

Abstract
Background
Recent studies of viral entry proteins from influenza, measles, human immunodeficiency virus, type 1 (HIV-1), and Ebola virus have shown, first with molecular modeling, and then X-ray crystallographic or other biophysical studies, that these disparate viruses share a coiled-coil type of entry protein.
Results
Structural models of the transmembrane glycoproteins (GP-2) of the Arenaviruses, lymphochoriomeningitis virus (LCMV) and Lassa fever virus, are presented, based on consistent structural propensities despite variation in the amino acid sequence. The principal features of the model, a hydrophobic amino terminus, and two antiparallel helices separated by a glycosylated, antigenic apex, are common to a number of otherwise disparate families of enveloped RNA viruses. Within the first amphipathic helix, demonstrable by circular dichroism of a peptide fragment, there is a highly conserved heptad repeat pattern proposed to mediate multimerization by coiled-coil interactions. The amino terminal 18 amino acids are 28% identical and 50% highly similar to the corresponding region of Ebola, a member of the Filovirus family. Within the second, charged helix just prior to membrane insertion there is also high similarity over the central 18 amino acids in corresponding regions of Lassa and Ebola, which may be further related to the similar region of HIV-1 defining a potent antiviral peptide analogue.
Conclusions
These findings indicate a common pattern of structure and function among viral transmembrane fusion proteins from a number of virus families. Such a pattern may define a viral transmembrane superfamily that evolved from a common precursor eons ago.
Background
Findings in a number of laboratories have indicated that the transmembrane (TM) proteins of a number of RNA viruses have common structural and functional elements critical for virus entry. These include a hydrophobic region designated a "fusion peptide", usually at or near the amino-terminus generated by cleavage of a precursor protein, together with fibrous structure defined by two antiparallel alpha helices. These general principles appear to apply to the Orthomyxoviruses, Paramyxoviruses, Retroviruses, Lentiviruses, and Filoviruses [1,2,3,4]. In some cases, such as between Ebola and Rous sarcoma viruses, there is considerable sequence identity to facilitate a comparison between two specific viruses [4]. In other cases, even within a single virus family such as the Retroviridae, both structural modeling and more limited sequence similarity must be combined to discern the relationship [3]. The finding of close sequence or structural similarity among otherwise disparate virus families has given rise to the concept of a viral TM superfamily sharing common structural and functional motifs [4]. Recent biophysical studies of entry protein structure have reinforced this concept [5,6].
In this respect, a general model of the Arenavirus glycoproteins, based on extensive study of lymphocytic choriomeningitis virus (LCMV) has been presented based on their overall similarity in functional organization to influenza and to other enveloped viruses. The GP-C precursor is proteolytically cleaved near a polybasic site to yield GP-1, a globular surface glycoprotein which contains receptor-binding sites, and GP-2, a TM protein forming the stalk of the complex via a coiled coil of amphipathic helices and responsible for virus entry by acid-dependent membrane fusion [7,8,9,10].
We present here a detailed model of GP-2 for Lassa fever virus, an Arenavirus associated with multiple epidemics of hemorrhagic fever with high morbidity and mortality in West Africa [11,12], and for the related lymphocytic choriomeningitis virus (LCMV) which has been associated with sporadic outbreaks of human disease in Europe and North America [12]. This model demonstrates that Arenaviruses share a number of specific sequence and structural motifs with other RNA viruses in the TM superfamily. Regions of Arenavirus GP-2 can be directly related to corresponding regions of Ebola, another agent of African hemorrhagic fever, and to HIV-1. Examination of the comparable regions of TM proteins from several virus families provides evidence suggesting divergence from a common ancestor.
Results and Discussion
The detailed model of LCM and Lassa fever virus GP-2 is shown in Figure 1. As shown previously for other members of the TM superfamily, both consist of two antiparallel helices separated by a disulfide-linked apex. The sequence of GP-2 contains a highly conserved hydrophobic sequence, LAGTFTWTL (in LCM) or LLGTFTWTL (in Lassa), in the vicinity of the post-translational cleavage site, with a canonical fusion tripeptide Gly-X-Phe. However, its candidacy as a functional fusion peptide, analogous to those of influenza, measles and HIV-1, has been questioned due to its weakly hydrophobic character and the fact that actual cleavage occurs, as shown in the models, not at the dibasic amino acids but within the hydrophobic site [13,14]. LCM virus does employ an acid dependent fusion event to enter the cell but virion-cell fusion is inactive above pH 6, and the Arenaviruses have never been demonstrated to exhibit cell-cell fusion. While this amino-terminus may not be a good candidate for a classical fusion peptide, its hydrophobic nature and position suggest that it may at least be the vestige of one.
Figure 1.
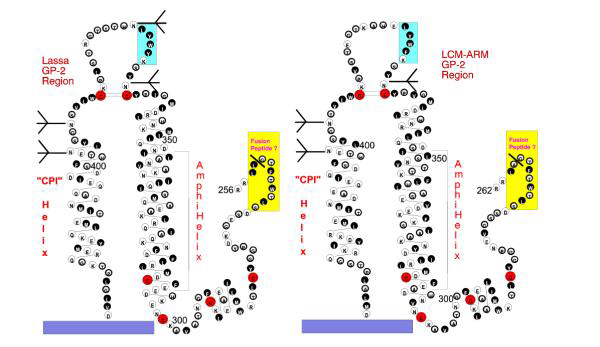
Models of the Transmembrane Proteins GP-2 of Lassa Virus and LCMV. Projections of the model structure for GP-2 of Lassa and LCMV are shown in parallel with one another, based on the consensus structures for algorithms of both viral amino acid sequences. Proposed helices are shown in helical net projection with sequential amino acids connected by solid lines. Proposed disulfide linkages are indicated by double lines. Hydrophobic amino acids are grouped with a solid background; neutral amino acids with a heavily outlined circle; hydrophilic amino acids with a light circle; glycosylation sites indicated by tridents. A heavy solid line indicates the point of proteolytic cleavage of the GP-C precursor protein to yield GP-1 and GP-2. Cysteines are highlighted by larger red circles. The surface membrane of the virus is indicated by a solid purple rectangle, the amino-terminal hydrophobic region by a yellow rectangle, and the conserved B-cell epitope by a blue rectangle. The two proposed antiparallel helices are labeled "AmphiHelix" for the extended heptad repeat, and "CPI Helix" for the charged, pre-insertion helix.
The region prior to the first helix consists first of a glycine-serine rich linker, and then a domain that is highly conserved among all Arenaviruses and contains four cysteines. Only the last of these four is conserved between the Filoviruses and Arenaviruses. We have not assigned disulfide linkages for these since there are neither data nor parallels with other viruses to permit such assignments. Since there is no disulfide cross-linkage with GP-1, these must participate in disulfide bonding within the same GP-2 protein, or in cross-linking GP-2 oligomers. The latter possibility is suggested by the kinetics of GP-2 association with experimental addition of reducing agent, indicating first a change in vitro from tetramers to dimers and then to monomers only after considerable additional reduction [8]. Whether the native multimeric form of GP-2 in the virion may be a trimer, as for the fusion glycoproteins of Retroviruses or Filoviruses, is yet to be determined.
The amino-terminal helices of both consist of extended amphipathic arrays with strong heptad repeats that have been previously noted [15], and are thought to form the backbone of the coiled-coil stalk of the viral glycoprotein complex [16] A peptide analogue of this extended heptad repeat in LCMV, GP-C 326-355, was examined by circular dichroism under different solvent conditions, as shown in Table 1. The peptide exhibited only limited helicity in aqueous solution, but 79% alpha helix in a neutral hydrophobic environment. This biophysical behavior is reminiscent of that of other similar peptides derived from the corresponding sequences of Paramyxoviral or Retroviral TM proteins [2,17].
Table 1.
Circular Dichroism Spectroscopy of Peptide GPC 326-355
| Θ222 (deg cm2 dmol-1) | % alpha helicity (100%=32,000) | ||
| Aqueous | Neutral | 4523 | 14.1 |
| Acid | 3777 | 11.8 | |
| TFE | Neutral | 25314 | 79.1 |
| Acid | 11565 | 36.1 |
Circular Dichroism and calculated per cent alpha helix for the peptide derived from the LCM sequence NKAALSKFKEDVESALHLFKTTVNSLISDQ, as described in Materials and Methods.
Comparison of the sequences of Lassa and LCM over this amphipathic heptad-repeat region (below) shows 31 identical of 58 amino acids, with the principal areas of conservation of sequence at the amino- and carboxy-terminal ends of the amphipathic helix.
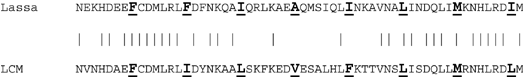
The middle 25 amino acids appear poorly conserved, with only 6 of 25 identical, yet the character of the amino acids substituted is generally conserved. In particular, while none of the central 4 heptad amino acids (underlined and in bold) are identical in each virus, in all cases the hydrophobic character of the heptad repeat is maintained.
The apical domain is the only region to be glycosylated, also in line with a number of TM proteins including that of HIV-1 and other Retroviruses. The apical sequence, particularly the peptide KFWYL in LCMV or KYWYL in Lassa, defines a broadly-cross reactive antibody epitope shared by these viruses [18] that is in precisely the same topographical location as the broadly-reactive apical epitope (positions 598-609, LGIWGCSGKLIC) that has been finely mapped in HIV-1 [19]. Also like that in HIV-1, it is responsive to multimer conformation, and increasingly exposed after receptor binding that results in release of the binding subunit, GP-1 [13].
The second helical region has properties similar to that of the Retroviruses and Filoviruses, in that it is highly charged (30%) and amphipathic, with its helicity possibly stabilized by multiple ion pairings of acidic and basic residues, as first noted for the corresponding region of HIV-1 [3]
Although Lassa fever and Ebola viruses represent different virus families, both helices share an unexpectedly high sequence homology. The first lies in the amino-terminal half of the extended amphipathic helix. As shown in a concentric helical wheel projection in Figure 2A, when the helices are oriented with respect to the exclusion of charge and the heptad repeats for each sequence, 9 identical or highly similar amino acids (50%) may be aligned in each sequence.
Figure 2.
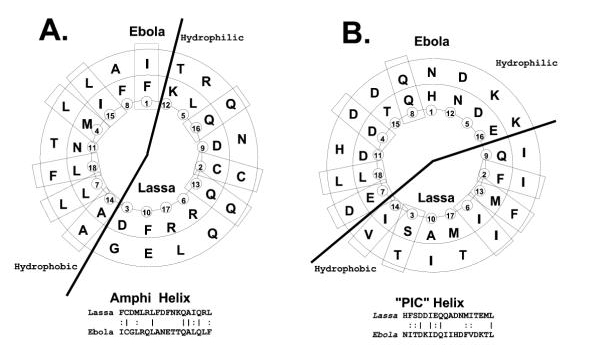
Concentric Helical Wheel Projections of Proposed Lassa and Ebola Helices. Helical wheel projections are shown for 18 amino acid segments of the proposed helices of Lassa and Ebola viruses. The projections are arranged concentrically to align the viral sequences, with the inner sequence that from Lassa virus, and the outer sequence from Ebola. Zaire virus (Genbank U31033). Rectangles indicate identical or highly similar residues in each sequence. Heavy lines indicate the hemicylinder of charge exclusion (hydrophobic) subtending an angle of 160° in each alignment. Below each wheel projection the linear sequences are also aligned to show the identities (solid lines) and high similarities (dotted lines). A. Concentric alignment of the amino terminal amphipathic helices ("lower amphi") of Lassa (amino acids 309-326, in wheel positions 1-18 respectively) and Ebola (555-572). B. Concentric alignment of the charged, pre-insertion helices ("CPI Helix") of Lassa (398-415) and Ebola (618-635).
The carboxy-terminal helical region also has properties in common with the similarly located helices in both Lassa and Ebola, shown as a concentric helical wheel projection in Figure 2B. Again, orienting the helix with respect to the hemicylindrical exclusion of charge, 9 identical or highly similar amino acids (50%) may be aligned. Furthermore, none of the amino acid differences represent a radical change of one sequence from the other.
Arenaviruses therefore share with a number of other virus families a fusion/entry protein GP-2 that appears to have the four cardinal structural features typical of proteins in the viral transmembrane entry protein superfamily. Our model of the extramembranal portion of GP-2 begins with a hydrophobic fusion peptide sequence, followed by two antiparallel extended helices, the first of which contains a strong heptad repeat sequence, which lie on either side of a disulfide-stabilized, glycosylated and strongly antigenic reverse turn. These features have been apparently maintained in spite of diversity in primary amino acid sequence within the Arenavirus family.
Conclusions
The most likely explanation for such high levels of similarity among Arenaviruses and Filoviruses would be divergence of both of these agents from a common viral ancestor. Since both virus families exhibit type variation over large areas coupled with stability among isolates within a more limited geographical area over considerable periods of time (the Arenaviruses being the more widespread) such divergence must have occurred eons ago. The potential importance of such apparent conservation in the biology of these agents is underscored by noting that of the corresponding peptide sequences within the TM superfamily of proteins, that for HIV-1 forms the center of a peptide analogue shown to inhibit fusion in the nanomolar range [20].
Modeling studies begun in the late 1980s have thus revealed a number of common and sequence motifs, subsequently shown in several cases to have homologous biological roles in infection, that were not otherwise apparent in studies of sequence homology. These models may lead to a common strategy of antiviral inhibition preventing entry of virus into host cells that is broadly applicable over a broad range of very diverse virus families.
Materials and Methods
Molecular Modeling
Sequences used for this analysis were LCMV - ARM (Genbank P09991) and Lassa, Josiah (Genbank P08669), and are numbered from the initiation methionine. Detailed models of the Arenavirus GP-1 proteins were determined by the methods of Gallaher et al. previously described [3,4,21] A consensus of several independent structural algorithms is used, and compared for different GP-2 sequences to test the consensus. The resulting model is an average consensus of the algorithms for these two sequences. Models are projected in helical net or helical wheel projections also as previously described.
Peptide Synthesis and Circular Dichroism
A peptide corresponding to amino acid positions 326-355 of LCMV-ARM-4 (Genbank VGXPLM) in single letter code, NKAALSKFKEDVESALHLFKTTVNSLISDQ, with an additional histidine at the N terminus was synthesized by standard BOC chemistry using double coupling and HF cleavage. The peptide was purified by reverse phase HPLC on a C-18 column and the peptide's weight confirmed by mass spectroscopy. The peptide was selected as predicted by the Lupas algorithm [22] to have a greater than 90% probability of forming a heptad repeat in the native protein structure. Peptide samples for circular dichroism (CD) were prepared at 0.1 mg/ml concentrations in either 1 mM NaCO3, pH 7.2 (Neutral) or in 100 mM MES, pH 5.5 (Acid). In spectra recorded with TFE, the TFE was present as 45% of solution volume. CD spectra were recorded from 300-180 nm with 0.5 nm steps with a pathlength of 0.1 cm and at 4°C. Final values were determined using the average of 15 spectra which were correlated with baseline spectra of buffer samples. A characteristic alpha helical spectrum was apparent for the peptide when placed in TFE with a positive peak at 195 nm (Θ = +43000) and a second minimum peak at 210 nm (Θ = -25000).
Acknowledgments
Acknowledgements
We thank Drs. Robert Garry, Ronald Luftig and Nicolas Bazan for their suggestions and encouragement, and Ms. Leonita G. Liu for help in preparing illustrations. This work was supported by grants from the U.S. National Institute of Dental Research DE 10862 (W.R.G.), an NIH training grant AI 07354 (C.D.) and an Emerging Research Center Grant from the NIH AI 39808 (M.J.B.).
Contributor Information
William R Gallaher, Email: wgalla@lsuhsc.edu.
Christopher DiSimone, Email: cxdisimone@yahoo.com.
Michael J Buchmeier, Email: buchm@scripps.edu.
References
- Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR., Jr Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc Nat Acad Sci USA. 1996;93:2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallaher WR, Ball JM, Garry RF, Griffin MC, Montelaro RC. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res Human Retroviruses. 1989;5:431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- Gallaher WR. Similar structural models of the transmembrane proteins of Ebola and avian sarcoma viruses. Cell. 1996;85:477–478. doi: 10.1016/s0092-8674(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W, Dessen A, Calder LJ, Harrison SC, Skehel JJ, Wiley DC. Structural basis for membrane fusion by enveloped viruses. Molecular Membrane Biology. 1999;16:3–9. doi: 10.1080/096876899294706. [DOI] [PubMed] [Google Scholar]
- Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Molecular Cell. 1998;2:605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- Wright KE, Spiro RC, Burns JW, Buchmeier MJ. Post-translational processing of the glycoproteins of lymphocytic choriomeningitis virus. Virology. 1990;177:175–83. doi: 10.1016/0042-6822(90)90471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JW, Buchmeier MJ. Protein-protein interactions in lymphocytic choriomeningitis virus. Virology. 1991;183:620–629. doi: 10.1016/0042-6822(91)90991-j. [DOI] [PubMed] [Google Scholar]
- Di Simone C, Buchmeier MJ. Kinetics and pH dependence of acid-induced structural changes in the lymphocytic choriomeningitis virus glycoprotein complex. Virology. 1995;209:3–9. doi: 10.1006/viro.1995.1225. [DOI] [PubMed] [Google Scholar]
- Borrow P, Oldstone MB. Characterization of lymphocytic choriomeningitis virus-binding protein(s): a candidate cellular receptor for the virus. J Virol. 1992;66:7270–7281. doi: 10.1128/jvi.66.12.7270-7281.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame JD, Baldwin JM, Jr, Gocke DJ, Troup JM. Lassa fever, a new virus disease of man from West Africa. I. Clinical description and pathological findings. Am J Trop Med Hyg. 1970;19:670–676. doi: 10.4269/ajtmh.1970.19.670. [DOI] [PubMed] [Google Scholar]
- Peters CJ, Buchmeier MJ, Rollin PE, Ksiazek TG. Arenaviruses (Chapter 50). In Virology; third Edition Edited by BN Fields, et al Lippincott-Raven, 1996. pp. 1521–1551.
- Di Simone C, Zandonatti MA, Buchmeier MJ. Acidic pH triggers LCMV membrane fusion activity and conformational change in the glycoprotein spike. Virology. 1994;198:455–465. doi: 10.1006/viro.1994.1057. [DOI] [PubMed] [Google Scholar]
- Lenz O, ter Meulen J, Feldmann H, Klenk H-D, Garten W. Identification of a novel consensus sequence at the cleavage site of the Lassa virus glycoprotein. J Virol. 2000;74:11418–11421. doi: 10.1128/jvi.74.23.11418-11421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers P, Pringle CR, Easton AJ. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J Gen Virol. 1990;71:3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- Burns J.W., Buchmeier M.J. Glycoproteins of the Arenaviruses. In The Arenaviridae Edited by MS Salvato Plenum Press, 1993. pp. 17–35.
- Wild C, Dubay JW, Greenwell T, Baird T, Jr, Oas TG, McDanal C, Hunter E, Matthews T. Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc Nat Acad Sci USA. 1994;91:12676–12680. doi: 10.1073/pnas.91.26.12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EL, Buchmeier MJ. Fine mapping of a peptide sequence containing an antigenic site conserved among arenaviruses. Virology. 1988;164:30–38. doi: 10.1016/0042-6822(88)90616-2. [DOI] [PubMed] [Google Scholar]
- Gnann JW, Jr, Nelson JA, Oldstone MB. Fine mapping of an immunodominant domain in the transmembrane glycoprotein of human immunodeficiency virus. J Virol. 1987;61:2639–2641. doi: 10.1128/jvi.61.8.2639-2641.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild C, Greenwell T, Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Human Retroviruses. 1993;9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- Gallaher WR, Ball JM, Garry RF, Martin-Amedee AM, Montelaro RC. A general model for the surface glycoproteins of HIV and other retroviruses. AIDS Res Human Retroviruses. 1995;11:191–202. doi: 10.1089/aid.1995.11.191. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]