

Abstract
The expression of several members of the FOX gene family is known to be altered in a variety of cancers. We show that in breast cancer, FOXF1 gene is a target of epigenetic inactivation and that its gene product exhibits tumor suppressive properties. Loss or downregulation of FOXF1 expression is associated with FOXF1 promoter hypermethylation in breast cancer cell lines and in invasive ductal carcinomas (IDCs). Methylation of FOXF1 in IDC (37.6% of 117 cases) correlated with high tumor grade. Pharmacologic unmasking of epigenetic silencing in breast cancer cells restored FOXF1 expression. Re-expression of FOXF1 in breast cancer cells with epigenetically silenced FOXF1 gene led to G1 arrest concurrent with or without apoptosis to suppress both in vitro cell growth and in vivo tumor formation. FOXF1-induced G1 arrest resulted from a blockage at G1-S transition of cell cycle through inhibition of the CDK2-RB-E2F cascade. Si-RNA-mediated depletion of FOXF1 in breast cancer cells led to increased DNA rereplication, suggesting that FOXF1 is required for maintaining the stringency of DNA replication and genomic stability. Further, expression profiling of cell-cycle regulatory genes showed that abrogation of FOXF1 function resulted in increased expression of E2F-induced genes involved in promoting progression of S and G2 phases. Therefore, our studies have identified FOXF1 as a potential tumor suppressor gene that is epigenetically silenced in breast cancer, which plays an essential role in regulating cell cycle progression to maintain genomic stability.
Keywords: FOXF1, breast cancer, epigenetic silencing
Introduction
The members of FOX gene family encode transcription factors that share a common, structurally related, DNA-binding domain called the forkhead domain (1, 2). Many members of this family have been found to play important roles in embryonic development and also in the control of cell cycle, cell growth, cellular metabolism, life span, and immune responses (3, 4). Genetic alteration and deregulation of members of several FOX subfamilies is linked to progression of certain types of cancers (5-8). A comprehensive understanding of expression profiles, genetic alterations and epigenetic changes of FOX family genes as well as their binding proteins and target genes is important to develop novel therapeutics and preventives for human diseases, especially for cancer.
The forkhead box-F1 (FOXF1) transcription factor plays a key role in regulating embryonic development. Foxf1−/− embryos exhibited defects in extraembryonic mesoderm development and died by day 8 post coitum (dpc) (9). On account of the lethal consequences of a complete loss of Foxf1 alleles, Foxf1+/− mice have been studied extensively for deciphering Foxf1 function. Haplo-insufficiency in Foxf1+/− mice resulted in various abnormalities in the mesenchyme of lung, liver, gallbladder, esophagus, and trachea (10-12). In humans, genome-wide array analysis of prostate cancer showed that FOXF1 mRNA expression was downregulated in tumors compared to normal prostate, providing a clue that FOXF1 may behave as a tumor suppressor gene (13).
In this study, we report that FOXF1 expression is aberrantly silenced in breast cancer through epigenetic mechanisms, and demonstrate that FOXF1 exerts tumor suppressor activity. Restoration of FOXF1 expression in breast cancer cells suppresses cell growth and tumorigenicity, whereas silencing of FOXF1 expression in FOXF1-expressing breast cancer cells by siRNA results in an increase in DNA rereplication, a sign of a defect in the stringent control of DNA replication. Therefore, we have identified FOXF1 as a putative tumor suppressor gene that is frequently epigenetically inactivated in breast cancer.
Materials and Methods
In silico analysis of FOXF1 gene expression
The Oncomine's Cancer Microarray Database (14) was used to perform in silico analysis of the FOXF1 gene expression in cancer versus the normal counterpart tissue.
Identification of the CpG island of FOXF1 gene
We obtained the genomic DNA sequence, including the upstream promoter sequence, of FOX1 gene from the GenBank Database of the National Center of Biotechnology Information (15). We identified the CpG-rich region at the FOXF1 gene locus by using the criteria and algorithm of online CpG Island Searcher (16).
Cell lines and tissue samples
Immortalized, nontumorigenic human mammary epithelial cells (HBL100) and ten breast cancer cell lines (obtained from the American Type Culture Collection) were cultured according to the ATCC online instructions. Mammary organoids were prepared from reduction mammoplasty specimens of normal women as previously described (17). We obtained fresh reduction mammoplasty specimens and frozen primary breast cancer samples from the Department of Pathology at the Johns Hopkins Hospital (Baltimore MD). All of human tissue specimens in this study were processed and used with prior approval from the JH Institutional Review Board.
Methylation-specific PCR and bisulfite sequencing analysis
Bisulfite modification of genomic DNA and methylation-specific PCR (MSP) amplification were performed as previously described (18, 19). The sequence information for MSP primers is provided in the Supplemental Experimental Procedures. Bisulfite sequencing analysis of the FOXF1 promoter was performed as previously described (19) by using a pair of external primers (primer sequences are available on request).
Conventional and quantitative reverse-transcription PCR (RT-PCR) analysis
Detailed methods are provided in the Supplemental Experimental Procedures.
Treatment with 5-aza-2′-deoxycytidine and/or trichostatin A
Breast cancer cell lines were treated with the regimen as previously described (19).
Immunohistochemistry assay
The immunohistochemical (IHC) analysis was performed using the avidin-biotin-peroxidase complex (ABC) method with the anti-FOXF1 antibody (Aviva antibody Corporation). See the Supplemental Experimental Procedures for details.
Statistical analysis
Comparison of clinicopathologic characteristics of invasive ductal carcinoma patients with methylation status of FOXF1 gene, and the association between FOXF1 methylation and FOXF1 protein expression was subjected to χ2 analysis using the SPSS system (version 11.5 for windows; SPSS Inc.). The Student's t-test was used to analyze the significance of difference between two groups of data. P < 0.05 was regarded as statistically significant.
Construction of the FOXF1 expression plasmid
Full-length cDNA fragments for FOXF1 gene were obtained by RT-PCR of RNA from normal mammary organoids and cloned into the pGEM-T-Easy vector (Promega). Sequence confirmed FOXF1 cDNA was subcloned into the pcDNA3.1 (−) vector (Invitrogen) with the haemagglutinin (HA)-tag coding sequence.
Transfection, Western blotting analysis and reporter assay
Details are provided in the Supplemental Experimental Procedures.
Colony formation assay
At 24 h after transfection, transfected cells were plated in 6-well plates at a density of 6,000-12,000 cells/well in complete medium containing 0.3-0.6 mg/ml geneticin (G418). After 2-3 weeks, colonies were fixed and then stained with 0.1% crystal violet. Unbound dye was washed out with water. Bound dye was then eluted with 10% acetic acid, and quantitated by measuring the absorbance at 590 nm.
Flow cytometry analysis
Detailed procedures are provided in the Supplemental Experimental Procedures.
Tumorigenicity assay
5×106 of G418-selected viable HA-FOXF1-transfected Hs578T cells were injected subcutaneously into the right rear flanks of six athymic nude mice (The Jackson Laboratory), and empty vector-transfected Hs578T cells were injected into the left rear flanks of the same mice as a control. Weekly measurements of the tumors were used to determine the tumor volume.
Immunofluorescence assay
Transfected cells on coverslips were fixed first and then were sequentially queried with primary (including anti-HA, anti-CDK2, anti-phospho-CDK2, anti-RB, anti-phospho-RB and anti-FOXF1 antibodies) and secondary (Alexa series, Invitrogen) antibodies according to the method detailed in the Supplemental Experimental Procedures.
siRNA transfection
siRNA transfections were performed with 20 nM of each siRNA (obtained from Dharmacon) using Oligofectamine™ RNAiMAX (Invitrogen) according to the instructions of the manufacturer. The FOXF1 siRNA sequence is: 5′-GAAAGGAGUUUGUCUUCUC-3′.
BrdU incorporation assay
Cells were BrdU-labeled, fixed, denatured and detected using the anti-BrdU antibody as described in the Supplemental Experimental Procedures.
Gene expression profiling of cell cycle regulatory genes
Gene expression profiling of cell cycle regulatory genes was performed using the RT Profiler PCR Array System (SABiosciences). See the Supplemental Experimental Procedures for details.
Results
Epigenetic silencing of FOXF1 gene expression in breast cancer
In silico meta-analysis of the Oncomine Cancer Microarray Database (14) was performed to examine whether FOXF1 gene expression is altered in cancer. The results showed that FOXF1 was underexpressed in various cancer types such as lung, prostate, bladder, ovarian and breast compared to their respective normal tissues (Supplemental Fig. 1 and Supplemental Table 1) (20-32). To validate these findings, we examined FOXF1 expression in a panel of breast cancer cell lines by quantitative RT-PCR (qRT-PCR). Immortalized human mammary epithelial cell line (HBL100) and three epithelial-enriched organoid samples from breast tissues were used as normal controls. Significant loss or downregulation of FOXF1 mRNA expression was observed in 9 out of 10 breast cancer cell lines compared to normal controls (Fig. 1A).
Figure 1.

Expression and methylation status of the FOXF1 gene in breast cancer cell lines. A, Quantitative RT-PCR analysis of FOXF1 expression in three mammary epithelial-enriched organoids, an immortalized HMEC line (HBL100), and 10 malignant breast cancer cell lines. B, Methylation-specific PCR (MSP) analysis of FOXF1 gene in breast cancer cell lines. The map of CpG dinucleotides in the exon 1 of FOXF1 gene and the genomic region upstream of exon 1 is shown on the top panel. The exon 1, MSP-amplified and bisulfite sequencing regions are indicated. The MSP result is shown on the bottom panel. U, unmethylated; M, methylated. C, Bisulfite sequencing analysis of FOXF1 promoter in normal breast epithelial and breast cancer cell lines. The amplified FOXF1 DNA region for sequencing analysis ranges from -209 to +62 nucleotide relative to the first nucleotide (set as the default +1) of exon 1 based on FOXF1 cDNA sequence (NM_001451). The methylation status of each CpG site is indicated by the open circle (unmethylated), partially filled circle (partially methylated) and completely filled circle (completely methylated). D, RT-PCR analysis of FOXF1 expression in breast cancer cell lines treated with 5-aza-2′-deoxycytidine (5-azaC), Trichostatin A (TSA) alone, or a combination (top panel). SKBR3 cells were treated with TSA at two different doses as indicated for 24 and 48 h (bottom panel). GAPDH expression is shown as an internal control.
As a first step to determine whether epigenetic mechanisms are involved in silencing FOXF1 we performed methylation-specific PCR (MSP). MSP detected hypermethylation of the CpG island in the FOXF1 promoter in 8 out of 10 breast cancer cell lines (Fig. 1B). MSP findings were further validated by bisulfite sequencing analysis. The FOXF1 promoter was fully methylated in MDA-MB-231 and MDA-MB-468, partially methylated in MDA-MB-435, MCF7 and T47D, and unmethylated in the normal controls and SKBR3 (Fig. 1C). With the exception of SKBR3, hypermethylation of FOXF1 showed an inverse correlation with mRNA expression in the same sample.
To further establish that epigenetic mechanisms play a role in downregulating FOXF1 expression, five hypermethylated cell lines were treated with epigenetic modifiers. Treatment of these lines with the demethylating agent, 5-aza-2′-deoxycytidine (5-azaC), or histone deacetylase inhibitor, trichostatin A (TSA), singly or in combination resulted in re-expression of FOXF1 mRNA, an effect that was enhanced in some cell lines (MDA-MB-231, MDA-MB-468 and T47D) by co-treatment with TSA (Fig. 1D). FOXF1 mRNA transcripts were also reexpressed in T47D cells by single treatment with TSA (Fig. 1D). Thus, promoter hypermethylation is the predominant mechanism of silencing gene expression in the breast cancer cell lines. TSA-treatment revived FOXF1 expression in SKBR3 cells (Fig. 1D), indicating that in this line, histone deacetylation, not DNA methylation, was responsible for silencing of FOXF1 expression.
To examine whether FOXF1 promoter hypermethylation also occurs in primary breast carcinomas, MSP analysis was performed on invasive ductal carcinomas (IDCs) (n=117), normal mammoplasty tissues (Mam) (n=8), organoids from normal reduction mammoplasty specimens (Org) (n=6) and peripheral white blood cells (WBC) (n=9) from normal individuals. The FOXF1 promoter was hypermethylated in 37.6% (44/117) of IDC tumors (Table 1), unmethylated in all normal breast tissue samples and organoids, and weakly methylated in two out of 9 WBC samples (Fig. 2A). Bisulfite sequencing analysis have further confirmed that IDC cases (IDC-4, IDC-6, IDC-8, IDC-11 and IDC-14) with positive MSP results had extensive CpG methylation throughout the analyzed genomic region compared to normal organoid and WBC DNA samples with an unmethylated CpG island (Fig. 2B). These five IDC samples consistently exhibited downregulation or loss of FOXF1 expression compared to the normal organoid (Fig. 2B). Hypermethylation of FOXF1 gene correlated with tumor grade (p=0.004) (Table 1). Thus hypermethylation of FOXF1 occurs commonly in primary breast tumors.
Table 1.
Correlation between the promoter hypermethylation of FOXF1 gene and clinicopathological characteristics of IDC tumors
| Clinicopathological characteristics1 | Meth (%)2 | p3 |
|---|---|---|
| Age | 0.483 | |
| < 50 (51) | 21 (41.2) | |
| ≥ 50 (66) | 23 (34.8) | |
| Size | 0.612 | |
| < 2 cm (46) | 16 (34.8) | |
| ≥ 2 cm (71) | 28 (39.4) | |
| Histologic grade | 0.004 | |
| G1 (15) | 1 (6.7) | |
| G2 (51) | 18 (35.3) | |
| G3 (51) | 25 (49.0) | |
| LV invasion | 0.904 | |
| Negative (55) | 21 (38.2) | |
| Positive (62) | 23 (37.1) | |
| Nodal involvement | 0.945 | |
| Negative (66) | 25 (37.9) | |
| Positive (51) | 19 (37.3) | |
| Stage | 0.557 | |
| I (34) | 13 (38.2) | |
| II (60) | 20 (33.3) | |
| III (23) | 11 (47.8) | |
| ER status | 0.829 | |
| Negative (57) | 22 (38.6) | |
| Positive (60) | 22 (36.7) | |
| PR status | 0.402 | |
| Negative (66) | 27 (40.9) | |
| Positive (51) | 17 (33.3) | |
| Her-2/neu status | 0.894 | |
| Negative (95) | 36 (37.9) | |
| Positive (22) | 8 (36.4) | |
| p53 status | 0.683 | |
| Negative (69) | 27 (39.1) | |
| Positive (48) | 17 (35.4) |
The IDC tumor sample number for each clinicopathological characteristic is shown in parenthesis. About classification systems for histologic grades and tumor stages and methods to determine the positive for ER (estrogen receptor), PR (progesterone receptor), Her-2/neu and p53 status were described in the “Supplemental Experimental Procedures” in detail.
The percentage of IDC cases with methylated FOXF1 gene is shown in parenthesis.
The p value with statistical significance is shown in bold.
Figure 2.
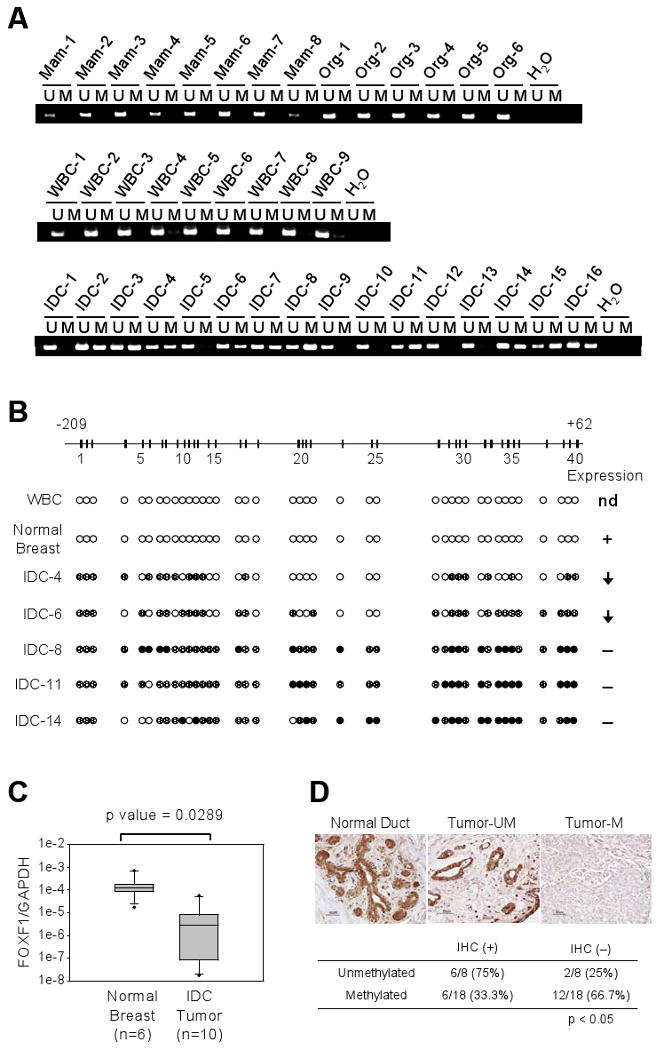
Epigenetic inactivation of FOXF1 in primary invasive breast tumors. A, MSP analysis was performed on normal mammoplasty tissues (Mam-1 to -8), normal organoids (Org-1 to -6), normal peripheral white blood cells (WBC-1 to -9) and primary invasive ductal carcinomas (IDC-1 to -16). B, Bisulfite sequencing analysis of the FOXF1 promoter in invasive ductal carcinomas. FOXF1 expression status in these primary IDC tumors is shown on the right side of the figure; arrow pointing down: underexpression, (+): expression, (−): no expression, nd: not determined. C, Quantitative RT-PCR analysis of FOXF1 expression in normal breast organoids vs. primary breast IDC tumors with methylated FOXF1 promoter. D, Immunohistochemistry analysis of FOXF1 protein expression in normal breast tissue and primary breast IDC tumors with or without methylated FOXF1 gene. A summarized result from 26 IDC samples is shown on the bottom panel.
To further explore the functional consequence of FOXF1 promoter hypermethylation, FOXF1 expression was examined in primary breast tumors. Quantitative RT-PCR analysis of 6 normal breast organoids (Org-1 to Org-6) and 10 IDC tumor tissues with methylated FOXF1 gene showed that FOXF1 mRNA was significantly underexpressed in IDC tumors (Fig. 2C) compared to normal organoids (p < 0.05). To further confirm the RT-PCR data, FOXF1 protein expression was examined by immunohistochemistry (IHC) assay in paraffin-embedded tissue sections of 26 tumors (Table 1) selected from the 117 IDC cases tested by MSP. Normal breast tissue exhibited positive immunostaining for FOXF1 protein in ductal epithelial cells (Fig. 2D). Of IDC tumors harboring methylated FOXF1 gene, 12/18 (66.7%) cases exhibited negative or focal, weak immunoreactivity for FOXF1 protein (less than 10% of total tumor cells) (Fig. 2D). Among the remaining cases, 6/18 (33.3%) showed heterogeneous positive immunostaining and were interpreted as IHC-positive. In contrast, in the unmethylated group, 6/8 (75%) were heterogeneous or homogeneous IHC-positive for FOXF1 (Fig. 2D). The inverse correlation between IHC reactivity for FOXF1 protein and methylation status for FOXF1 gene was statistically significant (p <0.05).
Collectively, evidence from promoter methylation, mRNA and protein expression studies strongly suggest that FOXF1 promoter hypermethylation correlates significantly with reduction or loss of FOXF1 expression.
FOXF1 re-expression in cancer cells inhibits cell proliferation in vitro and tumors in vivo
We elucidated the biological significance of epigenetic silencing of FOXF1 in breast cancer by performing a variety of assays in cells with enforced expression of HA-tagged FOXF1. Protein expression and nuclear localization of the CMV-HA-tagged FOXF1 construct were confirmed by Western blot and immunofluorescence (IF) analyses (data not shown). To test whether exogenously introduced FOXF1 functions as a transcription factor, reporter assays were performed using the reporter plasmid containing a minimal promoter with (p4XFREAC-Luc) or without (pApo-Luc) four tandem copies of the FOXF1 binding site (33). HA-FOXF1 protein induced reporter luciferase activity dramatically in p4XFREAC-Luc-transfected T47D and SKBR3 cells, but not in control pApo-Luc-transfected cells (Fig. 3A).
Figure 3.
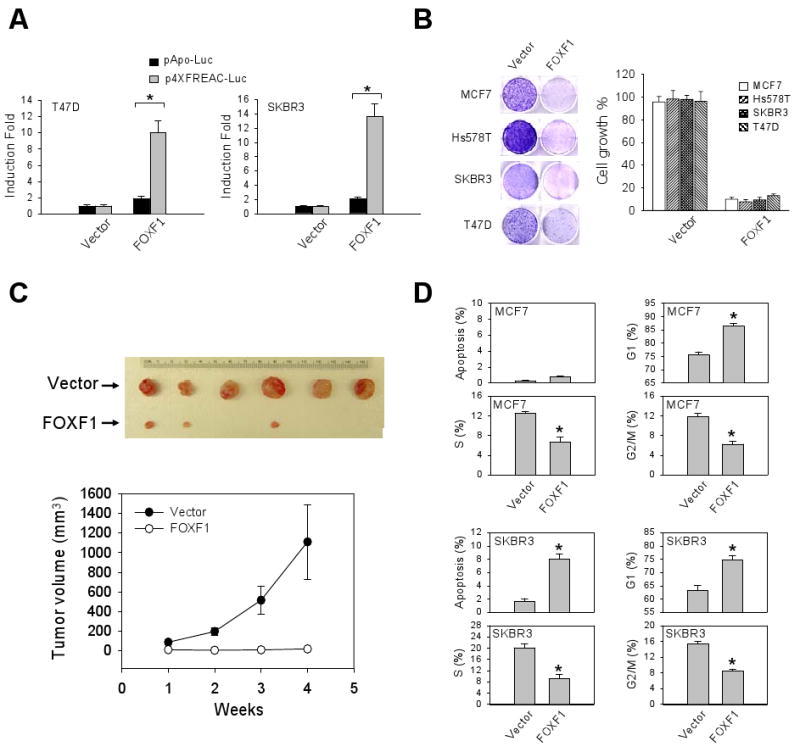
FOXF1 re-expression suppresses the growth and tumorigenicity of breast cancer cells by inducing G1 arrest and/or apoptosis. A, The transcriptional activity of HA-tagged FOXF1 in T47D and SKBR3 cells was measured by using the FOXF-responsive reporter plasmid (p4XFREAC-Luc) vs. its empty reporter control pApo-Luc. The data (mean ± SD) were generated from three independent experiments. The asterisk depicts statistical significance (p < 0.05). B, FOXF1 inhibits in vitro growth of multiple breast cancer cell lines. G418-resistant colonies were visualized by staining with crystal violet dye, shown on the left panel. Absorbance values at 590 nm of eluted binding dye are shown as the bar graph (on the right panel) based on three independent experiments. C, FOXF1 suppresses tumorigenicity of Hs578T cells. The measurements of tumor volume presented in the bottom panel are means and SD (n=6). D, Flow cytometry analysis of cell cycle profile and apoptotic status in FOXF1-transfected MCF7 and SKBR3 cells. Empty control vector or the HA-tagged FOXF1 expression plasmid DNA was contransfected with the green fluorescent protein (GFP) expression plasmid DNA into cells. The percentages of GFP-positive cells in sub-G1 (indicating apoptosis), G1, S, G2/M are mean ± SD of three independent experiments. The data indicated by an asterisk are statistically significant (p < 0.05).
To assess the effect of FOXF1 on growth of breast cancer cells, we transfected HA-tagged FOXF1-expressing plasmids into four FOXF1-negative breast cancer cell lines, including MCF7, Hs578T, SKBR3 and T47D cell lines. FOXF1 re-expression strongly suppressed growth of all these cell lines examined (Fig. 3B). Next, injection of FOXF1-expressing Hs578T cells s.c into athymic nude mice showed that tumor formation was significantly suppressed or completely blocked compared to mice injected with vector-transfected-Hs578T cells (Fig. 3C). These studies, taken together, strongly suggest that FOXF1 transcription factor possesses tumor suppressor characteristics.
To begin to decipher the mechanism whereby FOXF1 suppresses cell growth, cell cycle analysis was performed. The results showed a significant increase in the percentage of FOXF1-transfected MCF7 cells in G1 phase, indicating that FOXF1 induced G1 arrest (Fig. 3D). In addition to G1 arrest, apoptosis was concurrently observed in SKBR3 cells (Fig. 3D).
FOXF1 abrogates activation of the CDK2-RB-E2F cascade
To understand FOXF1-mediated G1 arrest, protein molecules involved in G1-S transition were examined. Phosphorylation of CDK2 at Thr-160 is known to activate CDK2 kinase activity, which in turn promotes G1-S transition (34). Immunofluorescence (IF) analysis showed that FOXF1 re-expression in MCF7 diminished phosphorylation of CDK2 at Thr-160 (Fig. 4A) and concurrently abrogated phosphorylation of RB, a downstream target of CDK2, at Ser-807/811 (Fig. 4B). FOXF1 had no significant effect on total levels of CDK2 protein (Fig. 4A), indicating FOXF1-mediated abrogation of CDK2 activity was not caused by downregulating protein levels of CDK2. In the IF analysis of total RB, a moderate decrease in total levels of RB protein was observed in FOXF1-transfected cells compared to non-transfected cells (Fig. 4B, the bottom panel), but even so, this effect could not completely account for dramatically diminishing RB phosphorylation by FOXF1 (Fig. 4B, the top panel). Further confirmation of IF data was provided by flow cytometric analysis through quantitative assessment of the overall staining intensities of phosphorylated CDK2 and phosphorylated RB in FOXF1-transfected MCF7 cells compared to vector-transfected control cells (Fig. 4C). These data, taken together, indicate that FOXF1 suppresses CDK2 activation and in turn inhibits CDK2-mediated phosphorylation of RB protein.
Figure 4.
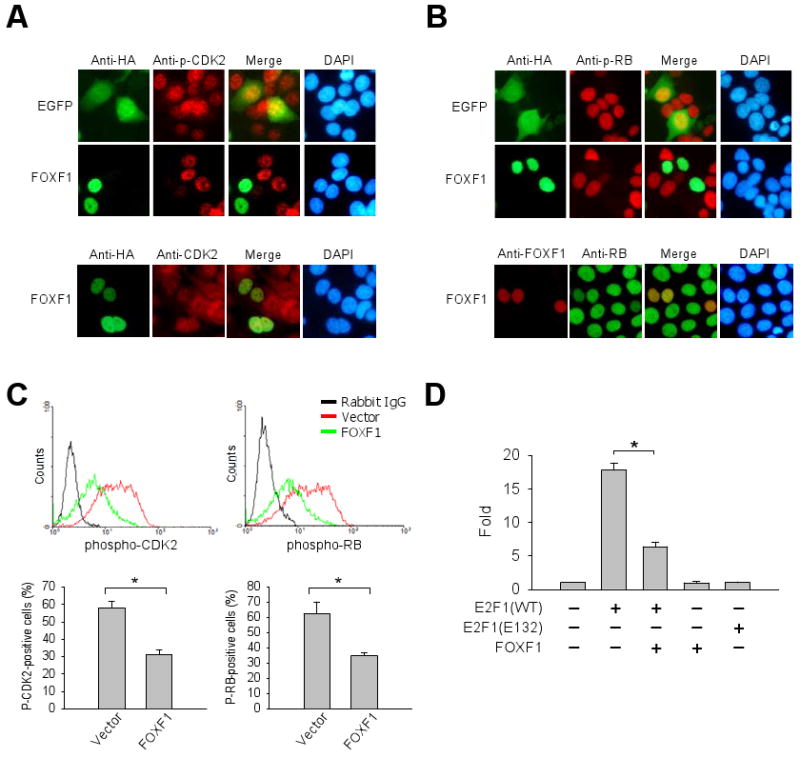
FOXF1 re-expression in breast cancer cells inhibits the CDK2-RB-E2F cascade pathway. A, Top panel: Immunofluorescence (IF) analysis of MCF7 cells, transfected with the GFP or HA-FOXF1 expression plasmid DNA, using anti-HA (green) and anti-phospho-CDK2 (Thr160) (red) antibodies. DNA was labeled with DAPI (blue). Bottom panel: IF of total CDK2 protein levels. B, IF analysis of the same cells as (A) with anti-phospho-RB (Ser807/811) (top panel) and anti-RB (bottom panel) antibodies. C, Flow cytometric analysis of the effect of FOXF1 on levels of phospho-CDK2 and phospho-RB proteins. The FACS histogram data are shown in the top panel. The bar graphs (mean ± SD) from three independent FACS experiments are shown in the bottom panel. The asterisk depicts a statistically significant difference (p < 0.05). D, FOXF1 attenuates E2F1 transcriptional activity. We transfected MCF7 cells with E2F-responsive luciferase reporter DNA and different combinations of expression plasmid DNA as indicated. The luciferase data were converted to fold activation relative to the empty vector control (set as 1). Error bars represent SD of three independent experiments. * p<0.05.
The E2F transcription factors are downstream targets of RB action. We therefore investigated the effect of FOXF1 on E2F1 transcriptional activity by the reporter assay using a promoter reporter plasmid containing E2F-responsive DNA polymerase α promoter upstream of the luciferase reporter gene (35). Wild-type E2F1, but not mutant E2F1 (E132) (36), induced an upregulation (almost 18-fold) of the promoter activity compared to the vector control (Fig. 4D). Co-transfection of FOXF1 with wild-type E2F1 significantly suppressed E2F1-mediated transactivation (67%) of the reporter activity (Fig. 4D). Suppression was E2F1-specific since FOXF1, in the absence of E2F1, had no significant effect on promoter-linked reporter activity (Fig. 4D). The IF data showing a consistent decrease in endogenous total RB protein levels in FOXF1-transfected cells (Fig. 4B) supported the in vitro reporter data since RB is one of the well-known E2F downstream target genes. These results, taken together, strongly suggest that FOXF1-induced inhibition of CDK2-RB-E2F cascade contributes to a blockage of G1-S transition of cell cycle.
FOXF1 knockdown leads to an increase in DNA rereplication and elevates expression of E2F-induced genes
Based upon the observed negative role for FOXF1 in G1-S progression, we investigated the effects of siRNA-mediated knockdown of FOXF1 on cell cycle progression of BT549 breast cancer cells. BrdU incorporation analysis was employed to examine the effect of FOXF1 depletion on DNA replication, cell cycle progression and apoptosis. As shown in Fig. 5A, FOXF1 siRNA depleted over 80% of endogenous FOXF1 mRNA levels. Flow cytometric analysis of BrdU-labeled, propidium iodide-stained cells revealed that FOXF1 knockdown resulted in a significant increase in the fraction of BT549 cells with greater than 4N of BrdU-incorporated DNA (6.7 ± 1.1% of control cells vs. 17.3 ± 2.8% of FOXF1-depleted cells, p < 0.05) (Fig. 5B). The polyploid cells formed upon FOXF1 depletion could still be labeled with BrdU, indicating that these cells were alive and actively replicating. In fact, the appearance of more BrdU-labeled polyploid cells indicates an increase in cells undergoing DNA re-replication (over-replication) which occurs when initiation of DNA replication fires more than once in each cell cycle (37). In addition to increased DNA re-replication in BT549 cells depleted of FOXF1, a three-fold induction of apoptosis was observed (Fig. 5B). The increase in apoptosis could be attributed to death of cells that had undergone DNA re-replication. These findings suggest that inactivation of FOXF1 results in a defect in the stringent control of DNA replication initiation, which in turn promotes more uncontrolled DNA replication in BT549 cells.
Figure 5.
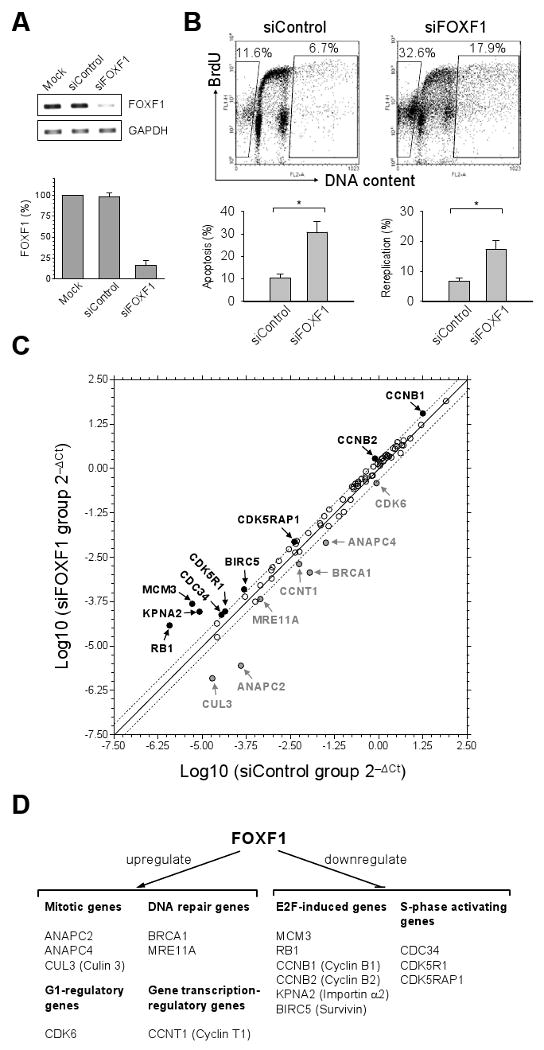
FOXF1 knockdown in cells leads to an increase in DNA rereplication, apoptosis and expression of E2F-induced genes. A, Analysis of the knockdown efficiency of FOXF1 siRNA. Gel-based and real-time-based RT-PCR results are shown on left and right panels, respectively. B, BrdU incorporation analysis of BT549 cells depleted of FOXF1. The FACS 2D dot plot data from BrdU incorporation experiments are shown in the top panel. The percentages of DNA-rereplicated (right gated cells) and apoptotic (left gated cells) cells are shown in 2D dot plots. The statistic bar graph data (mean ± SD) from three independent experiments are shown in the bottom panel. * (p<0.05). C, Expression profiling of cell cycle genes in BT549 cells depleted of FOXF1. The common logarithms of gene expression values from control siRNA-transfected cells were plotted against those from FOXF1 siRNA-transfected cells to make the scatter plot. The up-regulated (≥ 2 folds) and down-regulated (≤ -2 folds) genes in FOXF1 siRNA-transfected BT549 cells were indicated by the black and grey colors, respectively. D, Schematic summary of genes regulated by FOXF1.
To gain further insight into FOXF1's effects, expression profiling of 84 cell cycle genes was performed. Of 84 genes, 9 and 7 genes were found to be downregulated and upregulated, respectively, by FOXF1 in BT549 cells (Figs. 5C and D, and Supplemental Table 2). Surprisingly, 6 out of 9 FOXF1-downregulated genes were E2F-induced genes (38, 39), including MCM3, RB1, CCNB1, CCNB2, KPNA2 and BIRC5 (Fig. 5D). Among these genes, MCM3 encodes a protein participating in initiation of DNA replication, suggesting that FOXF1 might negatively regulate its expression to suppress DNA replication initiation. In addition, CDC34, required for ubiquitin-mediated degradation of cell cycle G1 regulators and for the initiation of DNA replication (40), is also a FOXF1-downregulated gene (Figs. 5C and D). Therefore, deregulation of MCM3 and CDC34 expression might contribute to DNA over-replication after FOXF1 function was silenced (Fig. 5B). Intriguingly, expression of two genes encoding CDK5-associated factors (CDK5R1 and CDK5RAP1) that are implicated in activation of CDK5 (41, 42) was elevated in FOXF1-depleted cells (Fig. 5C), implying that FOXF1 might abrogate CDK5 activation which is required for survival and proliferation of invasive breast cancer cells (43). FOXF1-upregulated genes included those that encode proteins involved in DNA repair (BRCA1, MRE11A), mitosis (ANAPC2, ANAPC4, and CUL3), transcriptional regulation (CCNT1) and G1 progression (CDK6) (Fig. 5C and D), suggesting that FOXF1 may regulate multiple biological processes. Upregulation of DNA repair genes BRCA1 and MRE11A by FOXF1 further supported the role of FOXF1 in maintaining genomic stability. ANAPC2, ANAPC4 and CUL3 are known to inhibit DNA replication and G2 progression during mitosis (44, 45); FOXF1, therefore, might inhibit DNA replication and G2 progression through upregulation of these mitotic genes. Moreover, it has been reported that expression patterns and function of CDK6 in breast cancer cells exhibited tumor-suppressive properties (46). Therefore, upregulation of CDK6 by FOXF1 might contribute to the growth-suppressive effect of FOXF1 in breast cancer cells.
Discussion
In this paper, we report the identification of FOXF1 gene as a novel epigenetic target in breast cancer. DNA hypermethylation was identified as the predominant underlying mechanism for silencing FOXF1 gene expression. Our studies showed that FOXF1 re-expression in breast cancer cell lines inhibited their growth and tumorigenicity, mainly by blocking G1-S transition of cell cycle, whereas inactivation of FOXF1 resulted in DNA over-replication, a risk factor leading to genomic instability. Our findings support the hypothesis that aberrant epigenetic inactivation of FOXF1 in mammary epithelial cells could contribute to mammary tumorigenesis.
Gene expression analysis in silico using the microarray database revealed that, compared to normal tissues, FOXF1 is underexpressed in a variety of cancers such as lung, prostate, bladder, ovarian, and breast. Hypermethylation of the FOXF1 promoter was found to be frequent (80% in cell lines, 40% in primary tumors) and is, in all likelihood, the predominant mechanism accounting for loss or downregulation of FOXF1 expression in breast cancer. FOXF1 promoter hypermethylation positively correlated with the tumor grade and appears to be an early event across both ductal and lobular breast cancers with the same frequency (data not shown). Therefore, FOXF1 gene is a common epigenetic target among various types of breast cancer. It will be important to decipher whether epigenetic mechanisms are also implicated in downregulating FOXF1 expression in other types of cancers.
Our studies have shed light on the mechanisms underlying FOXF1-mediated growth suppression. We found that enforced overexpression of FOXF1 led to G1 arrest concurrently with or without apoptosis dependent on the cell type examined. Inhibition of CDK2-RB-E2F signaling cascade by FOXF1 is most likely the mechanism accounting for the G1-arrest activity of FOXF1. Although mechanisms for FOXF1-mediated inactivation of the CDK2-RB-E2F cascade are still unclear, our siRNA knockdown studies provided some mechanistic clues. We found that knockdown of FOXF1 in cells led to an increase in DNA re-replication, a sign of a defect in the stringent control of DNA replication initiation. The stringency of DNA replication initiation is finely controlled by a sequential process involving DNA replication origin licensing, activation of origin to replicate DNA and disassembly of pre-replication complexes (47-49). Deregulation of these processes permits repeated firing of DNA replication origins, leading to DNA rereplication (also called over-replication) (48). Expression profiling analysis of cell cycle regulatory genes suggested that FOXF1 inhibits DNA replication initiation by downregulating expression of DNA replication initiation factor genes (e.g. MCM3 and CDC34) and by upregulating expression of mitotic genes (e.g. ANAPC2, ANAPC4 and CUL3) involved in negatively regulating G1-S transition. In addition, other FOXF1-regulated cell cycle genes such as CDK6, CDK5R1 and CDK5RAP1 might also contribute to this effect. These findings, taken together, suggest that gain or loss of FOXF1 function alters expression of these cell cycle regulatory genes, which in turn leads to these observed outcomes. In addition to MCM3, expression of five other E2F-induced genes was also elevated in cells depleted of FOXF1, suggesting that E2F transcriptional activity is activated upon silencing of FOXF1. These results are consistent with the data showing that FOXF1 suppresses E2F transcriptional activity through abrogation of the CDK2-RB-E2F cascade.
In addition to cell-cycle functions, the identified FOXF1-regulated genes are also implicated in multiple biological processes such as DNA repair, transcriptional regulation, nuclear transportation and cell survival control. For example, we found that BRCA1 is the downstream target of FOXF1 signaling. BRCA1 is a well-known tumor suppressor gene that plays multiple roles in transcription and DNA repair. BRCA1 is inactivated through genetic mutation or aberrant downregulation of its mRNA expression (50). It is tempting to speculate that epigenetic inactivation of FOXF1 is one of mechanisms leading to downregulation of BRCA1 expression in breast cancer. Collectively, our findings link the molecular roles of FOXF1 to cell cycle regulation and other biological processes.
In summary, our studies support a novel role for FOXF1 transcription factor in cell cycle regulation; a function that is abrogated through epigenetic silencing of FOXF1 during tumorigenesis. Therefore, FOXF1 and its downstream effectors might serve as novel molecular targets for detection and treatment of breast cancer and other human cancers.
Supplementary Material
Acknowledgments
We thank Dr. W. Kaelin, Dana-Farber Cancer Institute at Boston, for kindly providing the E2F-responsive reporter plasmid and E2F1 expression plasmids; and Dr. P. Carlsson, Göteborg University, Sweden, for kindly providing FOXF-responsive reporter plasmids. This work was supported by grants from the NIH SPORE-1 P50 CA088843-08, DOD Center of Excellence-W81XWH-04-1-0595 and the Avon Research Foundation to SS.
References
- 1.Lai E, Clark KL, Burley SK, Darnell JE., Jr Hepatocyte nuclear factor 3/fork head or “winged helix” proteins: a family of transcription factors of diverse biologic function. Proc Natl Acad Sci U S A. 1993;90:10421–3. doi: 10.1073/pnas.90.22.10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katoh M, Katoh M. Human FOX gene family (Review) Int J Oncol. 2004;25:1495–500. [PubMed] [Google Scholar]
- 3.Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1–23. doi: 10.1006/dbio.2002.0780. [DOI] [PubMed] [Google Scholar]
- 4.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 5.Kalinichenko VV, Major ML, Wang X, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–50. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll JS, Liu XS, Brodsky AS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Mani SA, Yang J, Brooks M, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–74. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev. 2007;7:847–59. doi: 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- 9.Mahlapuu M, Ormestad M, Enerback S, Carlsson P. The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development. 2001;128:155–66. doi: 10.1242/dev.128.2.155. [DOI] [PubMed] [Google Scholar]
- 10.Mahlapuu M, Enerback S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128:2397–406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- 11.Kalinichenko VV, Zhou Y, Bhattacharyya D, et al. Haploinsufficiency of the mouse Forkhead Box f1 gene causes defects in gall bladder development. J Biol Chem. 2002;277:12369–74. doi: 10.1074/jbc.M112162200. [DOI] [PubMed] [Google Scholar]
- 12.Kalinichenko VV, Bhattacharyya D, Zhou Y, et al. Foxf1 +/- mice exhibit defective stellate cell activation and abnormal liver regeneration following CCl4 injury. Hepatology. 2003;37:107–17. doi: 10.1053/jhep.2003.50005. [DOI] [PubMed] [Google Scholar]
- 13.Watson JE, Doggett NA, Albertson DG, et al. Integration of high-resolution array comparative genomic hybridization analysis of chromosome 16q with expression array data refines common regions of loss at 16q23-qter and identifies underlying candidate tumor suppressor genes in prostate cancer. Oncogene. 2004;23:3487–94. doi: 10.1038/sj.onc.1207474. [DOI] [PubMed] [Google Scholar]
- 14.Oncomine 4.3 Research Edition [database on the Internet] Ann Arbor (MI): Compendia Bioscience, Inc.; c2008-10 [cited 2008 Aug 12.] Available from: http://www.oncomine.org/ [Google Scholar]
- 15.National Center for Biotechnology Information [database on the Internet] Bethesda (MD): National Library of Medicine (US); c1988-2010 [updated 2005 Nov 7; cited 2008 Aug 16]. Available from: http://www.ncbi.nlm.nih.gov/ [Google Scholar]
- 16.CpG Island Searcher [analysis tool on the Internet] Los Angeles (CA): Keck School of Medicine of the University of Southern California; c2004 [cited 2008 Aug 25]. Available from: http://cpgislands.usc.edu/ [Google Scholar]
- 17.Bergstraesser LM, Weitzman SA. Culture of normal and malignant primary human mammary epithelial cells in a physiological manner simulates in vivo growth patterns and allows discrimination of cell type. Cancer Res. 1993;53:2644–54. [PubMed] [Google Scholar]
- 18.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo PK, Mehrotra J, D'Costa A, et al. Epigenetic suppression of secreted frizzled related protein 1 (SFRP1) expression in human breast cancer. Cancer Biol Ther. 2006;5:281–6. doi: 10.4161/cbt.5.3.2384. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778–89. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 21.Stearman RS, Dwyer-Nield L, Zerbe L, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol. 2005;167:1763–75. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharjee A, Richards WG, Staunton J, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–5. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lapointe J, Li C, Higgins JP, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101:811–6. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wachi S, Yoneda K, Wu R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 2005;21:4205–8. doi: 10.1093/bioinformatics/bti688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyrskjot L, Kruhoffer M, Thykjaer T, et al. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–8. doi: 10.1158/0008-5472.CAN-03-3620. [DOI] [PubMed] [Google Scholar]
- 27.Powell CA, Spira A, Derti A, et al. Gene expression in lung adenocarcinomas of smokers and nonsmokers. Am J Respir Cell Mol Biol. 2003;29:157–62. doi: 10.1165/rcmb.2002-0183RC. [DOI] [PubMed] [Google Scholar]
- 28.Yu YP, Landsittel D, Jing L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 29.Welsh JB, Sapinoso LM, Su AI, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–8. [PubMed] [Google Scholar]
- 30.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–82. [PubMed] [Google Scholar]
- 31.Lancaster JM, Dressman HK, Whitaker RS, et al. Gene expression patterns that characterize advanced stage serous ovarian cancers. J Soc Gynecol Investig. 2004;11:51–9. doi: 10.1016/j.jsgi.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Zhao H, Langerod A, Ji Y, et al. Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol Biol Cell. 2004;15:2523–36. doi: 10.1091/mbc.E03-11-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hellqvist M, Mahlapuu M, Samuelsson L, Enerback S, Carlsson P. Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J Biol Chem. 1996;271:4482–90. doi: 10.1074/jbc.271.8.4482. [DOI] [PubMed] [Google Scholar]
- 34.Fesquet D, Labbe JC, Derancourt J, et al. The MO15 gene encodes the catalytic subunit of a protein kinase that activates cdc2 and other cyclin-dependent kinases (CDKs) through phosphorylation of Thr161 and its homologues. EMBO J. 1993;12:3111–21. doi: 10.1002/j.1460-2075.1993.tb05980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sellers WR, Rodgers JW, Kaelin WG., Jr A potent transrepression domain in the retinoblastoma protein induces a cell cycle arrest when bound to E2F sites. Proc Natl Acad Sci U S A. 1995;92:11544–8. doi: 10.1073/pnas.92.25.11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407:645–8. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 37.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 38.Ishida S, Huang E, Zuzan H, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21:4684–99. doi: 10.1128/MCB.21.14.4684-4699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang Y, Saavedra HI, Holloway MP, Leone G, Altura RA. Aberrant regulation of survivin by the RB/E2F family of proteins. J Biol Chem. 2004;279:40511–20. doi: 10.1074/jbc.M404496200. [DOI] [PubMed] [Google Scholar]
- 40.Plon SE, Leppig KA, Do HN, Groudine M. Cloning of the human homolog of the CDC34 cell cycle gene by complementation in yeast. Proc Natl Acad Sci U S A. 1993;90:10484–8. doi: 10.1073/pnas.90.22.10484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee KY, Helbing CC, Choi KS, Johnston RN, Wang JH. Neuronal Cdc2-like kinase (Nclk) binds and phosphorylates the retinoblastoma protein. J Biol Chem. 1997;272:5622–6. doi: 10.1074/jbc.272.9.5622. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Ching YP, Lam WH, Qi Z, Zhang M, Wang JH. Identification of a common protein association region in the neuronal Cdk5 activator. J Biol Chem. 2000;275:31763–9. doi: 10.1074/jbc.M004358200. [DOI] [PubMed] [Google Scholar]
- 43.Goodyear S, Sharma MC. Roscovitine regulates invasive breast cancer cell (MDA-MB231) proliferation and survival through cell cycle regulatory protein cdk5. Exp Mol Pathol. 2007;82:25–32. doi: 10.1016/j.yexmp.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 45.Sumara I, Quadroni M, Frei C, et al. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell. 2007;12:887–900. doi: 10.1016/j.devcel.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 46.Lucas JJ, Domenico J, Gelfand EW. Cyclin-dependent kinase 6 inhibits proliferation of human mammary epithelial cells. Mol Cancer Res. 2004;2:105–14. [PubMed] [Google Scholar]
- 47.Nishitani H, Lygerou Z. DNA replication licensing. Front Biosci. 2004;9:2115–32. doi: 10.2741/1315. [DOI] [PubMed] [Google Scholar]
- 48.Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol. 2005;6:476–86. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kearsey SE, Cotterill S. Enigmatic variations: divergent modes of regulating eukaryotic DNA replication. Mol Cell. 2003;12:1067–75. doi: 10.1016/s1097-2765(03)00441-6. [DOI] [PubMed] [Google Scholar]
- 50.Turner NC, Reis-Filho JS, Russell AM, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26:2126–32. doi: 10.1038/sj.onc.1210014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.