

Abstract
In the present study we examine the effects of tissue inhibitor of metalloproteinases-2 (TIMP-2) on the phosphorylation status of specific phosphotyrosine residues on the vascular endothelial cell growth factor receptor-2 (VEGFR-2) cytoplasmic tail and examine the effects on associated downstream signaling pathways. In order to focus on metalloproteinase-independent mechanisms, we utilized the TIMP-2 analog known as Ala+TIMP-2 that is deficient in matrix metalloproteinase (MMP) inhibitory activity. Our experiments are designed to compare the effects of VEGF-A stimulation with or without Ala+TIMP-2 pretreatment, as well as basal responses in human microvascular endothelial cells. Our results show that Ala+TIMP-2 selectively alters the phosphorylation pattern of VEGFR-2 following VEGF-A stimulation and disrupts the downstream activation of PLC-γ, Ca+2 flux, Akt, and eNOS, as well as decreasing cGMP levels. Moreover, we observed an Ala+TIMP-2-induced reduction in cGMP levels typically elevated by exogenous NO donors implicating Ala+TIMP-2 in the direct activation of an isobutylmethylxanthine (IBMX)-sensitive cGMP phosphodiesterase activity. TIMP-2 suppression of endothelial mitogenesis and angiogenesis involves at least two mechanisms, one mediated by protein tyrosine phosphatase inhibition of VEGFR-2 activation and downstream signaling and a second mechanism involving direct activation of an IBMX-sensitive phosphodiesterase activity.
Keywords: Angiogenesis, endothelial cells, protein tyrosine phosphatase, phosphodiesterase, tissue inhibitors of metalloproteinases
Tumor angiogenesis is initiated by the hypoxic conditions related to rapid tumor growth that initiates tumor cell transcriptional activation of vascular endothelial growth factor (VEGF)-A (1). Tumor progression is also associated with remodeling of the extracellular matrix by members of the matrix metalloproteinase (MMP) family and concomitant release of proteolytic cleavage products of laminin, fibronectin and collagens, as well as sequestered growth factors including VEGF-A (2, 3). VEGF-A is well characterized as a potent inducer of endothelial cell proliferation, migration and survival associated with tumor progression.
Vascular endothelial growth factor receptor-2 (VEGFR-2) signaling is critical for the angiogenic response associated with cancer progression. The central role of the VEGF-A/VEGFR-2 receptor axis in pathologic angiogenesis is reflected in the development of multiple Federal Drug Administration approved human therapeutic strategies that selectively target VEGF-A/VEGFR-2. For example, Bevacisumab and Sunitinib are approved for cancer treatment, as well as Pegaptanib and Ranibizumad for treatment of patients with macular degeneration, and several other drugs are now in late stage clinical trials (4-6).
Similar to other receptor tyrosine kinases (RTKs) activation of VEGFR-2 signaling begins with ligand binding that results in receptor dimerization and autophosphorylation of specific tyrosine residues (2, 7). Phosphorylation of specific tyrosine residues is essential for the recruitment and activation of Src-homolgy-2 (SH2) binding proteins that propagate downstream intracellular signaling pathways. VEGFR-2 initiated signaling pathways regulate endothelial cell proliferation, migration, survival and vascular permeability.
Mammalian TIMPs are a family of four relatively small proteins that contain six disulfide bonds (8). TIMP-2 is unique in that it is the only member of the family that functions to both inhibit MMP activity, and facilitate the cell surface activation of pro-MMP-2 through formation of a tri-molecular complex with MT1-MMP (8). In addition, TIMP-2 is the only TIMP family member that is a potent inhibitor of human microvascular endothelial cell (hMVEC) mitogenesis in vitro and angiogenesis in vivo, and these effects are independent of MMP inhibitory activity, as demonstrated by the TIMP-2 analog, Ala+TIMP-2 that is devoid of MMP-inhibitory activity (9-12). The anti-angiogenic effect of TIMP-2 and Ala+TIMP-2 involves heterologous receptor inactivation. The specific mechanism involves binding of TIMP-2 or Ala+TIMP-2 to the α3β1 integrin, induction of protein tyrosine phosphatase (PTP) activity, and subsequent reduction in activation/phosphorylation of angiogenic RTKs, such as VEGFR-2 and FGFR-1 (10-12). Furthermore, studies using mice 80-90% deficient in the src-homology 2 (SH2) PTP activity (Shp-1) demonstrate that the in vivo anti-angiogenic activity of Ala+TIMP-2 is dependent Shp-1 activity (13). In addition to inhibition of endothelial cell mitogenesis also TIMP-2 inhibits endothelial cell migration, also via an Shp-1-dependent mechanism (14).
Based on our prior demonstration that all TIMP-2 and Ala+TIMP-2 effects are Shp-1-dependent we have not repeated these experiments here but choose to further our understanding of the anti-angiogenic mechanism of TIMP-2 through examining the effects on the pattern of VEGFR-2 phosphorylation in the cytoplasmic tail. To focus on MMP-independent effects of TIMP-2 experiments were conducted using the TIMP-2 analog, which we refer to as Ala+TIMP-2 that is deficient in MMP inhibitory activity (15). We show for the first time that Ala+TIMP-2 selectively alters VEGFR-2 phosphorylation, and diminished VEGF-A-mediated activation of downstream effectors. Also for the first time we observed that elevation of cGMP levels by administration of exogenous NO donors was directly down regulated by Ala+TIMP-2 through induction of isobutylmethyl xanthine (IBMX)-sensitive phosphodiesterase activity. These findings extend previous observations of the inhibitory effects of extracellular matrix proteins, specifically TIMP-2, on the proliferation and migration of hMVECs and provide novel insight into extracellular matrix regulatory signals that modulate cellular responses to VEGF-A.
Materials and Methods
Cell lines, growth factors, antibodies and reagents
Primary hMVECs and growth factors were purchased from commercial sources. Primary hMVECs were used between passages 3 to 6 for all experiments as previously described (11). Antibodies recognizing VEGFR-2 phosphorylated tyrosine residues: pY951, pY1059, and pY1214 were purchased from Biosource (Invitrogen, Carlsbad, CA); anti-pY996 and anti-pY1175 were from Cell Signaling Technology (Danvers, MA). Anti-PLC-γ-pY783 and anti-PLC-γ antibodies were from Biosource. Anti-phosho-AKT (S473), anti-AKT, anti-phospho-eNOS (S1177), and anti-eNOS antibodies were purchased from Cell Signaling Technology. Anti-VEGFR-2 antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Diethyltriamine NONOate (DETA/NO) and fura2-acetoxymethyl ester (fura-2, AM) were purchased from Cayman Chemical (Ann Arbor, MI), and Molecular Probes (Invitrogen, Carlsbad, CA), respectively. BAPTA-AM and 1H-[1,2,4] oxadiazole[4,3-a]quinoxalin-1-one (ODQ) were obtained from BIOMOL (Plymouth Meeting, PA). Sodium orthovanadate, okadaic acid, 8-Bromo (8Br)-cGMP, 4,5-diaminofluorescein diacetate (DAF-2DA), Nω-nitro-L-arginine methyl ester (L-NAME), and 3-isobutyl-1-metylxanthine (IBMX) were purchased form Sigma-Aldrich Chemical Co. (St. Louis, MO). Recombinant TIMP-2 and Ala+TIMP-2 were prepared as described, and endotoxin tested using the Limulus amoebocyte lysis assay (<2 EU/mg protein), as previously reported (15).
Cytosolic Ca2+ measurement
Increases in intracellular Ca2+ were measured using the Ca2+-sensitive fluorescent dye fura-2, AM using gelatin-coated 96 well black wall plates (BioCoat, BD Biosciences, San Jose, CA). hMVECs were incubated with 4 μM fura-2, AM for 30 min. at 37 °C, gently washed twice with Hank’s balanced salt solution (Invitrogen), and treated as indicated. The treated cells were then imaged using a BD Pathway 855 high content bioimager (BD Biosciences, San Jose, CA). Regions of interest in individual cells were gated and excited at 334 and 380 nm with emission at 520 nm. The 334/380-excitation ratio, which increases as a function of intracellular Ca2+ concentration, was captured at 4-sec. intervals.
Immunoblotting and immunoprecipitation
Following specified treatments, hMVECs were rinsed twice with ice cold phosphate buffered saline and lysed in buffer A (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.25% sodium deoxycholate, and 1% Triton X-100), supplemented with 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μg/mL pepstatin, and 100 μM Na3VO4, for 20 min on ice. Insoluble material was removed by centrifugation at 4 °C for 20 min at 14,000 rpm. Protein concentration in the lysates was determined with a BCA protein assay kit (Pierce, Rockford, IL). To extract total eNOS protein, cells were lysed in buffer B containing SDS rather than Triton X-100 (50 mM Tris-HCl, pH 7.4, 2% SDS, 25 mM β-glycerophosphate, 50 mM NaF, and 0.5 mM Na3VO4). hMVECs lysates were subjected to immunoprecipitation and Western Blot analysis as described previously (16). Chemiluminescent generated band intensities were integrated for quantification by the use of National Institutes of Health (NIH) Image J 1.34s software.
Nitric oxide measurements
Total NO was assayed using nitrate/nitrite fluorometric assay kit from Cayman Chemical Co. (Ann Arbor, MI). hMVECs were washed with phenol free-basal media and incubated with TIMP-2 (50 nM), Ala+TIMP-2 (50 nM), or L-NAME (1 mM) in the absence of and presence of VEGF-A. The nitrite and nitrate released into the media were measured fluorimetrically (excitation wavelength 355 nm: emission wavelength 460 nm), according to the manufacturer’s instructions (Cayman Chemical).
Intracellular NO was measured using the NO-specific fluorescence probe DAF-2DA as described (17). Total fluorescence of the treated and washed samples was determined (emission wavelength, 485 nm; excitation wavelength 538 nm) using the bottom-reading mode in a fluorescence plate reader (Tecan).
Intracellular cGMP measurement
Intracellular cGMP concentrations were determined as described previously (18). The experimental readings from each triplicate lysates falling within the linear region of the standard curve were evaluated with the Direct cGMP Assay Kit from Assay Designs, Inc (Ann Arbor, MI).
Cell proliferation assay
The number of hMVECs, following indicated in vitro treatments, were determined using Cell-Titer 96 Aqueous One Solution reagent (Promega) according to the manufacturer’s direction, as previously described (11).
Cell migration assay
To assess cell migration, the “scratch” wound assay was performed as previously described and quantified by measuring the width of the cell-free zone immediately before and 16 h after cell treatment using a computer-assisted microscope (Zeiss) at 3 distinct positions for each assay condition. The results are reported as the mean ± standard deviation of these triplicate measurements.
Statistical Analysis
All data were analyzed using the Prism Statistical Software package (Software MacKiev, Graphpad Software, La Jolla, CA) to perform two-sided student t test of statistical significance.
Results
TIMP-2 shows selective inhibition of VEGFR-2 tyrosine phosphorylation
We have previously reported that treatment of hMVECs with TIMP-2 results in a significant decrease in total VEGFR-2 tyrosine phosphorylation following VEGF-A stimulation (11), resulting in complete inhibition of VEGF-A-stimulated mitogenesis. This effect is independent of the ability of TIMP-2 to inhibit MMP activity, as shown by the use of a TIMP-2 derivative devoid of MMP inhibitory activity, Ala+TIMP-2. Furthermore, this effect was completely abrogated by the co-administration of the PTP-inhibitor sodium orthovanadate or genetic manipulation to reduce active Shp-1 levels (11, 12). TTo further elucidate the mechanisms through which TIMP-2 down regulates VEGF-A-mediated pro-angiogenic signaling, we examined the phosphorylation status of several tyrosine residues following treatment of hMVECs at 0, 2, 5 and 10 min. post VEGF-A stimulation with or without prior Ala+TIMP-2 treatment, Figure 1. It is well recognized that VEGFR-2 phosphorylation is maximal between 2-5 min following VEGF-A stimulation and decreases thereafter (19). Of the five tyrosine residues examined (Y951, Y996, Y1059, Y1175 and Y1214) VEGF-A treatment alone showed enhanced phosphorylation of these residues that peaked at 5 min. Pretreatment of the hMVECs with 50 nM Ala+TIMP-2 for 30 min. prior to VEGF-A stimulation resulted in minimal enhancement of Y1059 and Y1214 phosphorylation. These findings suggest that these residues are not targeted by the enhanced Shp-1 phosphatase activity previously observed following Ala+TIMP-2 pretreatment of VEGF-A-stimulated hMVECs (11, 12).
Figure 1.
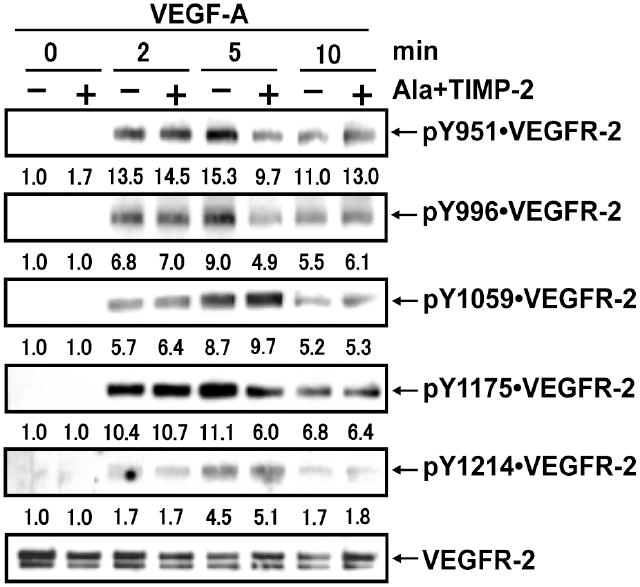
Ala+TIMP-2 suppresses VEGF-A-induced maximal phosphorylation of select tyrosine residues in VEGFR-2 in hMVECs. hMVECs stimulated with VEGF-A (10 ng/ml) for indicated time periods following pretreatment in the presence or absence of Ala+TIMP-2 (50 nM, 15 min) were subjected to Western blot analysis with commercial antibodies. The Western blots shown are representative of three independent experiments. Relative band intensities were measured by NIH image software and integrated densities were normalized against the VEGFR-2 loading control.
In contrast, Ala+TIMP-2 selectively suppressed maximal VEGF-A-stimulated phosphorylation of Y951, Y996 and Y1175 of the VEGFR-2 cytoplasmic domain and this effect peaked at 5 min post stimulation. These tyrosine residues have been specifically implicated in the regulation of hMVEC proliferation and migration, via initiation of critical downstream signaling pathways. Ala+TIMP-2 decreased phosphorylation of these specific tyrosine residues ranged from ~ 40% reduction for Y951 to ~50% reduction for both Y996 and Y1175.
Zymogram analysis of MMP-2 expression revealed that Ala+TIMP-2 or TIMP-2 (50 or 100 nM final concentration) treatment of hMVECs failed to alter expression or activation of this matrix metalloproteinase (data not shown).
TIMP-2 inhibits PLC-γ activation and Ca+2 flux
Since phosphorylation of Y951 and Y1175 are known to mediate activation of PLC-γ, we postulated that Ala+TIMP-2 reduction of phosphorylation at VEGFR-2 tyrosine residues Y951 and Y1175, would decrease both PLC-γ activation and cytosolic Ca+2 concentrations. As shown in Figure 2A, hMVECs treated with 50 nM Ala+TIMP-2 prior to VEGF-A stimulation demonstrated a significant (>65%) decrease in the association of PLC-γ with the VEGFR-2 at 5 min., as determined by immunoprecipitation analysis. We then examined the phosphorylation of PLC-γ on Y783 that has been associated with PLC-γ activation in vitro and in vivo (20). At 2 min. following treatment with VEGF-A, PLC-γ demonstrated a ~ 9.0-fold increase in Y783 phosphorylation that was not significantly attenuated by TIMP-2 pretreatment, Figure 2B. However, at 5 min. post treatment PLC-γ activation as determined by Y783 phosphorylation was decreased by 40 %. This observation suggest that Ala+TIMP-2 may not suppress maximal phosphorylation of PLC-γ at 2 min post VEGF-A stimulation, but does accelerate dephosphorylation as demonstrated by reduced phosphorylation at both the 5 and 10 min time points. To further examine the effect of this apparent Ala+TIMP-2-mediated reduction in PLC-γ activation at 5 min., we measured cytosolic Ca+2 levels, which are normally increased in response to PLC-γ activation by VEGFR-2 (21). Figure 2C demonstrates that VEGF-A stimulation results in a significant biphasic Ca+2 flux that starts at ~ 60 seconds and shows a maximal response at 120 seconds, followed by a gradual return to baseline at 300 seconds. In contrast, 50 nM Ala+TIMP-2 or TIMP-2 treatment severely attenuates the initial phase of the Ca+2 flux that starts a 60 seconds and completely abrogates the Ca+2 spike that occurs around 120 seconds post VEGF-A treatment. The calcium-chelating agent, BAPTA-AM (negative control), suppressed both phases of the intracellular Ca+2 increase usually observed following VEGF-A stimulation. These results are consistent with the Ala+TIMP-2-mediated decreased association of PLC-γ with VEGFR-2 demonstrated by co-immunoprecipitation experiments (Figure 2A), and decrease in PLC-γ phosphorylation/activation. In addition, these data also support the multiple modes of PLC-γ regulation and the recent observation that activation of PLC-γ requires overcoming auto-inhibition by the c-terminal SH2 domain, which is regulated by the split pleckstrin homology (sPHN) domain that occurs independently of Y783 phosphorylation (22). Accordingly these findings may account for the apparent dissociation of PLC-γ phosphorylation/activation and the suppression of Ca+2 cytosolic flux between 2 and 5 min.
Figure 2.

TIMP-2 accelerates VEGF-A-induced PLC-γ deactivation and suppresses intracellular Ca+2 influx. A, hMVECs were treated with Ala+TIMP-2 (50 nM) prior to VEGF-A stimulation for 5 min. The cells were immediately rinsed and lysed as described in materials and methods. B, hMVECs were stimulated with VEGF-A (10 ng/mL) in the absence or presence of Ala+TIMP-2 (50 nM) for the indicated time periods. Immunoblotting was performed using the antibody directed against pY783 PLC-γ and PLC-γ (loading control). Band intensities were integrated using NIH Image software and normalized using total PLC-γ at 0 min. Results are representative of three independent experiments. C, Intracellular Ca2+ influx was measured using fura-2. hMVECs were treated with VEGF-A (10 ng/ml) in the absence (black line) or presence of Ala+TIMP-2 (50 nM, blue line), TIMP-2 (50 nM, red line), or BAPTA-AM (10 μM, green line). Each tracing is the average response of ~ 60 cells. Experiments were performed in triplicate and shown are representative tracings.
Ala+TIMP-2 inhibits phosphorylation/activation of AKT and eNOS
Phosphorylation of Y1175 allows activation of the PI3K/AKT signaling pathway (23). To assess the effect of Ala+TIMP-2-mediated reduction of Y1175 phosphorylation on downstream signaling via the PI3K/AKT pathway, we determined the level of AKT activation by assaying the phosphorylation status of serine 473 (S473) (24). Ala+TIMP-2 significantly attenuated AKT phosphorylation on S473 at both 5 min. (60% inhibition) and 10 min. (40% inhibition) post VEGF-A stimulation (Figure 3A). The Ala+TIMP-2-mediated decrease in phosphorylation at this site was absent at the latter time point (30 min), further emphasizing the importance of suppressed AKT activation at the critical time point for activation of downstream pathways.
Figure 3.
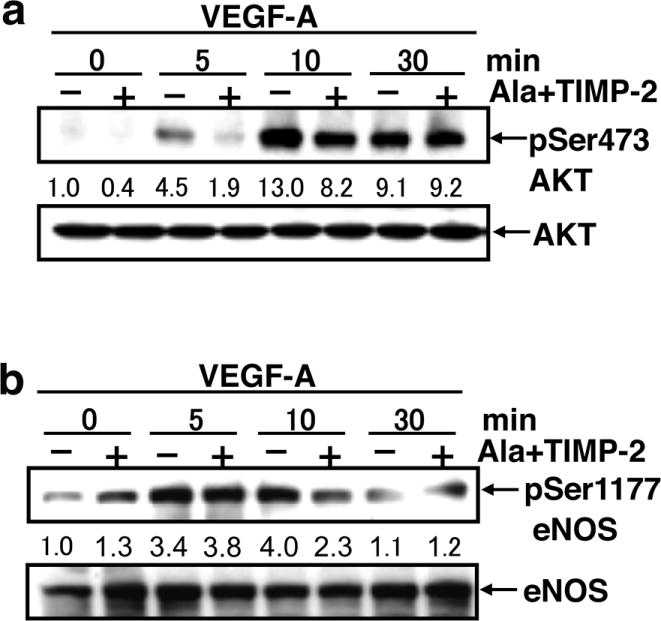
Ala+TIMP-2 suppresses Akt activation and maximal eNOS phosphorylation following VEGF-A stimulation. A, hMVECs were stimulated with VEGF-A (10 ng/mL) in the absence or presence of Ala+TIMP-2 (50 nM) and cell lysates isolated at the indicated times post-stimulation. The immunoblotting was performed using the antibodies against pS473 AKT and AKT. Band intensities were integrated using NIH Image software and normalized using total Akt at 0 min. Results shown are representative of three independent experiments. B, The activation status of eNOS was evaluated by immuoblots of pS1177eNOS. Band intensities were measured by NIH image software and normalized against total eNOS levels at time 0 min. Results shown are representative of three independent experiments.
PLC-γ associated intracellular Ca2+ flux is associated with calmodulin-mediated activation of eNOS (25), but eNOS activation may also be directly mediated by AKT phosphorylation of eNOS on S1177 in Ca2+-independent manner (26). Since we have previously shown that TIMP-2 significantly suppresses the PLC-γ-mediated cytosolic Ca+2 flux, we evaluated eNOS activation status by examining S1177 phosphorylation status. VEGF-A-induced eNOS phosphorylation on S1177 was inhibited 40% by Ala+TIMP-2 at 10 min. following stimulation (Figure 3B). Since maximal VEGF-A-induced phosphorylation of S1177 was observed at 10 min., these data are consistent with the suppression of maximal eNOS activation at 10 min. Collectively, these findings are consistent with suppression of maximal VEGFR-2 phosphorylation on Y951, and Y1175, in response to Ala+TIMP-2 treatment, resulting in suppression of both the PLC-γ and PI3K/AKT signaling pathways, and are consistent with the observed down regulation of eNOS activation. These results led us to investigate NO and further downstream signaling following Ala+TIMP-2 treatment and VEGF-A stimulation of hMVECs.
TIMP-2 inhibits VEGF-A-induced NO synthesis
Total NO levels were determined in VEGF-A stimulated hMVEC in the absence or presence of TIMP-2, Ala+TIMP-2, or the NOS inhibitor L-NAME, Figure 4. VEGF-A evoked a two-fold increase in total NO synthesis compared with basal conditions. In the presence of TIMP-2 or Ala+TIMP-2 we observed a statistically significant (p<0.001) inhibition of VEGF-A-stimulated total NO production close to basal levels and similar to those observed with L-NAME (p<0.01), Figure 4A. TIMP-2 and Ala+TIMP-2 both significantly reduced cytosolic NO levels close to basal levels (p<0.001), Figure 4B. The equipotent activity of TIMP-2 and Ala+TIMP-2 suggests that these effects were independent of MMP inhibition. These results are consistent with the reduced phosphorylation/activation of eNOS observed previously (Figure 3B) and suggest that the 40% reduction in eNOS phosphorylation, combined with reduced cytosolic Ca+2 flux, are sufficient to reduce eNOS activity below the threshold level necessary for elevation of either total or cytosolic NO levels.
Figure 4.

TIMP-2 restricts VEGF-A-mediated NO production. A, hMVECs were incubated in the presence (+) or absence (Basal) of VEGF-A (10 ng/ml), as well as with VEGF-A following 15 min pretreatment with TIMP-2 (50 nM), Ala+TIMP-2 (50 nM), or L-NAME (1 mM) for 5 min. Total NO (nitrite and nitrate) released from cells in the medium was measured described in “Materials and Methods” and normalized for the amount of total cellular protein (mg). Data are expressed as mean ±SD of triplicate. **P<0.01 ***P<0.001 versus VEGF-A treatment alone. B, hMVECs were loaded with the fluorescent indicator DAF-2DA as described, and then treated as described in Figure 2A. Fluorescence was measured in a fluorescence plate reader. Data are expressed as mean ±SD of triplicate. **P<0.01 ***P<0.001 versus VEGF-A treatment alone.
TIMP-2 inhibits VEGF-A-stimulated increase in cellular cGMP
Conformational changes resulting from NO binding to the sixth position of the heme ring of soluble guanylyl cyclase (sGC) leads to a 200-fold increase in enzyme activation of the enzyme, and subsequent increase in cytosolic cGMP levels (27). In the current study, we show that VEGF-A stimulation leads to a maximal 4-fold increase in cGMP synthesis at 10 min, Figure 5A. Alternatively, in the presence of 50 nM concentrations of Ala+TIMP-2 (or TIMP-2), the normal VEGF-A-induced cGMP increase was suppressed to levels similar to those observed in the presence of the eNOS inhibitor L-NAME, Figure 5A. The suppression of cGMP production in the presence of Ala+TIMP-2, TIMP-2 and L-NAME are all statistically significant (p< 0.01) at the 10 min. time point, which was the point of maximal cGMP response to VEGF-A.
Figure 5.
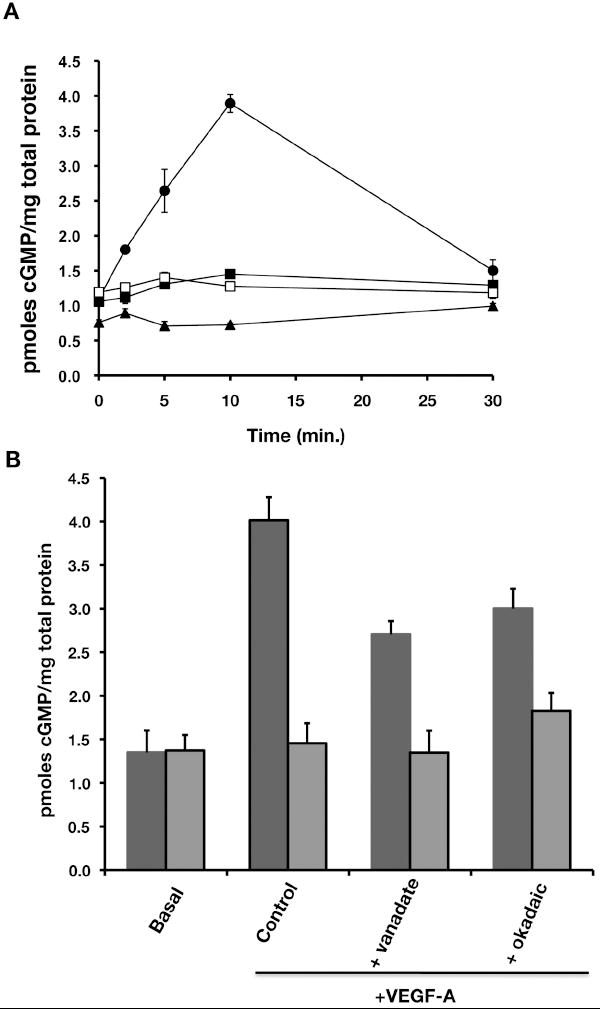
TIMP-2 inhibits cGMP synthesis evoked by VEGF-A leading to the inhibition of VEGF-A-induced cell proliferation and cell migration. A, hMVECs were treated with VEGF-A for the indicated time course in the absence (➂) or presence of TIMP-2 (50 nM, ➄), Ala+TIMP-2 (50 nM, ➅), or L-NAME (1 mM, ➉). The intracellular cGMP was determined as described in “Materials and Methods”. The data shown reflect the mean intracellular cGMP level from triplicates determinations and are representative of three independent experiments. B, hMVECs were pretreated with either orthovanadate (1 μM) or okadaic acid (100 nM) for 30 min. and then stimulated with VEGF-A in the presence (blue bars) or absence of Ala+TIMP-2 (red bars) for 10 min. The intracellular cGMP was determined as described in “Materials and Methods”. The shown data are the mean of intracellular cGMP level from triplicates determinations. *p<0.01 versus control; **p<0.001 versusVEGF-A alone in the absence of Ala+TIMP-2.
These findings are consistent with our prior observations that TIMP-2 or Ala+TIMP-2 decreased both VEGFR-2 phosphorylation and eNOS activity. We have previously shown that the effects of TIMP-2 on hMVEC growth and migration are sensitive to the nonspecific PTP inhibitor orthovanadate (Na3VO4) and expression of dominant negative Shp1 (dnShp1)(11, 12, 28). Given that Okadaic acid suppresses the serine/threonine phosphatase activity of protein phosphatase 2a (PP2a) that modulates phosphorylation/activation of eNOS on S1177 we examined whether inhibition of either PTP or PP2a activities would reverse the TIMP-mediated suppression of VEGF-A-induced cGMP synthesis through alteration of VEGFR-2 or eNOS phosphorylation (29). To this end, we determined the effects of both Na3VO4 and okadaic acid on cellular cGMP levels following VEGF-A stimulation with or without Ala+TIMP-2 treatment. Both orthovanadate and okadaic acid slightly inhibited VEGF-A-induced cGMP synthesis in hMVECs without TIMP-2 treatment, Figure 5B. However, the addition of either orthovanadate (1 μM) or okadaic acid (100 nM) was unable to reverse the essentially complete suppression of VEGF-A-induced cGMP synthesis in hMVEC following Ala+TIMP-2 treatment, Figure 5B. These findings imply that the suppression of increased VEGF-A-induced cGMP levels observed following Ala+TIMP-2 treatment is not mediated solely via induction of Shp-1 or PP2a. Therefore, it is reasonable to assume that modulation of cGMP levels by Ala+TIMP-2 occurs via additional mechanisms that are independent of the Ala+TIMP-2-mediated decrease in VEGFR-2 phosphorylation and subsequent suppression of eNOS activation.
TIMP-2 suppresses cGMP production in response to exogenous NO donors
To further examine this novel TIMP-2 signaling effect, we determined the ability of Ala+TIMP-2 to inhibit cGMP synthesis initiated by exogenous NO-donors. DETA/NO or SNAP are exogenous NO donors that can directly activate soluble guanylyl cyclase (sGC) and increase cytosolic cGMP levels (30). Treatment of hMVECs with 10 μM of DETA/NO or SNAP for 5 min resulted in a significant increase in cGMP over basal levels, Figure 6A, respectively. DETA/NO gave ~5.5 fold increase, while SNAP gave a slightly lower, but still statistically significant ~3.5 fold increase over basal levels. Somewhat surprisingly, Ala+TIMP-2 (or TIMP-2) inhibited DETA/NO- or SNAP-induced cGMP synthesis in hMVECs in a statistically significant fashion (p<0.001) comparable to levels observed with the selective guanylyl cyclase inhibitor ODQ (Figure 6A).
Figure 6.
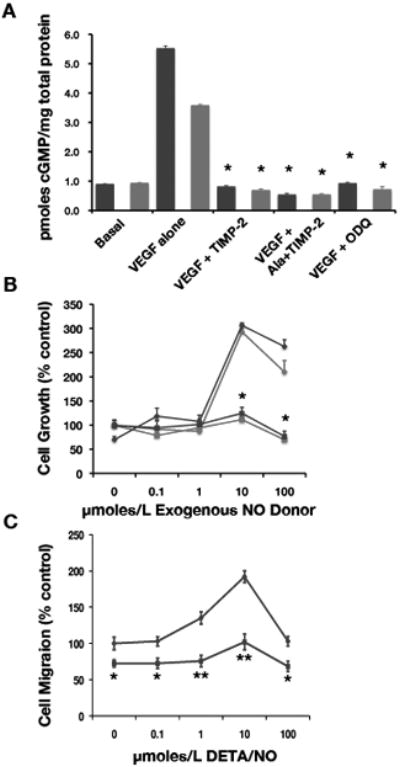
TIMP-2 inhibits exogenous NO-induced cGMP synthesis and subsequent hMVEC proliferation and migration. A, Serum starved hMVECs were treated with 10 μM of DETA/NO (blue bars) or SNAP (red bars) for 5 min. in the absence or presence of TIMP-2 (50 nM), Ala+TIMP-2 (50 nM), or ODQ (10 μM). The intracellular cGMP was determined as described in “Materials and Methods”. The data are presented as the mean of triplicate determinations and the results are representative of three independent experiments. *P<0.001 versus DETA/NO or SNAP in control (VEGF-A alone) cells. B, hMVECs were serum-starved for overnight and incubated for 24 hrs in the presence of the indicated concentrations of DETA/NO (blue) or SNAP (red) with (➄) or without (◇) Ala+TIMP-2. Cell proliferation was evaluated as described in “Materials and Methods”. The data are presented as the mean of triplicate determinations. *P<0.001 untreated versus DETA/NO or SNAP treated at each concentration. C. hMVECs were grown to confluence and scratched in the monolayer using pipette tip. After incubation for 18 hrs with DETA/NO, at the indicated doses, and in the presence ( ) or absence (
) or absence ( ) of Ala+TIMP-2 (50 nM), the relative migration distance of treated cells into the monolayer defect was measured. The data are presented as the mean of triplicate determinations. *P<0.01, **P<0.001 versus untreated (0 μM DETA/NO, in the presence of Ala+TIMP-2).
) of Ala+TIMP-2 (50 nM), the relative migration distance of treated cells into the monolayer defect was measured. The data are presented as the mean of triplicate determinations. *P<0.01, **P<0.001 versus untreated (0 μM DETA/NO, in the presence of Ala+TIMP-2).
Previous studies show that NO effects on endothelial cell proliferation and migration are concentration dependent. DETA/NO at concentrations below 100 μM stimulate proliferation and migration of endothelial cells, but inhibit these cellular responses at higher concentrations (30). Likewise, treatment of hMVECs with 10 or 100 μM of DETA/NO or SNAP results in a two- to three-fold increase in cell proliferation compared with basal levels. In the current study, we show that Ala+TIMP-2 suppresses hMVEC proliferation following either 10 or 100 μmole/L of DETA/NO or SNAP in a statistically significant manner (p<0.001), Figure 6B. Although treatment with 10 μmole/L DETA/NO showed a two-fold increase in spontaneous cell migration of hMVECs compared with basal migration, addition of Ala+TIMP-2 showed statistically significant inhibition of NO-stimulated cell migration at all concentrations tested, Figure 6C. In fact, Ala+TIMP-2 inhibited spontaneous hMVEC migration at 0 μM DETA/NO, consistent with out previous findings (28).
These studies demonstrate that Ala+TIMP-2 (or TIMP-2) directly down regulates exogenous NO donor-mediated increase in cGMP, as well as downstream suppression of cGMP-mediated increased hMVEC proliferation (DETA/NO and SNAP, Figure 6B) and migration DETA/NO, Figure 6C). It is possible that Ala+TIMP-2 (and TIMP-2) modulate cGMP levels by either suppressing sGC activity or enhancing phosphodiesterase (PDE) activity. Indeed, we showed that VEGF-A, Ala+TIMP-2 (and TIMP-2) suppress exogenous NO-induced hMVEC mitogenesis to near basal levels (Figure 6B), and our earlier data show that Ala+TIMP-2 suppresses eNOS activation and elevation of cytosolic cGMP levels. Given that NO directly activates sGC, this enzyme may be a target of the Ala+TIMP-2 anti-angiogenic signal. On the other hand, an alternative mechanism for the Ala+TIMP-2 suppression of cGMP levels may involve activation of cGMP PDE activity. In the latter case, if PDE activation significantly contributes to the mechanism of Ala+TIMP-2 suppression increased cGMP, inhibition of PDE activity should reverse these effects.
To evaluate this hypothesis, we measured cGMP levels in the presence of an exogenous NO donor (DETA/NO) with or without Ala+TIMP-2 following pretreatment with a non-selective phosphodiesterase inhibitor isobutyl methyl xanthine (IBMX), Table 1. Pretreatment of hMVECs with IBMX, led to a 1.4-fold increase in basal level of cGMP. The cells treated with NO donors in the presence of Ala+TIMP-2 caused a 7.8-fold increase in cGMP levels while the cells treated with NO donors resulted in a 1.8-fold increase in cGMP compared with cells not treated with IBMX. Two-way ANOVA test of these data show that both the effects of the Ala+TIMP-2 treatment and the IBMX are highly significant (p<0.001) when compared with the control and DETA/NO alone groups. These results clearly suggest that the TIMP-2-mediated signaling is capable of inducing activity of a PDE isoform. However, the identification of the specific PDE is beyond the scope of the present study.
Table 1.
Effects of PDE Inhibition on cGMP levels in VEGF-A and Ala+TIMP-2 treated hMVECs.
| Total cytosolic cGMP levels (pmole/mg total protein) | |||
|---|---|---|---|
| Basal | DETA/NO | Ala+TIMP-2+DETA/NO | |
| -IBMX | 1.0±0.4 | 4.8±0.07 | 0.8±0.07 |
| +IBMX | 1.4±0.03 | 8.5±0.04 | 6.2±0.15 |
| Fold increase | 1.4 | 1.8 | 7.8* |
Two-way ANOVA test of these data show that both the effects of the Ala+TIMP-2 treatment and the IBMX are highly significant (p<0.001) when compared with the control and DETA/NO alone groups.
Discussion
The anti-mitogenic effects of TIMP-2 in both neoplastic and endothelial cells are independent of the ability to inhibit MMP activity (9, 11, 31). Furthermore, the anti-mitogenic activity of TIMP-2 and the Ala+TIMP-2 analog are mediated by a mechanism of heterologous receptor inactivation involving the binding of Ala+TIMP-2 (or TIMP-2) to the α3β1 integrin receptor enhancing PTP activity resulting in reduced activation of the cognate RTKs (11). Murine genetic models, siRNA knockdown and biochemical experiments have been used to demonstrated the requirement for α3 and β1 integrin subunits, and definitively identified the PTP activity as the protein tyrosine phosphatase Shp-1 (11, 12, 31). Subsequent work demonstrated that Ala+TIMP-2 could disrupt FGF-2-mediated activation of mitogen-activated protein kinase pathway (MAPK) via an integrin α1 and Shp-1 dependent mechanism (10).
In the present report we have extended these studies to show that Ala+TIMP-2 suppresses maximal phosphorylation of specific tyrosine residues in the cytoplasmic domain of VEGFR-2, that diminished receptor activation is manifest in disruption of downstream signaling as evidenced by the abatement of PLCγ activation and associated cytosolic Ca+2 flux, as well as curtailment of PI3K/AKT activation. We also demonstrate that Ala+TIMP-2 abates the maximal activation of eNOS and increased NO levels observed following VEGF-A stimulation, as well as the subsequent elevation of cGMP levels, associated with an enhanced mitogenesis and migration of endothelial cells. Our study also shows that Ala+TIMP-2 regulation of cGMP levels can occur independently of PTP activity via regulation of PDE activity. Although we do not investigate the precise mechanism of PDE activation, prior work from our laboratory has implicated cyclic nucleotides in TIMP-2-mediated cell growth regulation (32), and recent work has confirmed TIMP-2 mediated elevation of cAMP levels (Seo and Stetler-Stevenson, unpublished) consistent with the differential regulation of endothelial barrier function by PDE isoforms (33).
Ala+TIMP-2 reduced phosphorylation of selective tyrosine residues on the cytoplasmic domain of VEGFR-2. Ala+TIMP-2 suppressed maximal phosphorylation at three tyrosine residues, Y951, Y996 and Y1175. Y951 and Y996 are located in the tyrosine kinase insert domain of VEGFR-2. Y951 phosphorylation results in enhanced association of this receptor with the adaptor molecule VRAP/TSAd, and increases endothelial cell migration and proliferation via both PLC-γ and PI3K (7, 34). Ala+TIMP-2 also reduced phosphorylation of Y1175, which is associated with the direct binding and activation of PLC-γ, as well as activation of PI3K downstream signaling pathways, both resulting in enhanced endothelial cell migration and proliferation via the MAPK pathway (21).
Both Y951 and Y1175 of VEGFR-2 are implicated in VEGF-induced activation of eNOS, but the role of VEGFR-2 Y996 has not been definitively determined. The VEGFR-2 mutant Y951F suppresses NO release through inhibition of phosphorylation of eNOS at S1177 (35). VEGFR-2 Y1175 mediates AKT activation, also implicated in phosphorylation of eNOS on S1177 (23). In addition, PLC-γ activation through phosphorylation of Y1175 binding generates diacylglycerol and elevates intracellular Ca+2, which as previously noted is associated with calmodulin-mediated activation of eNOS (25). Therefore, we speculate that Ala+TIMP-2 combined suppression of both these mechanisms cooperates to inhibit NO production. In summary, Ala+TIMP-2 suppression of maximal VEGFR-2 phosphorylation on Y951 and Y1175 effectively eliminated all of the downstream signaling effects noted above as demonstrated either by reduced effector activity (PLC-γ, PI3K, AKT or eNOS activity) or decreased second messenger levels (Ca+2 levels, NO levels and cGMP levels), resulting in suppression of hMVEC proliferation and migration.
We have previously shown that Ala+TIMP-2 reduced phosphoryltion of VEGFR-2, EGFR and FGFR-1 via an increased association of Shp-1 with these receptors (10, 11, 31). This led us to examine the phosphorylation of VEGFR-2 cytoplasmic tail following Ala+TIMP-2 treatment. We speculate that the specificity of this heterologous receptor inactivation mechanism of Ala+TIMP-2 inhibition of RTK phosphorylation is mediated by creation of receptor SH2-domains created following activation of the receptor kinase domain. This hypothesis would explain the observation that phosphorylation of Y1059 on VEGFR-2 following Ala+TIMP-2 is unchanged, as this site is central for kinase activity. This finding also contrasts with a recent report describing autoregulation of VEGFR-2 phosphorylation by Shp-1 following VEGF-A binding in which Y1059 decreases, but Y951 is not effected (36). This difference suggests that heterologous vs. autologous down regulation of VEGFR-2 are distinct, and one consequence is the alteration of cell motility probably regulated by Y951.
In summary our present findings expand on previous reports demonstrating that the anti-angiogenic effects of TIMP-2 involve as Shp-1-dependent mechanism of heterologous receptor inactivation. We demonstrate that Ala+TIMP-2 selectively down regulates maximal phosphorylation of VEGFR-2 on Y951, Y996 and Y1175, with concomitant suppression of PI3K and PLCγ signaling, resulting in abrogation of VEGF-A stimulated hMVEC proliferation and migration. In addition, we observed that Ala+TIMP-2 enhanced activity of an IBMX-sensitive PDE that suppresses the cGMP increase following treatment with exogenous NO donors. Moreover, our results support the development of TIMP-2 as a novel, endogenous anti-angiogenic therapy that may complement those therapies already in clinical use or in preclinical trials.
Acknowledgments
The authors also thank Drs. Jensen-Taubman and Guedez for their critical reading of the manuscript. Current address for Dr. Lee: Shingyeong University, Room 206, Namyang-dong, Hwaseong-si, Gyeonggi-do, Republic of Korea 445-741
This study was supported by intramural research funds from the National Cancer Institute, Center for Cancer Research Project # Z01SC 009179.
Footnotes
Disclosure/Duality of Interest The authors have no conflict or duality of interest regarding this work to disclose.
References
- 1.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 2.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3(6):422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 4.Doggrell SA. Pegaptanib: the first antiangiogenic agent approved for neovascular macular degeneration. Expert Opin Pharmacother. 2005;6(8):1421–1423. doi: 10.1517/14656566.6.8.1421. [DOI] [PubMed] [Google Scholar]
- 5.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3(1):24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 6.Regillo CD, Brown DM, Abraham P, Yue H, Ianchulev T, Schneider S, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol. 2008;145(2):239–248. doi: 10.1016/j.ajo.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7(5):359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 8.Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477(1-2):267–283. doi: 10.1016/s0167-4838(99)00279-4. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez CA, Butterfield C, Jackson G, Moses MA. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): loop 6 is a novel angiogenesis inhibitor. Journal of Biological Chemistry. 2003;278(42):40989–40995. doi: 10.1074/jbc.M306176200. [DOI] [PubMed] [Google Scholar]
- 10.Seo DW, Kim SH, Eom SH, Yoon HJ, Cho YR, Kim PH, et al. TIMP-2 disrupts FGF-2-induced downstream signaling pathways. Microvascular Research. 2008;76(3):145–151. doi: 10.1016/j.mvr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, et al. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114(2):171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 12.Seo DW, Li H, Qu CK, Oh J, Kim YS, Diaz T, et al. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J Biol Chem. 2006;281(6):3711–3721. doi: 10.1074/jbc.M509932200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73(7):1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 14.Oh J, Diaz T, Wei B, Chang H, Noda M, Stetler-Stevenson WG. TIMP-2 upregulates RECK expression via dephosphorylation of paxillin tyrosine residues 31 and 118. Oncogene. 2006;25(30):4230–4234. doi: 10.1038/sj.onc.1209444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, et al. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J Biol Chem. 1999;274(30):21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- 16.Lee JS, Kang Decker N, Chatterjee S, Yao J, Friedman S, Shah V. Mechanisms of nitric oxide interplay with Rho GTPase family members in modulation of actin membrane dynamics in pericytes and fibroblasts. Am J Pathol. 2005;166(6):1861–1870. doi: 10.1016/S0002-9440(10)62495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saini R, Patel S, Saluja R, Sahasrabuddhe AA, Singh MP, Habib S, et al. Nitric oxide synthase localization in the rat neutrophils: immunocytochemical, molecular, and biochemical studies. Journal of Leukocyte Biology. 2006;79(3):519–528. doi: 10.1189/jlb.0605320. [DOI] [PubMed] [Google Scholar]
- 18.Guo D, Tan YC, Wang D, Madhusoodanan KS, Zheng Y, Maack T, et al. A Rac-cGMP signaling pathway. Cell. 2007;128(2):341–355. doi: 10.1016/j.cell.2006.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dougher M, Terman BI. Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene. 1999;18(8):1619–1627. doi: 10.1038/sj.onc.1202478. [DOI] [PubMed] [Google Scholar]
- 20.Poulin B, Sekiya F, Rhee SG. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc Natl Acad Sci U S A. 2005;102(12):4276–4281. doi: 10.1073/pnas.0409590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. Embo J. 2001;20(11):2768–2778. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeBell K, Graham L, Reischl I, Serrano C, Bonvini E, Rellahan B. Intramolecular regulation of phospholipase C-gamma1 by its C-terminal Src homology 2 domain. Molecular & Cellular Biology. 2007;27(3):854–863. doi: 10.1128/MCB.01400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, et al. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem. 2004;279(21):22267–22275. doi: 10.1074/jbc.M312729200. [DOI] [PubMed] [Google Scholar]
- 24.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 25.Busse R, Mulsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265(1-2):133–136. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- 26.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 27.Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33(18):5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 28.Oh J, Seo DW, Diaz T, Wei B, Ward Y, Ray JM, et al. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res. 2004;64(24):9062–9069. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- 29.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr (495) regulates Ca(2+)/calmodulin-dependnet endothelilal nitic oxide synthetase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 30.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD. Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A. 2005;102(37):13141–13146. doi: 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoegy SE, Oh HR, Corcoran ML, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. Journal of Biological Chemistry. 2001;276(5):3203–3214. doi: 10.1074/jbc.M008157200. [DOI] [PubMed] [Google Scholar]
- 32.Corcoran ML, Stetler-Stevenson WG. Tissue inhibitor of metalloproteinase-2 stimulates fibroblast proliferation via a cAMP-dependent mechanism. Journal of Biological Chemistry. 1995;270(22):13453–13459. doi: 10.1074/jbc.270.22.13453. [DOI] [PubMed] [Google Scholar]
- 33.DeFouw LM, DeFouw DO. Differential phosphodiesterase activity contributes to restrictive endothelial barrier function during angiogenesis. Microvasc Res. 2001;62(3):263–270. doi: 10.1006/mvre.2001.2340. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto T, Bohman S, Dixelius J, Berge T, Dimberg A, Magnusson P, et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. Embo J. 2005;24(13):2342–2353. doi: 10.1038/sj.emboj.7600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmad S, Hewett PW, Wang P, Al-Ani B, Cudmore M, Fujisawa T, et al. Direct evidence for endothelial vascular endothelial growth factor receptor-1 function in nitric oxide-mediated angiogenesis. Circ Res. 2006;99(7):715–722. doi: 10.1161/01.RES.0000243989.46006.b9. [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharya R, Kwon J, Wang E, Mukherjee P, Mukhopadhyay D. Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1 (SHP-1) dephosphorylates VEGF Receptor-2 and attenuates endothelial DNA synthesis, but not migration. J Mol Signal. 2008;3:8–20. doi: 10.1186/1750-2187-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]