

Abstract
Myotonic muscular dystrophy (DM1) is the most common inherited neuromuscular disorder in adults and is considered the first example of a disease caused by RNA toxicity. Using a reversible transgenic mouse model of RNA toxicity in DM1, we provide evidence that DM1 is associated with induced NKX2-5 expression. Transgene expression resulted in cardiac conduction defects, increased expression of the cardiac-specific transcription factor NKX2-5 and profound disturbances in connexin 40 and connexin 43. Notably, overexpression of the DMPK 3′ UTR mRNA in mouse skeletal muscle also induced transcriptional activation of Nkx2-5 and its targets. In human muscles, these changes were specific to DM1 and were not present in other muscular dystrophies. The effects on NKX2-5 and its downstream targets were reversed by silencing toxic RNA expression. Furthermore, using Nkx2-5+/− mice, we show that NKX2-5 is the first genetic modifier of DM1-associated RNA toxicity in the heart.
DM1, an autosomal dominant disorder mapping to chromosome 19q, is the most common inherited neuromuscular disorder in adults, with an incidence of 1:8,000 globally. Common features of this multi-systemic disease in adults include myotonia, progressive skeletal muscle loss, cardiac conduction abnormalities, smooth muscle dysfunction, hypersomnolence, cataracts, testicular atrophy, frontal balding and insulin resistance1. DM1 is caused by an expansion of a (CTG)n triplet repeat in the 3′ UTR of the gene encoding DM protein kinase (DMPK) from a normal range (n = 5 to ~30) to greater than several thousand repeats (n = 50 to >2,000)2. Individuals with DM1 have a proclivity for cardiac conduction abnormalities, with about 60%–70% developing substantial problems that often result in sudden death3. These characteristic conduction disturbances consist primarily of sinoatrial and atrioventricular blocks of varying degrees of severity, resulting in slow and irregular heartbeats, and they are often accompanied by fibrosis, fatty infiltration and atrophy of the conduction system and the myocardium.
As the mutation responsible for DM1 lies in a noncoding region of DMPK, several pathogenic mechanisms have been proposed (reviewed in ref. 4), but accumulating data suggest that mutant DMPK mRNAs have a role in many DM1 phenotypes5–9. The DM1 mutation results in an mRNA that is sequestered in the nucleus10. DM2, another almost identical clinical disorder, is caused by an expansion of a (CCTG)n sequence in the first intron of ZNF9 and also results in ribonuclear inclusions11. This discovery gave strong credence to the RNA toxicity hypothesis of myotonic dystrophy pathogenesis, wherein the mutant transcripts alter the function of RNA splicing factors, primarily members of the muscleblind-like (MBNL) and CUG-BP and ETR-3-like factor (CELF) family of RNA-binding proteins, either by sequestering them as part of the ribonuclear inclusions (as for MBNL1) or by increasing their expression (as for CUG-BP1). These proteins are RNA splicing factors, and altering their functional levels in adult tissues results in aberrant splicing and reversion to embryonic splicing patterns for various target mRNAs that are normally regulated under a mutual antagonism of MBNL1 and CUG-BP1 (ref. 12).
The mechanisms underlying the cardiac conduction defects in DM1 are unknown. Thorough molecular analysis of tissues from individuals with myotonic dystrophy has not been performed owing to the inherent difficulties associated with obtaining cardiac tissues from living donors. The analysis of the few available autopsy samples is also complicated by the fact that the individuals usually have had a long history of disease and associated comorbidities that could result in secondary effects. Based on mouse models, haploinsufficiency of DMPK had been posited as a potential pathogenic mechanism13,14. However, with the discovery of the mutation causing DM2 and the postulation of a common mechanism, attention has been placed on the RNA toxicity pathogenesis. Several aberrant splicing events resulting in a reversion to embryonic splicing patterns have been identified in hearts from individuals with DM1, but their relationship to DM1 pathology is unclear15.
We recently developed an inducible mouse model of RNA toxicity in DM1 in which we have clearly demonstrated the reversible nature of the DMPK 3′ UTR mRNA toxicity16. These strains (named 5-313 and 5-336) express a green fluorescent protein (GFP) gene fused with the DMPK 3′ UTR (denoted GFP-DMPK 3′ UTR) under the control of a tetracycline-inducible human DMPK promoter. Notably, upon induction, the mice overexpress a normal DMPK 3′ UTR mRNA with only (CUG)5 and have no apparent ribonuclear inclusions or evidence of reported RNA splicing defects in the heart16. They rapidly develop myotonia (the cardinal feature of myotonic dystrophy) and progressive heart block (Fig. 1a), a phenotypic combination seen only in individuals with myotonic dystrophy. This model represents the first clear demonstration of RNA toxicity resulting in cardiac conduction abnormalities. Here we explore the molecular basis of this phenomenon and report how this led to the unanticipated finding of induced expression of NKX2-5 in cardiac and skeletal muscle and describe the identification of NKX2-5 as a genetic modifier of the cardiotoxicity caused by the DMPK 3′ UTR mRNA.
Figure 1.

Cardiac conduction abnormalities. (a) ECG abnormalities in induced transgenic mice. P (atrial) and R (ventricular) waves are indicated. * denotes missing R waves in second-degree block. Note the lack of P waves in the complete heart block example. (b) Immuno-histochemistry of a cross-section of mouse hearts, showing CX43 (green), CX40 (red) and CX40 CX43 coexpression (orange). Note the thinning and loss of CX40 staining in the conduction system (Purkinje fibers) of induced mice. (c) CX43 expression is lower in ventricular myocardium of induced mice than in uninduced mice, as detected by immunohistochemistry (IHC), RT-PCR (mean ± s.d.) and protein blotting. Numbers above lanes in protein blot represent the number days of transgene induction. Numbers below lanes represent relative CX43 expression compared to wild-type mice. Numbers below days 6–17 represent the average of those samples. GAPDH was included as a loading control.
RESULTS
Connexin 40 and 43 are affected by RNA toxicity
Normal cardiac conduction is mediated by orderly propagation of electrical impulses from one cardiomyocyte to the next. Connexin proteins are essential components of gap junctions that are required for this propagation, and their importance in cardiac development and conduction abnormalities has been borne out by a number of mouse models17. Deficiencies in connexin 43 (CX43) and connexin 40 (CX40) result in cardiac conduction defects18. In adult mice, CX43 is expressed by cardiomyocytes throughout the myocardium. The expression of CX40 is restricted to the atria and the cardiac conduction system (the His bundle, its two branches and the Purkinje fibers)17, regions that are prime targets for the type of abnormalities seen in individuals with DM1 and in our transgenic mice.
Using the 5-313 transgenic mice, we evaluated the expression of CX40 and CX43 in the heart by fluorescent immunohistochemistry. Doxycycline-treated mice (which we refer to hereafter as ‘induced mice’) that had cardiac conduction defects showed substantial reductions in CX40 in the His bundle, its branches and the Purkinje fibers of the sub-endocardium, compared with CX40 expression in untreated mice (which we refer to hereafter as ‘uninduced mice’) (Fig. 1b). CX40 in the blood vessels of the heart or the atria did not appear to be affected. CX43 also seemed to show a generalized reduction in the ventricular myocardium, especially in mice with severe conduction abnormalities (Fig. 1c). We confirmed reductions in CX43 by protein blotting and quantitative RT-PCR (Fig. 1c). CX43 expression declined rapidly beginning 5 d after transgene induction, corresponding to the changes in cardiac conduction.
NKX2-5 is induced by RNA toxicity in the heart
The cardiac transcription factor NKX2-5 is thought to be a key factor in the regulation of connexin expression and in the maintenance of normal cardiac conduction17,19. NKX2-5 is a member of the NK homeobox family; it acts as a specific transcriptional activator or repressor and interacts with other transcription factors such as GATA4, TBX5 and SRF19. NKX2-5 is expressed predominantly in cardiac tissues from early embryonic development through adulthood. In humans, dominant mutations in NKX2-5 cause heart block, ranging from prolonged PR intervals (first-degree block) to complete (third-degree) block20, exactly like the cardiac effects observed in our mice. Furthermore, depending on genetic background, some heterozygous Nkx2-5+/− mice develop prolonged PR intervals (a measure of cardiac conduction speed from the sinoatrial node to the ventricle)21,22, and transgenic mice overexpressing a mutant NKX2-5 (with amino acid change I183P) develop progressive heart block and demonstrate substantial downregulation of CX40 and CX43 (refs. 23,24).
Therefore, we hypothesized that NKX2-5 expression was reduced in the hearts of induced mice. Immunofluorescence microscopy showed that NKX2-5 was expressed in cardiomyocytes (as assessed by double detection of dystrophin and NKX2.5; Supplementary Fig. 1 online). We were surprised, however, to find that on average, the induced mice had significantly more nuclei with detectable NKX2-5, compared with uninduced mice (306 ± 70 versus 185 ± 50, P = 0.002; Fig. 2a), although there were no obvious differences in cardiac size, weight or morphology. In 5-313 mice heterozygous for the GFP-DMPK 3′ UTR transgene locus (hereafter termed ‘5-313 heterozygotes’) co-expressing a Nkx2-5LacZ allele, we did not find any difference in the number of nuclei expressing β-galactosidase between induced mice and uninduced mice (205 ± 37 versus 192 ± 13, respectively; P = 0.24), suggesting that the higher numbers of NKX2-5 nuclei shown by immunohistochemistry are due to increased expression rather than an increase in the number of cardiomyocytes. Quantitative RT-PCR (Fig. 2a) and RNA blotting showed a corresponding increase in Nkx2-5 mRNA expression in the hearts of mice expressing the transgene (Fig. 2b), suggesting that the DMPK 3′ UTR mRNA may upregulate Nkx2-5 transcription. These effects on Nkx2-5, Gia5 (Cx40) and Gia1 (Cx43) are probably not due to alternative splicing defects, as they each consist of only two exons.
Figure 2.

NKX2-5 expression is induced by RNA toxicity in the heart. (a) Immunohistochemistry for NKX2-5 in ventricular myocardium demonstrates that induced transgenic mice have more nuclei with detectable NKX2-5, compared to uninduced mice (shown graphically in the lower left panel; mean ± s.d.; P = 0.002). RT-PCR demonstrates that Nkx2-5 mRNA expression in heart is also higher in induced mice (lower right panel; mean ± s.d.; P = 0.03). (b) RNA blotting confirmed RT-PCR results (numbers below the blot refer to expression level relative to uninduced mice). GAPDH was used as a loading control.
RNA toxicity in skeletal muscle induces NKX2-5 expression
During embryonic development, NKX2-5 is expressed primarily in the heart and sparsely in a few extracardiac tissues, including pharyngeal and laryngeal tissues, liver, spleen, stomach, occipital muscles and tongue25,26. Expression in extracardiac tissues is not evident in adult tissues25,26. Specifically, there is no evidence for Nkx2-5 expression in skeletal muscle or myoblasts. However, a recent study suggested that there may be low expression of NKX2-5 in C2C12 mouse skeletal myoblasts and that overexpression of NKX2-5 in those cells results in defects in myogenic differentiation27, a phenotype seen in cell culture models of DMPK 3′ UTR mRNA toxicity6 and in DM1 myoblasts28.
This led us to consider the possibility of induced NKX2-5 expression in skeletal muscle of transgenic mice expressing the toxic RNA. Immunofluorescence assays for NKX2-5 in skeletal muscle of mice overexpressing the GFP-DMPK 3′ UTR showed strong staining for NKX2-5 in myonuclei (Fig. 3a). We observed Nkx2-5 mRNA (as detected by RT-PCR; data not shown) and NKX2-5 protein only in skeletal muscle of mice expressing the toxic RNA (Fig. 3b). We did not observe expression of Nkx2-5 mRNA or NKX2-5 protein in the skeletal muscles of wild-type or uninduced mice, mdx mice (a mouse model for Duchenne muscular dystrophy) or mice overexpressing GFP29 (data not shown). The effect on Nkx2-5 seemed specific, as we did not detect induced expression by microarray analysis, RT-PCR or immunohistochemistry of Tbx5, Tbx20 or Gata4 in skeletal muscle of mice expressing the toxic RNA (data not shown).
Figure 3.

NKX2-5 is ectopically induced in skeletal muscle. (a) Transgene expression in induced mice. DAPI staining marks nuclei; GFP expression indicates transgene expression. Immunohistochemistry for NKX2-5 (red) shows that there is no expression in uninduced skeletal myonuclei but markedly increased expression in muscle from induced mice. (b) Protein blotting for NKX2-5 in skeletal muscle. GAPDH was used as a loading control.
NKX2-5 downstream targets are affected
To evaluate the biological relevance of induced Nkx2-5 expression in cardiac and skeletal muscle, we assessed the expression of reported NKX2-5 downstream targets19 by RT-PCR in uninduced and induced 5-313 and 5-336 mice. In cardiac tissue from induced 5-313 mice in addition to the effects on Gia1 (Fig. 1c), we found higher expression of Nppb (brain natriutretic peptide, also known as Bnp) and Ankrd1 (cardiac ankryin repeat protein, formerly known as Carp) and lower expression of Gia5 (Fig. 4a). In skeletal muscle of induced 5-313 mice, we found induced expression of Nppb, Ankrd1 and adenosine A1 receptor (Adora1) (Fig. 4b). In skeletal muscle of induced 5-336 mice, we observed even higher expression of these downstream targets: Nppb expression increased 645-fold (P = 0.01), Ankrd1 expression increased 116-fold (P = 0.007) and Adora1 expression increased 13-fold (P = 0.01). Expression of Nppa (atrial natriuretic peptide, also known as Anp) mRNA was about nine times higher in the induced 5-313 mice, although this did not reach statistical significance (P = 0.09), and it was 24 times higher in induced 5-336 mice (P = 0.003). Expression of Nppa and Nppb, which are both well-characterized transcriptional targets of NKX2-5 and are highly expressed in cardiomyocytes, was barely detectable, if at all, in skeletal muscle of wild-type FVB mice, mice overexpressing GFP and mdx mice (data not shown). Protein blotting for CARP expression in skeletal muscle confirmed the RT-PCR results (Fig. 4c). In addition, expression of CX43 was decreased in skeletal muscle of induced mice (Fig. 4c).
Figure 4.

NKX2-5 downstream targets are affected by induction of transgene expression. (a) Real-time RT-PCR results for Nppb, Ankrd1 and Gia5 mRNA in hearts of 5-313 transgenic mice, relative to uninduced 5-313 mice (mean ± s.d.). (b) Real-time RT-PCR results for Nppb, Ankrd1 and Adora1 mRNA in skeletal muscle of 5-313 transgenic mice, relative to uninduced 5-313 mice (mean ± s.d.). Note reversion toward normal levels with transgene silencing. (c) Protein blots of downstream targets in skeletal muscle extracts demonstrate that expression of CARP is induced and CX43 is downregulated by RNA toxicity, and normal levels are restored in reverted mice. Lanes 1–2: uninduced mice; 3–5: induced mice; 6–8: reverted mice. GAPDH was used as a loading control.
NKX2-5 is sufficient to affect its targets in myoblasts
NKX2-5 is known to interact with a number of partners to affect its targets. To directly test if overexpression of NKX2-5 was sufficient to affect its downstream targets in skeletal myoblasts, we used transient overexpression of Nkx2-5 in C2C12 mouse myoblasts. This resulted in dose-dependent upregulation of Nppa and Nppb (Fig. 5), both of which are considered to be expressed normally only in the heart in adults.
Figure 5.
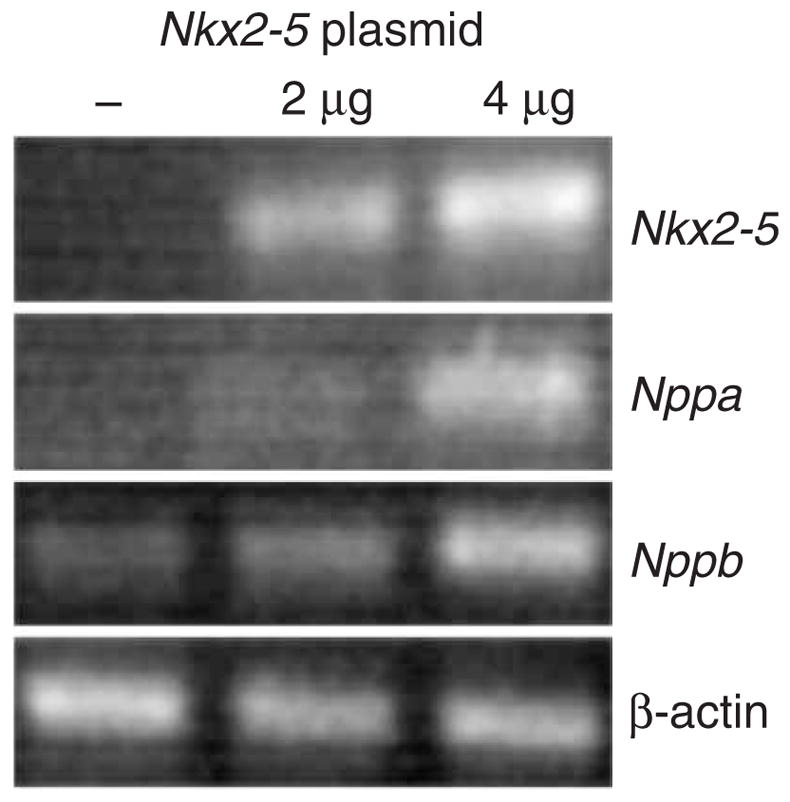
NKX2-5 is sufficient to transcriptionally activate downstream targets in skeletal myoblasts. RT-PCR of transient transfection assays demonstrates that Nkx2-5 expression in C2C12 skeletal myoblasts is sufficient to drive dose-dependent expression of cardiac-specific downstream targets Nppa and Nppb. β-actin is a loading control. Leftmost lane is a negative control (no plasmid).
NKX2-5 is induced in DM1
To assess the potential clinical relevance of this finding, we performed immunofluorescence staining for NKX2-5 in human skeletal muscle samples. Normal muscle had no detectable NKX2-5, whereas skeletal muscle from an individual with adult-onset DM1 showed clear expression of NKX2-5 in myonuclei (Fig. 6a). We confirmed these results by protein blotting (Fig. 6b) and by RT-PCR (Fig. 6c). We analyzed skeletal muscle samples from five different individuals with DM1, seven controls without muscular dystrophy and four individuals with other muscle diseases (three with Duchenne muscular dystrophy and one with X-linked myopathy) and found that expression of NKX2-5 was specific to muscles of individuals with DM1. Although we had a limited number of samples, NKX2-5 expression seemed to be twice as high in hearts from individuals with DM1 as in hearts from individuals without DM1 (Fig. 6b). We observed expression of cardiac-specific downstream targets of NKX2-5 (NPPA, NPPB) in total RNA extracts from skeletal muscle of individuals with DM1 (Fig. 6c). These effects on NKX2-5, NPPA and NPPB were present only in muscles from individuals with DM1 and not in individuals with other muscular dystrophies or in controls without muscular dystrophy (Fig. 6c). Thus, the induction of NKX2-5 and its downstream effects in skeletal muscle are not a nonspecific stress response to myopathic changes; instead, they seem to be a specific response to the mutant DMPK mRNA.
Figure 6.

NKX2-5 is expressed in muscle tissue from individuals with DM1. (a) DAPI staining for myonuclei and immunohistochemistry for NKX2-5 in skeletal muscle from individuals with or without DM1. NKX2-5 expression is present in muscle from individuals with DM1 (note green nuclear staining). (b) Protein blotting of muscle protein extracts shows NKX2-5 in tissue from individuals with DM1 (lanes 1–4) but not in normal muscle (lanes 5, 6), in muscle from individuals with Duchenne muscular dystrophy (lane 7) or in muscle from individuals with X-linked myopathy (lane 8). Lane 9: positive control (normal human heart). GAPDH was used as a loading control. NKX2-5 expression seems higher in hearts from individuals with DM1 than in hearts from individuals without DM1. (c) RT-PCR shows that expression of NKX2-5 and its downstream targets NPPA and NPPB is specific to muscles from individuals with DM1. GAPDH was used as a loading control. MD, muscular dystrophy.
Silencing toxic RNA reverses effects of NKX2-5 induction
We have found that RNA toxicity and cardiac conduction abnormalities are reversible in many of the induced mice16 upon silencing transgene expression by withdrawing doxycyline. These mice are termed ‘reverted mice’. We found it remarkable that CX40 expression was restored in the His bundle and the Purkinje fibers of reverted mice (Fig. 7a). RT-PCR showed restoration of Gia5 mRNA expression (Fig. 4a). Protein blotting and RT-PCR showed restoration of CX43 expression in cardiac tissues (Fig. 7b) and in skeletal muscles (Fig. 4c). In the heart, the number of NKX2-5-positive nuclei (Fig. 7c) and Nkx2-5 mRNA expression reverted to levels found in uninduced mice, as assessed by RNA blotting (Fig. 2b) and RT-PCR (Fig. 7c). We observed similar results for NKX2-5 in skeletal muscle by immunofluorescence and protein blotting (Fig. 7d). None of the reverted mice showed any skeletal muscle expression of Nkx2-5 mRNA, as assessed by RT-PCR (data not shown). Effects on downstream target genes of NKX2-5 were also reversed in both skeletal and cardiac muscle (Fig. 4a–c).
Figure 7.

Reversal of RNA toxicity and molecular changes. (a) CX40 expression (red) is reduced in the cardiac conduction system of induced mice but restored to more normal levels in reverted mice. Insets show differences in CX40 (red) and CX43 (green) expression in uninduced and induced ventricular myocardium. (b) Upper panel: protein blot shows that CX43 expression is restored in hearts of reverted mice. Lanes 1–3: uninduced mice; 4–6: induced mice; 7–9: reverted mice. Numbers below lanes indicate relative expression. Lower panel: RT-PCR shows that Gia1 (Cx43) mRNA expression is restored in reverted mice. (c) Reverted mice have normal Nkx2-5 expression in cardiac muscles, as shown by a count of nuclei and by RT-PCR. (d) NKX2-5 in skeletal muscle is undetectable by immunohistochemistry (upper panel) or by protein blotting (lower panel) in reverted mice. Lanes 1–2: uninduced mice; 3–5: induced mice; 6–8: reverted mice. GAPDH was used as a loading control. Graphs in b and c show mean ± s.d.
NKX2-5 is a genetic modifier of cardiotoxicity
If RNA toxicity induces NKX2-5 expression, and this has a role in DM1 phenotypes, then reducing NKX2-5 expression might result in modulation of the phenotypes. Unfortunately, NKX2-5-null mice die during embryonic development. However, Nkx2-5 heterozygote mice (Nkx2-5LacZ/+) are viable and develop little or no phenotype in an FVB background21. Thus, we bred Nkx2-5LacZ/+ mice21 with 5-313 mice homozygous for the GFP-DMPK 3′ UTR transgene locus (hereafter termed ‘homozygous 5-313 mice’). The resulting progeny were either Nkx2-5LacZ/+ 5-313 heterozygotes (n = 29) or Nkx2-5+/+ 5-313 heterozygotes (n = 25). After weaning, at approximately 4 weeks of age, we performed electromyograms (EMGs) and electrocardiograms (ECGs). None of the mice had myotonia. The average PR intervals for the two groups were 0.029 s and 0.031 s, respectively, and only one Nkx2-5+/+ 5-313 heterozygote had a PR interval of 0.04 s. There was no statistical difference in the PR intervals between the two groups (P = 0.08). Subsequently, we induced expression of the GFP-DMPK 3′ UTR transgene with doxycycline and then performed EMGs and ECGs on a weekly basis. We were not surprised to find that all the mice developed myotonia by 3 weeks, as myotonia has previously been shown to result from splicing defects in Clcn-1 mRNA processing8. However, Nkx2-5 haploinsufficiency had a clear cardioprotective effect (Fig. 8a). By 2 weeks after induction of toxic RNA production, the average PR interval in Nkx2-5+/+ 5-313 heterozygotes was 0.041 s, with 15 of 25 mice having PR intervals of ≥0.04 s (range, 0.04–0.0575 s), whereas the average PR interval in Nkx2-5LacZ/+ 5-313 heterozygotes was 0.034 s, with only 3 of 29 mice having PR intervals ≥0.04 s (two had an interval of 0.04 s, and one had an interval of 0.045 s). This difference was statistically significant (P = 0.00003). The cardioprotective effects of Nkx2-5 haploinsufficiency correlated with the reduced expression of Nkx2-5 mRNA and NKX2-5 protein (Fig. 8b,c). At the time this manuscript was written, these differences persisted after 3 months of induction. In addition, preliminary experiments using the 5-336 mice showed the same trends (data not shown). Notably, in 5-313 homozygous mice, Nkx2-5 haploinsufficiency did not have a protective effect; all of those mice developed myotonia and complete heart block within 2 weeks, irrespective of their Nkx2-5 genotype. The 5-313 homozygote mice produced much more toxic RNA and concomitantly showed a more severe phenotype than the 5-313 heterozygote mice, an outcome that may be analogous to repeat-dosage effects seen in individuals with DM1. The protective effect of reduced NKX2-5 was overwhelmed in these mice and is consistent with the concept of Nkx2-5 as a modifier of RNA toxicity.
Figure 8.

Nkx2-5 haploinsufficiency protects against cardiac conduction defects induced by DMPK 3′ UTR mRNA. (a) PR intervals in Nkx2-5+/+ 5-313 heterozygotes increase much more than in Nkx2-5LacZ/+ 5-313 heterozygotes after induction of DMPK 3′ UTR mRNA toxicity (mean ± s.d.). * indicates that PR intervals are not significantly different before doxycycline induction (P = 0.08). ** indicates that PR intervals are significantly different after doxycycline induction (P = 0.00003). (b) RT-PCR confirms that Nkx2-5 mRNA expression is much lower in Nkx2-5LacZ/+ 5-313 heterozygote hearts than in Nkx2-5+/+ 5-313 heterozygote hearts (mean ± s.d.). (c) Representative protein blot demonstrating that NKX2-5 expression is lower in Nkx2-5LacZ/+ 5-313 heterozygote hearts than in Nkx2-5+/+ 5-313 heterozygote hearts (+/− indicates Nkx2-5LacZ/+ mice). Numbers below blot show relative NKX2-5 expression compared to uninduced Nkx2-5+/+ 5-313 heterozygotes. Uninduced skeletal muscle (sk.m.) extract was used as a negative control for NKX2-5. At least five mice were analyzed for each group for RT-PCR and protein blotting. Dox, doxycycline.
DISCUSSION
NKX2-5, RNA toxicity and cardiac conduction defects
What is the potential relevance of these findings? Clearly, perturbations in the delicately balanced expression of NKX2-5 have deleterious effects on the heart19. NKX2-5 overexpression and its consequent effects on CX40 and CX43 provide a plausible molecular mechanism for cardiac conduction defects in DM1. Based on prevailing data from humans and mouse models, we had predicted that NKX2-5 expression might be decreased in the hearts of mice expressing the toxic RNA. Instead, we observed increased NKX2-5 expression. Although compensatory increases in NKX2-5 have been demonstrated in animal models in which cardiac hypertrophy is induced19, there is no known human inherited disorder associated with NKX2-5 overexpression. However, considerable data suggest that NKX2-5 overexpression has deleterious effects. In humans, trisomy of the distal region of chromosome 5q (where the NKX2-5 gene resides) has been reported to result in severe congenital heart defects, suggesting that overexpression of NKX2-5 during human embryonic development is deleterious30. Notably, in our ongoing, preliminary experiments examining the developmental aspects of RNA toxicity in our mice, we have observed substantial neonatal demise and generalized edema (data not shown). In addition, one group has reported that they have been unable to generate viable transgenic mice over-expressing NKX2-5 under the control of a β–myosin heavy chain (MHC) promoter owing to various heart defects and growth retardation during embryonic development23. However, others have generated transgenic mice overexpressing NKX2-5 under the control of an α-MHC promoter that is primarily active in the postnatal heart31. These mice rapidly develop progressive cardiac conduction defects and die by 2 to 4 months of age. These phenotypes are very similar to observations in our inducible mouse model of RNA toxicity and in individuals with DM1.
Deficiencies in CX43 and CX40 clearly result in cardiac conduction defects18. Based on data from Gia5 (Cx40) promoter analysis, Gia5 has been identified as a downstream target of NKX2-5 (refs. 32,33). However, recent data from Nkx2-5+/− mice34 and from a mouse with a knockout of NKX2-5 retricted to the cardiac ventricles35 have demonstrated that deficiencies in NKX2-5 have no major effects on CX40 in individual cardiomyocytes. In another study, transgenic mice overexpressing a mutant NKX2-5 (I183P) developed progressive heart block within weeks after birth and demonstrated downregulation of CX40 and CX43 (refs. 23,24). This mutation was thought to be dominant negative. However, the authors note that the endogenous Nkx2-5 gene is upregulated, especially in postnatal hearts, suggesting a potential for a gain of function of the endogenous alleles23. In a follow-up study, they tested this and showed that overexpression of NKX2-5 (I183P) in adult cardiomyocytes does not affect Gia5 or Gia1 expression and, in transgenic mice, results in a late onset (>1 year of age) of milder cardiac conduction defects31. However, in transgenic mice, the authors found that overexpressing wild-type NKX2-5 leads to rapid and progressive heart block within weeks after birth and to profound downregulation of CX40 and CX43 in the heart31,36. This effect on CX40 and CX43 in mice over-expressing NKX2-5 was confirmed by overexpression of NKX2-5 in isolated adult cardiomyocytes, but the precise molecular mechanism for this remains unknown31,37. Thus, there is clear evidence from mouse models and cell culture experiments that overexpression of NKX2-5 is sufficient to cause the effects we see in our mouse model of RNA toxicity.
Implications of NKX2-5 induction in DM1 skeletal muscle
NKX2-5, BNP and ANP are normally considered cardiac-specific proteins, with no known reports of substantial expression in adult skeletal muscle, and they are commonly used as cardiac-specific markers. Thus, we were surprised by the clear and consistent expression of these genes in skeletal muscle from individuals with DM1. In skeletal muscle, the induced expression of NKX2-5 seems specific to DM1 and thus may be a clinically useful diagnostic and potential prognostic marker. NKX2-5 in skeletal muscle seems to have transcriptional effects on downstream target genes in our mouse model (Fig. 4b,c), in skeletal myoblasts (Fig. 5) and in muscles from individuals with DM1 (Fig. 7). Proteins such as BNP and ANP that are induced only in DM1 muscles may prove to be useful markers for monitoring disease status and therapeutic response. Of particular clinical significance, elevated serum BNP is used very frequently as a highly specific marker for cardiac volume overload (for example, in congestive heart failure), but given our findings in muscles from individuals with DM1, this may need to be reconsidered.
The expression of NKX2-5 in skeletal muscle may also have pathologic consequences. The decrease in CX43 expression in skeletal muscles of induced mice may be relevant to muscle regenerative defects seen in DM1. Studies in myoblasts38 and conditional tissue-specific Gia1 knockout mouse models39 have highlighted the importance of appropriate temporal upregulation of Gia1 in myoblast fusion and muscle regeneration. We are in the process of testing the effects of NKX2-5 overexpression directly in skeletal muscle using a mouse model. Although overexpression of NKX2-5 in mouse or human skeletal muscle and its effects has not yet been directly evaluated, increased NKX2-5 expression in myoblasts clearly has adverse effects on myogenic differentiation27. Notably, the amounts of MyoD and myogenin, key myogenic transcription factors needed for proper differentiation, are markedly reduced by NKX2-5 overexpression—exactly the same result as in a myoblast cell culture model of RNA toxicity40 and in DM1 myoblasts41.
New pathogenic mechanisms of RNA toxicity
Our studies provide evidence for a previously undescribed mechanism of RNA toxicity in DM1: namely, induced NKX2-5 expression. This expands the mode of RNA toxicity in DM1 beyond RNA splicing defects and may provide explanations for pathogenic effects not accounted for by RNA mis-splicing. Furthermore, the results from Nkx2-5-haploinsufficient mice provide clear genetic evidence that NKX2-5 acts as a modifier of RNA toxicity in the heart.
It is clear that NKX2-5 transcription is induced by the DMPK 3′ UTR mRNA. The transcriptional elements driving Nkx2-5 expression have both enhancer and repressor regions that respond in a time- and tissue-specific manner to various factors, including Smad proteins, BMPs, GATA-4 and perhaps NKX2-5 itself 42–46. Even after 10 years of extensive research, the complicated transcriptional control of Nkx2-5 is not well understood. Nonetheless, this now represents an amenable target for understanding the pathways connecting the toxic RNA to its downstream effects.
METHODS
Transgenic mice
Transgenes and phenotypic characterization of transgenic mice are described elsewhere16. We used two different transgenic lines in an FVB background (5-336 and 5-313) in this study. Results using 5-336 mice mirror those from the 5-313 mice. Both lines express the GFP-DMPK 3′ UTR transgene under the control of a tetracycline-responsive human DMPK promoter. Because of the inherent leakiness of the transgene, the 5-336 mice develop myotonic dystrophy without transgene induction and progress rapidly to conduction abnormalities and death upon transgene induction. The 5-313 mice are not ‘leaky’ and are normal before transgene induction. All experiments in this paper have used this line, unless otherwise indicated. Expression of the transgene was induced with 0.2% (wt/vol) doxycycline in drinking water. The generation and characterization of Nkx2-5LacZ/+ mice is reported elsewhere47.
ECGs and EMGs
Detailed protocols for EMGs are described elsewhere16. Mice were anesthesized with intraperitoneal valium (5 mg per kg body weight) and ketamine (100–150 mg per kg body weight) and were kept under a warming lamp during the entire procedure. Three lead ECGs were performed using a BioAmp/Powerlab from ADInstruments; data were collected on a computer for later analysis. Mice revived in about 30–40 min without complications. All protocols were approved by the relevant institutional committees for animal care and use.
Immunohistochemistry and β-galactosidase staining
All microscopy was done using an Olympus IX 50 inverted microscope with fluorescent attachments; images were captured with a CCD camera and were presented using Photoshop 5.5. Tissues were collected in isopentane and were flash-frozen in liquid nitrogen. We prepared serial 6-μm cryostat sections of hearts and assessed up to 20 sagittal sections through the interventricular septum and atrioventricular nodal region for each mouse to ensure that the cardiac conduction system was adequately visualized. Representative pictures are shown. Cryostat sections of skeletal muscles were similarly prepared. For NKX2-5 quantification in the heart, we captured ten visual fields of ventricular myocardium by CCD camera for each mouse and counted NKX2-5-positive nuclei. We analyzed at least six mice for each group (uninduced, induced and reverted). Similar experiments were performed using cardiac tissues from Nkx2-5LacZ/+ 5-313 heterozygotes stained for β-galactosidase. Two-tailed homoscedastic t-tests were used to determine statistical significance. Protocols for tissue fixation and immunohistochemistry are described elsewhere48.
We used antibodies specific to the following proteins: CX43 (Sigma, C6219, 1:1,000), CX40 (Santa Cruz, SC20466, 1:200), NKX2-5 (for mouse tissues: Santa Cruz, SC8697, 1:200; for human tissues: Santa Cruz, SC14033, 1:200) and dystrophin (Santa Cruz, SC7461). Secondary antibodies were from Molecular Probes (1:400).
RNA analyses
Total RNA was extracted from tissues that were collected in isopentane and flash-frozen in liquid nitrogen49. RNA blots of 5–10 μg of total RNA separated on 1% glyoxal gels were probed with Nkx2-5 or Gapdh using standard procedures. For RT-PCR, all RNAs were treated with DNase I (Ambion) and were checked by PCR for β-actin to ensure that they were not contaminated with DNA before further analysis. Real-time RT-PCR was done using the BioRad iCycler and BioRad kits using the manufacturer’s protocols, with detection by SYBRGreen dye. Primer sequences and PCR conditions can be found in Supplementary Methods online. All assays were done in duplicate, and results were normalized to results from an uninduced mouse50. At least five mice for each group and RNA target were analyzed. Two-tailed homoscedastic t-tests were used to determine statistical significance. All results are expressed relative to uninduced 5-313 mice with P values for comparison (for example, uninduced versus induced, or induced versus reverted). All data are presented graphically as mean ± s.d.
Protein blotting
We used standard protocols to prepare total protein extracts from frozen tissues in RIPA buffer (50 mM Tris-HCl, pH. 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) and protease inhibitor (Roche). We used 50–100 μg of total protein to detect NKX2-5 in human muscle and heart extracts using a 1:500 dilution of antibody to NKX2-5 (anti-NKX2-5) (SC14033). For mouse heart extracts, anti-NKX2-5 (Santa Cruz, SC8697 was used at a 1:500 dilution, with blots incubated for 2 h in PBS with 5% (wt/vol) milk. To detect mouse NKX2-5 and CARP, 100 μg of skeletal muscle extracts were incubated overnight at 4 °C in PBS with 5% milk, with a 1:1,000 dilution of monoclonal anti-NKX2-5 (Abnova, clone 1E4-G5) or a 1:500 dilution of anti-CARP (Santa Cruz, SC30181), respectively. To detect CX43, protein blots of 20 μg of heart extracts were incubated for 1 h with anti-CX43 (1:8,000 dilution) in PBS with 3% BSA. Protein blots of 100 μg of skeletal muscle extracts were incubated for 2 h with anti-CX43 (1:1,000 dilution) in PBS with 5% BSA. Glyceraldehyde phosphate dehydrogenase (GAPDH), detected with anti-GAPDH (Ambion, 4300), was used as a loading control. Blots were scanned, and relative protein expression was quantified using ImageQuant 5.1.
C2C12 transfections
C2C12 myoblasts were grown under standard conditions6 and transfected with 2 or 4 μg of pcDNA3.1-HisC-Nkx2-5 (the Nkx2-5 ORF) or 4 μg of pcDNA3.1 (Invitrogen) and with 0.2 μg of pmax-GFP (Amaxa) as a transfection monitor. Cells were transfected using the Nucleofector kit (Amaxa) according to the manufacturer’s instructions (using solution V and program T20). Transfection efficiency was greater than 80%–90%. Cells were harvested 24 h later and RNA extracted. Transfection experiments were repeated at least three times.
Supplementary Material
Acknowledgments
We thank P. Mahadevan for critically reading the manuscript. Human tissues were purchased from the University of Miami Brain and Tissue Bank. Transgenic mice were generated by the University of Wisconsin-Madison Transgenic Core Facility. All studies were done under the auspices of the University of Virginia Animal Care and Use Committee and Institutional Review Board. This work was supported by the Muscular Dystrophy Association (grant 3889) and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR052771).
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS
M.S.M. discovered NKX2-5 induction and performed mouse phenotypic analyses, immunohistochemistry and protein blotting. R.S.Y. performed molecular analyses and transfection assays. Q.Y. managed the mouse colony and performed mouse phenotypic analyses and tissue collection. C.D.F.-M. performed RT-PCR assays and analyses, and V.S. performed mouse experimental work and tissue staining. J.P. and C.A.T. provided human tissues. A.L.T. provided ECG machines and assistance in performing and analyzing ECGs. O.W.P. and R.P.H. provided Nkx2-5LacZ/+ mice and gave experimental advice, critical insight and comments. M.S.M. was responsible for conceptual design and execution.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
References
- 1.Harper PS. Myotonic Dystrophy. W.B. Saunders; London: 1989. [Google Scholar]
- 2.Mahadevan M, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 3.Pelargonio G, Dello Russo A, Sanna T, De Martino G, Bellocci F. Myotonic dystrophy and the heart. Heart. 2002;88:665–670. doi: 10.1136/heart.88.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tapscott SJ, Thornton CA. Biomedicine. Reconstructing myotonic dystrophy. Science. 2001;293:816–817. doi: 10.1126/science.1063517. [DOI] [PubMed] [Google Scholar]
- 5.Mankodi A, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 6.Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum Mol Genet. 1999;8:1975–1984. doi: 10.1093/hmg/8.11.1975. [DOI] [PubMed] [Google Scholar]
- 7.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 8.Charlet BN, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 9.Mankodi A, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 10.Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liquori CL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 12.Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 2004;74:793–804. doi: 10.1086/383590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berul CI, et al. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J Clin Invest. 1999;103:R1–R7. doi: 10.1172/JCI5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berul CI, Maguire CT, Gehrmann J, Reddy S. Progressive atrioventricular conduction block in a mouse myotonic dystrophy model. J Interv Card Electrophysiol. 2000;4:351–358. doi: 10.1023/a:1009842114968. [DOI] [PubMed] [Google Scholar]
- 15.Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA. Nuclear RNA foci in the heart in myotonic dystrophy. Circ Res. 2005;97:1152–1155. doi: 10.1161/01.RES.0000193598.89753.e3. [DOI] [PubMed] [Google Scholar]
- 16.Mahadevan MS, et al. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaussin V. Offbeat mice. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:1022–1026. doi: 10.1002/ar.a.20074. [DOI] [PubMed] [Google Scholar]
- 18.Lo CW. Role of gap junctions in cardiac conduction and development: insights from the connexin knockout mice. Circ Res. 2000;87:346–348. doi: 10.1161/01.res.87.5.346. [DOI] [PubMed] [Google Scholar]
- 19.Akazawa H, Komuro I. Cardiac transcription factor Csx/Nkx2–5: Its role in cardiac development and diseases. Pharmacol Ther. 2005;107:252–268. doi: 10.1016/j.pharmthera.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Schott JJ, et al. Congenital heart disease caused by mutations in the transcription factor NKX2–5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 21.Biben C, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2–5. Circ Res. 2000;87:888–895. doi: 10.1161/01.res.87.10.888. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka M, et al. A mouse model of congenital heart disease: cardiac arrhythmias and atrial septal defect caused by haploinsufficiency of the cardiac transcription factor Csx Nkx2.5. Cold Spring Harb Symp Quant Biol. 2002;67:317–325. doi: 10.1101/sqb.2002.67.317. [DOI] [PubMed] [Google Scholar]
- 23.Kasahara H, et al. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2.5 homeoprotein. J Clin Invest. 2001;108:189–201. doi: 10.1172/JCI12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wakimoto H, Kasahara H, Maguire CT, Izumo S, Berul CI. Developmentally modulated cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of DNA nonbinding mutant Nkx2.5. J Cardiovasc Electrophysiol. 2002;13:682–688. doi: 10.1046/j.1540-8167.2002.00682.x. [DOI] [PubMed] [Google Scholar]
- 25.Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development. 1993;119:419–431. doi: 10.1242/dev.119.2.419. [DOI] [PubMed] [Google Scholar]
- 26.Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac and extracardiac expression of Csx/Nkx2.5 homeodomain protein. Circ Res. 1998;82:936–946. doi: 10.1161/01.res.82.9.936. [DOI] [PubMed] [Google Scholar]
- 27.Riazi AM, Lee H, Hsu C, Van Arsdell G. CSX/Nkx2.5 modulates differentiation of skeletal myoblasts and promotes differentiation into neuronal cells in vitro. J Biol Chem. 2005;280:10716–10720. doi: 10.1074/jbc.M500028200. [DOI] [PubMed] [Google Scholar]
- 28.Furling D, Lemieux D, Taneja K, Puymirat J. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul Disord. 2001;11:728–735. doi: 10.1016/s0960-8966(01)00226-7. [DOI] [PubMed] [Google Scholar]
- 29.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 30.Chen CP, et al. Molecular cytogenetic analysis of de novo dup(5)(q35.2q35.3) and review of the literature of pure partial trisomy 5q. Am J Med Genet A. 2006;140:1594–1600. doi: 10.1002/ajmg.a.31329. [DOI] [PubMed] [Google Scholar]
- 31.Kasahara H, et al. Nkx2.5 homeoprotein regulates expression of gap junction protein connexin 43 and sarcomere organization in postnatal cardiomyocytes. J Mol Cell Cardiol. 2003;35:243–256. doi: 10.1016/s0022-2828(03)00002-6. [DOI] [PubMed] [Google Scholar]
- 32.Bruneau BG, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 33.Linhares VL, et al. Transcriptional regulation of the murine Connexin40 promoter by cardiac factors Nkx2–5, GATA4 and Tbx5. Cardiovasc Res. 2004;64:402–411. doi: 10.1016/j.cardiores.2004.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jay PY, et al. Nkx2–5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest. 2004;113:1130–1137. doi: 10.1172/JCI19846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pashmforoush M, et al. Nkx2–5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373–386. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 36.Wakimoto H, et al. Cardiac electrophysiological phenotypes in postnatal expression of Nkx2.5 transgenic mice. Genesis. 2003;37:144–150. doi: 10.1002/gene.10236. [DOI] [PubMed] [Google Scholar]
- 37.Teunissen BE, et al. Analysis of the rat connexin 43 proximal promoter in neonatal cardiomyocytes. Gene. 2003;322:123–136. doi: 10.1016/j.gene.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Gorbe A, et al. Transient upregulation of connexin43 gap junctions and synchronized cell cycle control precede myoblast fusion in regenerating skeletal muscle in vivo. Histochem Cell Biol. 2005;123:573–583. doi: 10.1007/s00418-004-0745-2. [DOI] [PubMed] [Google Scholar]
- 39.Araya R, et al. Expression of connexins during differentiation and regeneration of skeletal muscle: functional relevance of connexin43. J Cell Sci. 2005;118:27–37. doi: 10.1242/jcs.01553. [DOI] [PubMed] [Google Scholar]
- 40.Amack JD, Reagan SR, Mahadevan MS. Mutant DMPK 3′-UTR transcripts disrupt C2C12 myogenic differentiation by compromising MyoD. J Cell Biol. 2002;159:419–429. doi: 10.1083/jcb.200206020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Timchenko NA, Iakova P, Cai ZJ, Smith JR, Timchenko LT. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol. 2001;21:6927–6938. doi: 10.1128/MCB.21.20.6927-6938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown CO, III, et al. The cardiac determination factor, Nkx2–5, is activated by mutual cofactors GATA-4 and Smad1/4 via a novel upstream enhancer. J Biol Chem. 2004;279:10659–10669. doi: 10.1074/jbc.M301648200. [DOI] [PubMed] [Google Scholar]
- 43.Liberatore CM, Searcy-Schrick RD, Vincent EB, Yutzey KE. Nkx-2.5 gene induction in mice is mediated by a Smad consensus regulatory region. Dev Biol. 2002;244:243–256. doi: 10.1006/dbio.2002.0604. [DOI] [PubMed] [Google Scholar]
- 44.Lien CL, et al. Control of early cardiac-specific transcription of Nkx2–5 by a GATA-dependent enhancer. Development. 1999;126:75–84. doi: 10.1242/dev.126.1.75. [DOI] [PubMed] [Google Scholar]
- 45.Prall OW, et al. An Nkx2–5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwartz RJ, Olson EN. Building the heart piece by piece: modularity of cis-elements regulating Nkx2–5 transcription. Development. 1999;126:4187–4192. doi: 10.1242/dev.126.19.4187. [DOI] [PubMed] [Google Scholar]
- 47.Elliott DA, et al. A tyrosine-rich domain within homeodomain transcription factor Nkx2–5 is an essential element in the early cardiac transcriptional regulatory machinery. Development. 2006;133:1311–1322. doi: 10.1242/dev.02305. [DOI] [PubMed] [Google Scholar]
- 48.Mankodi A, et al. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54:760–768. doi: 10.1002/ana.10763. [DOI] [PubMed] [Google Scholar]
- 49.Langlois MA, Lee NS, Rossi JJ, Puymirat J. Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol Ther. 2003;7:670–680. doi: 10.1016/s1525-0016(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 50.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.