224. doi: 10.1021/jm049696n. [DOI] [PubMed] [Google Scholar]
- 24.Rahnasto M, Wittekindt C, Juvonen RO, Turpeinen M, Petsalo A, Pelkonen O, Poso A, Stahl G, Holtje HD, Raunio H. Pharmacogenom J. 2008;8:328. doi: 10.1038/sj.tpj.6500481. [DOI] [PubMed] [Google Scholar]
- 25.Sellers EM, Ramamoorthy Y, Zeman MV, Djordjevic MV, Tyndale RF. Nicotine Tobacco Res. 2003;5:891. doi: 10.1080/14622200310001615231. [DOI] [PubMed] [Google Scholar]
- 26.Zhang W, Kilicarslan T, Tyndale RF, Sellers EM. Drug Metab Dispos. 2001;29:897. [PubMed] [Google Scholar]
- 27.Ono S, Hatanaka T, Hotta H, Satoh T, Gonzalez FJ, Tsutsui M. Xenobiotica. 1996;26:681. doi: 10.3109/00498259609046742. [DOI] [PubMed] [Google Scholar]
- 28.Koenigs LL, Trager WF. Biochemistry. 1998;37:13184. doi: 10.1021/bi981198r. [DOI] [PubMed] [Google Scholar]
- 29.Tantcheva-Poor I, Servera-Llaneras M, Scharffetter-Kochanek K, Fuhr U. Br J Dermatol. 2001;144:1127. doi: 10.1046/j.1365-2133.2001.04233.x. [DOI] [PubMed] [Google Scholar]
- 30.Nides M. Am J Med. 2008;121:S20. doi: 10.1016/j.amjmed.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Buchhalter AR, Fant RV, Henningfield JE. Drugs. 2008;68:1067. doi: 10.2165/00003495-200868080-00005. [DOI] [PubMed] [Google Scholar]
- 32.Hucker HB, Gillette JR, Brodie BB. J Pharmacol Exp Ther. 1960;129:94. [PubMed] [Google Scholar]
- 33.Murphy PJ. J Biol Chem. 1973;248:2796. [PubMed] [Google Scholar]
- 34.Nguyen TL, Gruenke LD, Castagnoli N. J Med Chem. 1976;19:1168. doi: 10.1021/jm00231a018. [DOI] [PubMed] [Google Scholar]
- 35.Peterson LA, Castagnoli N. J Med Chem. 1988;31:637. doi: 10.1021/jm00398a024. [DOI] [PubMed] [Google Scholar]
- 36.Kyerematen GA, Vesell ES. Drug Metab Rev. 1991;23:3. doi: 10.3109/03602539109029754. [DOI] [PubMed] [Google Scholar]
- 37.Neurath GB. Clin Investig. 1994;72:190. doi: 10.1007/BF00189309. [DOI] [PubMed] [Google Scholar]
- 38.Tricker AR. Toxicology. 2003;183:151. doi: 10.1016/s0300-483x(02)00513-9. [DOI] [PubMed] [Google Scholar]
- 39.Hecht SS, Hochalter JB, Villalta PW, Murphy SE. Proc Natl Acad Sci U S A. 2000;97:12493. doi: 10.1073/pnas.220207697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peterson LA, Trevor A, Castagnoli N. J Med Chem. 1987;30:249. doi: 10.1021/jm00385a004. [DOI] [PubMed] [Google Scholar]
- 41.Brandänge S, Lindblom L. Biochem Biophys Res Commun. 1979;91:991. doi: 10.1016/0006-291x(79)91977-6. [DOI] [PubMed] [Google Scholar]
- 42.Gorrod JW, Hibberd AR. Eur J Drug Metab Pharmacokinet. 1982;7:293. doi: 10.1007/BF03189632. [DOI] [PubMed] [Google Scholar]
- 43.Obach RS. Drug Metab Dispos. 2004;32:89. doi: 10.1124/dmd.32.1.89. [DOI] [PubMed] [Google Scholar]
- 44.Ogliaro F, Harris N, Cohen S, Filatov M, de Visser SP, Shaik S. J Am Chem Soc. 2000;122:8977. [Google Scholar]
- 45.Harris N, Cohen S, Filatov M, Ogliaro F, Shaik S. Angew Chem Int Ed. 2000;39:2003. doi: 10.1002/1521-3773(20000602)39:11<2003::aid-anie2003>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 46.Shaik S, Cohen S, Visser SPd, Sharma PK, Kumar D, Kozuch S, Ogliaro F, Danovich D. Eur J Inorg Chem. 2004:207. [Google Scholar]
- 47.Li C, Wu W, Kumar D, Shaik S. J Am Chem Soc. 2006;128:394. doi: 10.1021/ja055987p. [DOI] [PubMed] [Google Scholar]
- 48.Wang Y, Kumar D, Yang C, Han K, Shaik S. J Phys Chem B. 2007;111:7700. doi: 10.1021/jp072347v. [DOI] [PubMed] [Google Scholar]
- 49.Li D, Wang Y, Yang C, Han K. Dalton Trans. 2009:291. doi: 10.1039/b810767j. [DOI] [PubMed] [Google Scholar]
- 50.Molinie R, Kwiecien RA, Paneth P, Hatton W, Lebreton J, Robins RJ. Arch Biochem Biophys. 2007;458:175. doi: 10.1016/j.abb.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 51.McCoy GD, Demarco GJ, Koop DR. Biochem Pharmacol. 1989;38:1185. doi: 10.1016/0006-2952(89)90267-0. [DOI] [PubMed] [Google Scholar]
- 52.Williams DE, Shigenaga MK, Castagnoli N., Jr Drug Metab Dispos. 1990;18:418. [PubMed] [Google Scholar]
- 53.Jones JP, Trager WF, Carlson TJ. J Am Chem Soc. 1993;115:381. [Google Scholar]
- 54.von Weymarn LB, Brown KM, Murphy SE. J Pharmacol Exp Ther. 2006;316:295. doi: 10.1124/jpet.105.091306. [DOI] [PubMed] [Google Scholar]
- 55.Huang XQ, Zheng F, Crooks PA, Dwoskin LP, Zhan CG. J Am Chem Soc. 2005;127:14401. doi: 10.1021/ja052681+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ogliaro F, Cohen S, de Visser SP, Shaik S. J Am Chem Soc. 2000;122:12892. [Google Scholar]
- 57.de Visser SP, Ogliaro F, Sharma PK, Shaik S. J Am Chem Soc. 2002;124:11809. doi: 10.1021/ja026872d. [DOI] [PubMed] [Google Scholar]
- 58.Kumar D, de Visser SP, Sharma PK, Cohen S, Shaik S. J Am Chem Soc. 2004;126:1907. doi: 10.1021/ja039439s. [DOI] [PubMed] [Google Scholar]
- 59.Hirao H, Kumar D, Thiel W, Shaik S. J Am Chem Soc. 2005;127:13007. doi: 10.1021/ja053847+. [DOI] [PubMed] [Google Scholar]
- 60.Yoshizawa K, Kagawa Y, Shiota Y. J Phys Chem B. 2000;104:12365. [Google Scholar]
- 61.Yoshizawa K, Kamachi T, Shiota Y. J Am Chem Soc. 2001;123:9806. doi: 10.1021/ja010593t. [DOI] [PubMed] [Google Scholar]
- 62.Lee C, Yang W, Parr RG. Physical Review B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 63.Becke AD. J Chem Phys. 1992;96:2155. [Google Scholar]
- 64.Becke AD. J Chem Phys. 1992;97:9173. [Google Scholar]
- 65.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 66.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. revision C.02. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 67.Hay JP, Wadt WR. J Chem Phys. 1985;82:299. [Google Scholar]
- 68.Friesner RA, Murphy RB, Beachy MD, Ringnalda MN, Pollard WT, Dunietz BD, Cao Y. J Phys Chem A. 1999;103:1913. [Google Scholar]
- 69.Gonzalez C, Schlegel HB. J Chem Phys. 1989;90:2154. [Google Scholar]
- 70.Gonzalez C, Schlegel HB. J Phys Chem. 1990;94:5523. [Google Scholar]
- 71.de Visser SP, Ogliaro F, Sharma PK, Shaik S. Angew Chem Int Ed. 2002;41:1947. [PubMed] [Google Scholar]
- 72.Sousa SrF, Fernandes PA, Ramos MJo. J Phys Chem A. 2007;111:10439. doi: 10.1021/jp0734474. [DOI] [PubMed] [Google Scholar]
- 73.Paier J, Marsman M, Kresse G. J Chem Phys. 2007;127:024103. doi: 10.1063/1.2747249. [DOI] [PubMed] [Google Scholar]
- 74.Harvey JN. Annu Rep Prog Chem, Sect C: Phys Chem. 2006;102:203. [Google Scholar]
- 75.Harvey JN. Struct Bond. 2004;112:115. [Google Scholar]
- 76.Salomon O, Reiher M, Hess BA. J Chem Phys. 2002;117:4729. [Google Scholar]
- 77.Reiher M, Salomon O, Artur Hess B. Theor Chem Acc. 2001;107:48. [Google Scholar]
- 78.Fouqueau A, Mer S, Casida ME, Daku LML, Hauser A, Mineva T, Neese F. J Chem Phys. 2004;120:9473. doi: 10.1063/1.1710046. [DOI] [PubMed] [Google Scholar]
- 79.Fouqueau A, Casida ME, Daku LML, Hauser A, Neese F. J Chem Phys. 2005;122:044110. doi: 10.1063/1.1839854. [DOI] [PubMed] [Google Scholar]
- 80.Harvey JN. J Am Chem Soc. 2000;122:12401. [Google Scholar]
- 81.Smith DMA, Dupuis M, Straatsma TP. Mol Phys. 2005;103:273. [Google Scholar]
- 82.Shaik S, Kumar D, de Visser SP, Altun A, Thie W. Chem Rev. 2005;105:2279. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 83.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 84.Altun A, Thiel W. J Phys Chem B. 2004;109:1268. doi: 10.1021/jp0459108. [DOI] [PubMed] [Google Scholar]
- 85.Alvarez-Idaboy JR, Galano A, Bravo-Perez G, Ruiz ME. J Am Chem Soc. 2001;123:8387. doi: 10.1021/ja010693z. [DOI] [PubMed] [Google Scholar]
- 86.Nguyen TL, Gruenke LD, Castagnoli N. J Med Chem. 1979;22:259. doi: 10.1021/jm00189a008. [DOI] [PubMed] [Google Scholar]
- 87.Carlson TJ, Jones JP, Peterson L, Castagnoli N, Iyer KR, Trager WF. Drug Metab Dispos. 1995;23:749. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.