

Abstract
Safely eradicating prions, amyloids and preamyloid oligomers may ameliorate several fatal neurodegenerative disorders. Yet, whether small-molecule drugs can directly antagonize the entire spectrum of distinct amyloid structures or ‘strains’ that underlie distinct disease states is unclear. Here, we investigated this issue using the yeast prion protein Sup35. We have established how epigallocatechin-3-gallate (EGCG) blocks synthetic Sup35 prionogenesis, eliminates preformed Sup35 prions, and disrupts inter- and intra-molecular prion contacts. Unexpectedly, these direct activities were strain selective, altered the repertoire of accessible infectious forms and facilitated emergence of a new prion strain that configured original, EGCG-resistant intermolecular contacts. In vivo, EGCG cured and prevented induction of susceptible but not resistant strains, and elicited switching from susceptible to resistant forms. Importantly, 4,5-bis-(4-methoxyanilino)phthalimide directly antagonized EGCG-resistant prions and synergized with EGCG to eliminate diverse Sup35 prion strains. Thus, synergistic small-molecule combinations that directly eradicate complete strain repertoires likely hold considerable therapeutic potential.
Small molecules that safely eliminate self-templating amyloids and prions as well as their cytotoxic oligomeric precursors are needed to treat, prevent or delay many deadly afflictions that plague humankind, including Alzheimer's disease and prion disorders1-3. Yet, disrupting the exceptionally stable protein-protein interfaces of amyloids poses daunting challenges for small molecules4,5. Nonetheless, candidates have emerged that inhibit6-9 and even reverse amyloidogenesis10,11. Typically, amyloidogenic proteins fold into multiple structurally distinct amyloid forms or ‘strains’, which confer distinct phenotypes12-16. Beyond sharing the ‘cross-β’ amyloid conformation where the β-sheet strands run orthogonal to the fiber axis, however, little is known about the underlying atomic structures of these distinct strains or how structural polymorphism enciphers distinct phenotypes or disease states. This conformational diversity severely complicates the development of small molecule therapies, particularly because small molecules are only likely to be administered after substantial accumulation of diverse amyloid conformers. A key unaddressed issue is whether small molecules can directly antagonize entire repertoires of structurally distinct misfolded forms.
For mammalian prions, this issue is recalcitrant due to immense difficulties in generating prion strains from solely pure protein de novo that infect wild-type animals and cause transmissible disease12. Thus, the direct effects of small molecules on distinct infectious PrP conformers remains unclear17, despite advances in protein misfolding cyclic amplification18,19 and promising leads from cell culture20-22. Furthermore, it cannot be excluded that small molecule effects in cell culture are indirect or reflect secondary alterations in proteostasis23,24. Therefore, how small molecules directly affect the folding, formation and integrity of pure mammalian prion strains remains uncertain.
We sought a defined system to study different prion strains comprised of pure protein. Hence, we employed Sup35, a translation termination factor, which forms infectious amyloids that transmit heritable reductions in translation termination fidelity and comprise the yeast prion [PSI+]25. Sup35 is one of the best-studied amyloidogenic proteins, with analytical tools that are not yet available for other amyloids11,26,27. These tools provide unique opportunities to deduce new mechanistic insights concerning how small molecules affect amyloid structure. Furthermore, distinct infectious strains are readily generated using pure Sup3515,26, allowing the effects of small molecules on their folding, formation and integrity to be dissected.
We employed Sup35's prion domain, termed NM (Fig. 1a), which spontaneously forms infectious amyloids after a lag phase15,26 (Fig. 1b). During lag phase, NM rapidly partitions between monomeric (∼90% of total NM) and oligomeric (∼10% of total NM) species11,28-31 (Fig. 1b, step 1). NM monomers are predominantly random coil and rapidly sample multiple transient conformations32. However, the specific intermolecular contacts required for prionogenesis ultimately occur in structurally fluid NM oligomers26,30,31. NM monomers within molten oligomers gradually rearrange (Fig. 1b, step 2) to form (Fig. 1b, step 3) amyloidogenic oligomers, which have a conformation distinct to fibers31,33. The intermolecular contacts that distinguish fibers form very rapidly after the appearance of these obligate, transient intermediates26,30,31,33 (Fig. 1b, step 4). Fibers then seed their own rapid assembly (Fig. 1b, step 5). Fibers elongate at both ends by capturing and converting monomers to the cross-β form34 (Fig. 1b, step 5). Short prion recognition elements within the N-terminal domain (N), termed ‘Head’ and ‘Tail’ are proposed to make homotypic intermolecular contacts. Thus, fibers consist of alternating Head-to-Head and Tail-to-Tail contacts that are separated by a central core16,26,27 (Fig. 1a-c). The Head and Tail regions can nucleate assembly, although the Head nucleates more rapidly16,26,27.
Figure 1. Model of Sup35 prion assembly and chemical structure of EGCG.

(a) Sup35 is composed of a C-terminal GTPase domain (amino acids 254-685, black) that confers translation termination activity, a highly charged middle domain (M, amino acids 124-253, dark grey), and a prionogenic N-terminal domain (N, amino acids 1-123, light grey) enriched in Gln, Asn, Tyr and Gly. Within N, prion recognition elements termed the ‘Head’ (red) and ‘Tail’ (orange), which flank a ‘Central Core’ (blue), are proposed to play important roles in prion formation26,27.
(b) Mechanism of Sup35 prionogenesis in vitro. Only the Head (red), Central Core (blue), and Tail (orange) regions of N are depicted. During lag phase, an equilibrium is rapidly established between monomers (∼90% of total NM) and structurally labile, molten oligomers (∼10% of total NM) (step 1). The monomers within molten oligomers gradually rearrange (step 2) until they access the amyloidogenic oligomer conformation (step 3). This transient intermediate rapidly converts to the amyloid form at the end of lag phase (step 4). Amyloid fibers then assemble rapidly as they recruit and convert monomers at their ends (step 5). The prion recognition elements within N are proposed to make homotypic intermolecular contacts such that Sup35 prion fibers are held together by an alternating sequence of ‘Head-to-Head’ (red) and ‘Tail-to-Tail’ (orange) contacts. The Central Core (blue) is sequestered by intramolecular contacts. This schematic is not meant to imply precise details of molten oligomer or amyloidogenic oligomer structure.
(c) Schematic of different Sup35 prion strain structures. NM25 fibers form after assembly at 25°C. NM25 fibers also form at 4°C when NM is chemically crosslinked, with BMB (a flexible 11Å crosslinker) in the Tail region (orange). NM4 fibers form after assembly at 4°C. NM4 fibers also form at 25°C when NM is chemically crosslinked with BMB in the Head region (red). Note that the Central Core (blue) and Tail (orange) are comprised of different amino acids in the NM25 and NM4 fiber conformations. We must note that the atomic structure of Sup35 prion strains remains uncertain and alternative models of Sup35 prion structure have been proposed1,16,26,27,35,38-40.
(d) Weak [PSI+] (pink), [psi-] (red) and strong [PSI+] (white) yeast growing on 25% YPD. These cells carry a premature stop codon in their ADE1 gene, do not make functional Ade1 and accumulate a red metabolite provided Sup35 is fully functional. Thus, colony color gives an indication of the extent of Sup35 aggregation and contingent loss-of-function. Color ranges from red in [psi-] colonies through pink in weak [PSI+] colonies to white in strong [PSI+] colonies as Sup35 aggregation and loss-of-function increases. Transformation of NM25 fibers into [psi-] cells yields mostly weak [PSI+], whereas transformation of NM4 fibers into [psi-] cells yields mostly strong [PSI+].
(e) Chemical structure of EGCG, DAPH-12, EGC, gallic acid, and DAPH-6.
(f) Space-filling model of EGCG reveals a nonplanar structure.
NM assembles into distinct ensembles of infectious amyloid strains at 25°C versus 4°C, termed NM25 and NM415,26,35 (Fig. 1c). Strain biases can also be controlled by specifically crosslinking NM monomers in the Head or Tail region (Fig. 1c), using the flexible 11Å crosslinker 1,4-bis-maleimidobutane (BMB), to create covalent NM dimers26. Strain biases created by crosslinking overcome those created by temperature26. Thus, NM that is BMB-crosslinked in the Head forms NM4 at 4°C and 25°C, whereas NM that is BMB-crosslinked in the Tail forms NM25 at 4°C and 25°C26 (Fig. 1c). Infecting [psi-] cells (which lack Sup35 prions) with NM4 induces predominantly ‘strong’ [PSI+] strains and NM25 induces predominantly ‘weak’ [PSI+]15,26 (Fig. 1d). [PSI+] strains are distinguished using an ade1 nonsense reporter, which allows their (weak or strong) translation termination defect to be determined25. Thus, [psi-] colonies are red and require adenine, whereas [PSI+] colonies range from pink (weak) to white (strong) depending on the extent of Sup35 aggregation and contingent inactivation36,37 (Fig. 1d).
NM4 and NM25 strains possess different intermolecular contacts and sequester overlapping, but distinct portions of N in their amyloid core26,35 (Fig. 1c). The length of the central core and position of the Tail-to-Tail contact are markedly different between different strains (Fig. 1c). Residues N-terminal to the Head are also organized differently in NM4 and NM2526,35. The atomic structures of Sup35 prion strains remain unknown and several models have been advanced1,16,26,27,35,38-40. Nonetheless, different strains are readily distinguished, even at the resolution of spatial arrangements of individual amino acids26,35. Thus, Sup35 provides an unparalleled opportunity to explore precisely how small molecules directly affect prion strains.
Here, we focused on epigallocatechin-3-gallate (EGCG; 1) (Fig. 1e, f), which inhibits the de novo fibrillization of polyglutamine, Aβ42 and α-synuclein6,7. We found that Sup35 folds into a spectrum of infectious conformations with differing sensitivities to EGCG. By combining EGCG with another small molecule, 4,5-bis-(4-methoxyanilino)phthalimide (DAPH-12; 2) (Fig. 1e), which directly antagonizes Sup35 prionogenesis in a distinct manner11, we have uncovered how small molecules can synergize to directly inhibit or reverse prionogenesis and antagonize a broader spectrum of prion strains.
Results
EGCG directly inhibits Sup35 prionogenesis in a strain-selective manner
EGCG potently inhibited NM fibrillization at 25°C as assessed by Congo Red (CR) binding (Fig. 2a), Thioflavin-T (ThT) fluorescence (Supplementary Fig. 1a) and sedimentation (Supplementary Fig. 1b). By contrast, EGCG fragments (Fig. 1e): epigallocatechin (EGC; 3) or gallic acid (4) had no effect (Fig. 2a and Supplementary Fig. 1a-c). Similar results were obtained when NM fibrillization was conducted at 37°C (Supplementary Fig. 1d). Electron microscopy (EM) revealed that fibers were scarce in the presence of EGCG at 25°C (Fig. 2b). Instead, oligomeric forms of NM persisted (Fig. 2b).
Figure 2. EGCG inhibits assembly of select Sup35 prion strains.
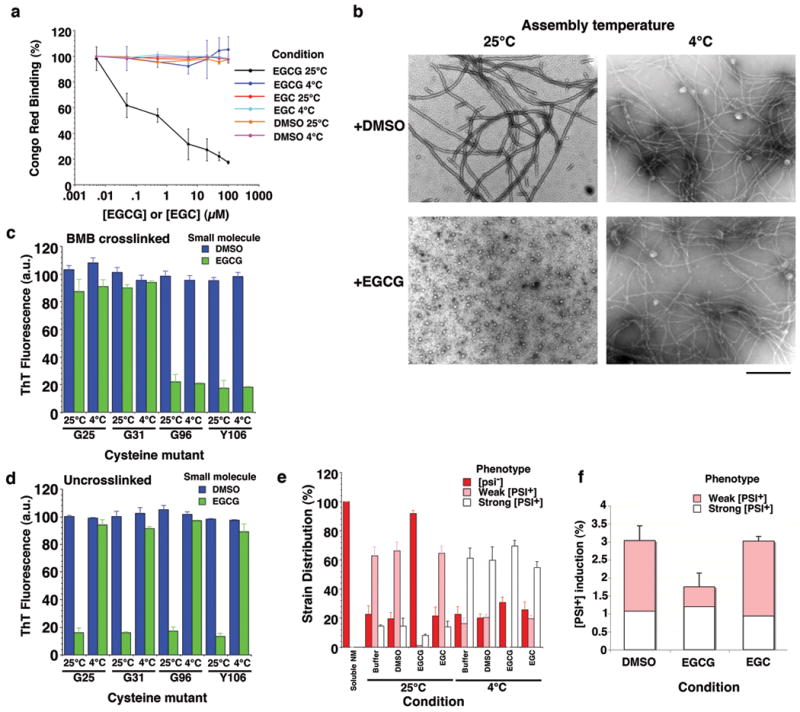
(a) Spontaneous, agitated NM (5μM) fibrillization after 4h at 25°C or 4°C in the presence of EGCG, EGC (0-100μM) or DMSO (0-1%). Fibrillization was measured by CR binding and 100% reflects assembly in the absence of EGCG, EGC or DMSO. Values represent means±SD (n=3-8).
(b) NM was assembled as in (a) at 25°C or 4°C in the presence of DMSO (1%) or EGCG (20μM) and processed for EM. Bar, 0.5μm.
(c, d) NM cysteine variants were crosslinked (c) under denaturing conditions with a flexible 11Å BMB crosslink at position 25, 31, 96 or 106. (d) Alternatively, NM cysteine variants were left uncrosslinked. The indicated NM protein (5μM) was then assembled with agitation at 25°C or 4°C for 4h in the presence of DMSO (1%) or EGCG (20μM). Fibrillization was measured by ThT fluorescence. Values represent means±SD (n=3).
(e) NM (5μM) was incubated with agitation at 25°C or 4°C for 4h in the absence or presence of DMSO (1%), EGCG or EGC (20μM). Reactions were dialyzed to remove unbound small molecule, concentrated, sonicated and transformed into [psi-] cells. The proportion of [psi-], weak [PSI+] or strong [PSI+] transformants was then determined. Values represent means±SD (n=3).
(f) NM-YFP was overexpressed in [psi-] Δpdr5 cells for 12h in the presence of DMSO (1%), EGCG or EGC (125μM). Cells were then plated on 25% YPD and the proportion of weak and strong [PSI+] colonies was determined. Values represent means±SD (n=3).
By contrast, NM fibrillization at 4°C was insensitive to EGCG (Fig. 2a, b and Supplementary Fig. 1a, b). Very similar results were obtained when NM fibrillization was conducted at 2°C (Supplementary Fig. 1d). Importantly, full-length Sup35 amyloid assembly was also inhibited by EGCG at 25°C but not 4°C (Supplementary Fig. 1e, f). These data establish that EGCG did not interfere with CR or ThT assays by some non-specific mechanism.
How could EGCG work at 25°C but not 4°C? EGCG was stable for 24h at both 4°C and 25°C under our conditions (Supplementary Fig. 1g). Furthermore, EGCG inhibited Aβ42 fibrillization at both 4°C and 25°C (Supplementary Fig. 1h). Thus, EGCG was stable and could inhibit amyloidogenesis at 4°C. Moreover, NM reversibly bound equal amounts of EGCG at 25°C and 4°C (Supplementary Fig. 1i).
EGCG might selectively inhibit NM25 formation regardless of the assembly temperature (Fig. 1c). To test this hypothesis, we employed NM single cysteine mutants that were crosslinked with BMB26 in either the Head (to generate NM4) or the Tail (to generate NM25) (Fig. 1c). NM proteins that were crosslinked in the Head were insensitive to EGCG at 4°C and 25°C, whereas those crosslinked in the Tail were inhibited by EGCG at both temperatures (Fig. 2c). By contrast, uncrosslinked NM was only sensitive to EGCG at 25°C (Fig. 2d). BMB-crosslinked NM G96C or NM Y106C fibrillization showed similar sensitivity to EGCG concentration at 4°C and 25°C (Supplementary Fig. 1j). Thus, EGCG was equally potent at 25°C and 4°C in selectively preventing the formation of fiber conformations that nucleate from the Tail, i.e. NM25 (Fig. 1c).
EGCG selectively antagonizes early folding events of NM25
How is selective inhibition achieved and how does NM evade EGCG at 4°C? We excluded that EGCG inhibited molten oligomer formation (Fig. 1b, step 1) or diverted the majority of NM into assembly-incompetent oligomers (Supplementary Results and Supplementary Fig. 2a-d). Rather, EGCG prevented the maturation of NM oligomers (Fig. 1b, steps 2 and 3; Supplementary Results and Supplementary Fig. 2e, f). Early folding events within NM oligomers (Fig. 1b, step 2) are essential for fibrillization and can be tracked using single cysteine NM mutants labeled with acrylodan11,26,30. Sequestration of labeled sites from solvent causes increases in acrylodan fluorescence. Specific portions of N (residues ∼21-106 at 25°C and residues ∼21-96 at 4°C) become solvent inaccessible in molten oligomers prior to fibrillization26 (Fig. 1b, steps 1 and 2). We employed NM with acrylodan attached to cysteines introduced at: N21, Q38, G86, G96 or Y106. These mutated and labeled variants retain wild-type assembly kinetics and infectivity11,26. After 15min, these mutants were in lag phase (Supplementary Fig. 2g). At 4°C, EGCG had no effect on acrylodan fluorescence (Supplementary Fig. 2h). By contrast, at 25°C, EGCG inhibited increases in acrylodan fluorescence at positions 86, 96 and 106, but not at 21 and 38 (Supplementary Fig. 2h). Thus at 25°C, EGCG altered the folding of the amyloid core region of N (residues ∼21-106 at 25°C), such that C-terminal portions of the Tail (Fig. 1c) remained solvent accessible and failed to enter the molten interior of NM oligomers.
Nucleation of NM fibers (Fig. 1b, step 4) occurs shortly after the appearance of an obligate oligomeric intermediate30,31 (Fig. 1b, step 3), which is detected using a conformation-specific antibody31,41. EGCG prevented formation of this intermediate at 25°C but not at 4°C, explaining why fibrillization was selectively inhibited at 25°C (Supplementary Fig. 2i). Circular dichroism (CD) confirmed that EGCG trapped NM in a predominantly unstructured state at 25°C and prevented the acquisition of β-sheet structure (Supplementary Fig. 2j). NM oligomers isolated after 4h in the presence of EGCG at 25°C were enriched in random coil with little β-sheet content (Supplementary Fig. 2j). At 4°C and 25°C, EGCG bound directly to several regions within the unstructured N domain (Supplementary Fig. 2k and Supplementary Results). EGCG binding precluded origination of certain fiber strains but not others. Specifically, EGCG prevented the conformational rearrangements within molten oligomers required for NM25 nucleation (Fig. 1b, steps 2 and 3).
EGCG selectively inhibits induction of weak [PSI+]
Did EGCG preclude specific infectious conformations? NM conformers formed at 25°C in the presence of EGCG (Fig. 2b) failed to effectively seed fibrillization (Supplementary Fig. 3a) or transform [psi-] cells to [PSI+] (Fig. 2e). Transformation of [psi-] cells with NM25 yielded a mixture of ∼80% weak and 20% strong [PSI+] variants (Fig. 2e). EGCG eliminated the appearance of weak [PSI+] and mildly reduced the proportion of strong [PSI+] colonies (Fig. 2e). NM fibers formed at 4°C in the presence of EGCG seeded NM fibrillization (Supplementary Fig. 3a) and induced exclusively strong [PSI+] (Fig. 2e). By contrast, NM4 yielded ∼20% weak and 80% strong [PSI+] (Fig. 2e). These strain-selective inhibitory effects were corroborated using BMB-crosslinked NM G25C and NM Y106C (Supplementary Fig. 3b). Thus, NM can access an EGCG-resistant prion form. Moreover, assembly in the presence of EGCG selected for the drug-resistant strain.
Similar selectivity occurred in vivo. Overexpressing NM-YFP in [psi-] Δpdr5 cells (which lack, Pdr5, an ABC transporter that expels small molecules from the cell11) induced ∼70% weak and 30% strong [PSI+] (Fig. 2f). EGCG reduced [PSI+] induction and formation of NM-YFP foci in a manner that selectively antagonized weak [PSI+] ∼4-fold (Fig. 2f and Supplementary Fig. 3c). Induction of strong [PSI+] was unaffected (Fig. 2f). Thus, EGCG selectively antagonized prions that encode weak [PSI+].
EGCG prevents select prion recognition events
To gain mechanistic insights into small molecule action, we determined how EGCG affected interactions between NM and short peptides thought to represent prion recognition elements27. The peptides comprised amino acids that overlapped with the Head (5-25 and 20-38), the core region (41-71) and the Tail (88-106) (Fig. 1c). At 25°C, NM did not interact with 41-71, but did interact with the 5-25, 20-38 and 88-106 peptides (Fig. 3a). These interactions resisted 2% SDS indicating that they were sites of prionogenesis27. Indeed, ThT-reactive fibers were recovered (Supplementary Fig. 4). Strikingly, EGCG prevented these interactions at 25°C (Fig. 3a). At 4°C, NM did not interact with 41-71, but did interact with the 5-25, 20-38 and 88-106 peptides in an SDS-resistant manner that was altered by EGCG (Fig. 3a). Thus, some Sup35 prion recognition events were EGCG- resistant, while others were EGCG-sensitive.
Figure 3. EGCG prevents inter- and intramolecular contact formation of select Sup35 prion strains.

(a) The indicated N peptides were immobilized on nitrocellulose and incubated for 16h at either 25°C or 4°C with NM-his (1μM) in the presence of DMSO (1%) or EGCG (20μM). The amount of NM-his bound to each peptide was then determined by immunoblot using an anti-his antibody, followed by densitometry and comparison to known amounts of NM-his. Values represent means±SD (n=3).
(b) Proximity analysis assessed by excimer fluorescence. NM proteins (5μM) carrying pyrene labels at the indicated single sites were incubated for 4h with agitation at 25°C or 4°C in the presence of DMSO (1%) or EGCG (20μM). The ratio of excimer fluorescence to non-excimer fluorescence (I465nm/I375nm) is plotted. Note that EGCG causes distinct intersubunit interfaces to form at 4°C and prevents their formation at 25°C.
(c) NM (5μM) labeled with pyrene at positions 69 and 79 was incubated for 4h with agitation at 25°C in the absence or presence of DMSO (1%), EGCG or EGC (20μM). The ratio of excimer fluorescence to non-excimer fluorescence (I476nm/I384nm) is plotted. Values represent means±SD (n=3).
(d) A new fiber strain, NM4E, forms in the presence of EGCG at 4°C. Note that the Head-to-Head contact and the Tail-to-Tail contact is more N-terminal relative to NM4 and NM25 fibers.
To analyze how EGCG affects intermolecular contact formation (Fig. 1b, steps 4 and 5), we used pyrene-labeled single cysteine NM mutants, which retain wild-type assembly kinetics and infectivity, indicating that pyrene does not significantly alter fiber structure11,26. Upon intermolecular contact formation, pyrene molecules at select positions, in the Head or the Tail (Fig. 1b, steps 4 and 5, and Fig. 1c), form excimers (excited-state dimers) that produce a red shift in fluorescence26. EGCG prevented contact formation at 25°C, but not 4°C (Fig. 3b). To determine whether EGCG prevented intramolecular contacts in the central core (Fig. 1c), we used a double pyrene-labeled NM mutant (Y69C, Y79C), which behaves like wild-type NM (Supplementary Fig. 5a, b), but forms excimers exclusively in NM25 because of a strain-specific intramolecular contact between residues 69 and 79 in the central core11 (Fig. 1c and Supplementary Fig. 5c). EGCG precluded this contact (Fig. 3c). Thus, EGCG appeared to prevent the inter- and intramolecular contacts that distinguish NM25.
A new prion strain appears to form in the presence of EGCG
Unexpectedly, NM fibers assembled at 4°C in the presence of EGCG configured subtly distinct contacts to those formed in its absence (Fig. 3b). EGCG caused the Head and Tail contact sites to shift toward the N-terminus: positions 21, 73 and 79 showed greater excimer fluorescence, whereas positions 38 and 96 showed reduced excimer fluorescence (Fig. 3b). This difference was also apparent from the peptide arrays. At 4°C in the presence of EGCG, NM interacted more strongly with 5-25 than 20-38 and less strongly with 88-106 (Fig. 3a). We term this novel fiber conformation, NM4E (Fig. 3d). This emergence of novel drug-resistant prion strains might have implications for drug design.
EGCG directly remodels specific Sup35 prion strains
Did EGCG remodel preformed prions? NM4 and NM25 fibers were incubated with EGCG for 2h or 24h at 25°C. After 24h, but not 2h, EGCG effectively remodeled NM25 fibers and to a lesser extent NM4 fibers (Fig. 4a and Supplementary Fig. 6a-c). NM25 fibers were scarce after 24h with EGCG, and were converted to oligomers, which ranged in size from 10-35nm with a mean diameter of ∼25nm (Fig. 4b and Supplementary Fig. 6d). By contrast, NM4 fibers were largely unaffected although more oligomers were evident (Fig. 4b). EGCG solubilized NM25 fibers as determined by sedimentation (Supplementary Fig. 6e). However, only ∼20% of the total NM in NM25 fibers was released as monomers, as determined by passage through a 50kDa filter (Supplementary Fig. 6f) and size-exclusion chromatography (data not shown). Therefore, EGCG converted NM25 fibers to mostly soluble oligomers, which retained some β-sheet structure (Supplementary Fig. 6g), but were not detected by conformation-specific antibodies that recognize: amyloidogenic oligomers31,33,41 or mature fibers31,42 (Supplementary Fig. 6h). After 24h (but not 2h), EGCG remodeled NM25 fibers to species with diminished seeding and [PSI+]-inducing activity (Fig. 4c and Supplementary Fig. 6i-k). Thus, EGCG converted NM25 fibers to non-templating forms. The few [PSI+] colonies that developed were strong [PSI+] (Fig. 4c). NM4 fibers treated with EGCG did not induce weak [PSI+] and induction of strong [PSI+] was only mildly reduced (Fig. 4c). Similar selectivity was observed with full-length Sup35 (Supplementary Fig. 7a, b). Thus, EGCG preferentially dismantled amyloid structures that encode weak [PSI+].
Figure 4. EGCG disrupts preformed inter- and intramolecular contacts of select Sup35 prion strains.
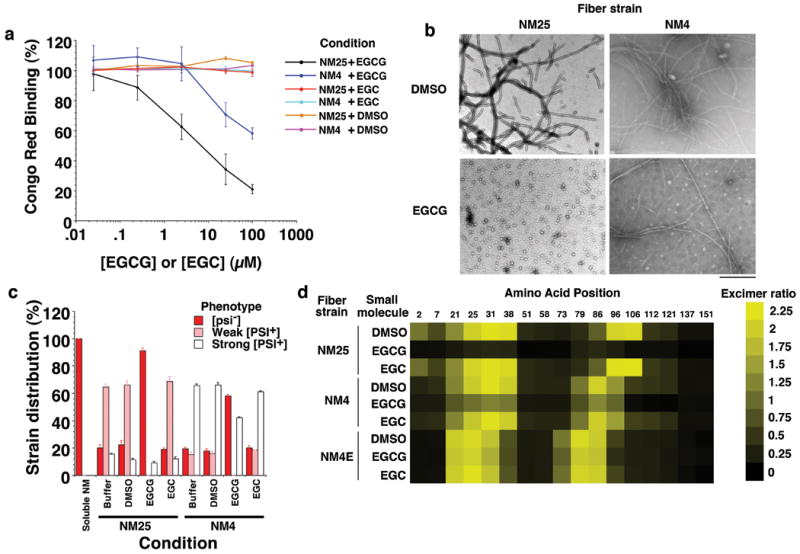
(a) NM25 or NM4 fibers (2.5μM monomer) were incubated for 24h at 25°C with DMSO (1%), EGCG or EGC (0.025-100μM). Fiber integrity was then determined by CR binding. Values represent means±SD (n=3).
(b) NM25 or NM4 fibers (2.5μM monomer) were incubated with DMSO (1%) or EGCG (20μM) for 24h and processed for EM. Bar, 0.5μm.
(c) NM25 or NM4 fibers (2.5μM monomer) were incubated with or without DMSO (1%), EGCG or EGC (20μM) for 24h. Reactions were then dialyzed to remove unbound small molecule, concentrated, sonicated and transformed into [psi-] cells. The proportion of [psi-], weak [PSI+] or strong [PSI+] transformants was then determined. Values represent means±SD (n=3).
(d) NM proteins (5μM) carrying pyrene labels at single sites were assembled into fibers by incubation for 16h with agitation at 25°C, 4°C or at 4°C in the presence of EGCG (20μM). Assembled NM25, NM4 or NM4E fibers (2.5μM monomer) were then treated with DMSO (1%), EGCG or EGC (25μM) for 24h. The ratio of excimer fluorescence to non-excimer fluorescence (I465nm/I375nm) is displayed.
Did EGCG remodel intermolecular contacts of preformed prions? To answer this question, we assembled NM4, NM4E and NM25 (Fig. 3d), using pyrene-labeled, single cysteine NM mutants, and treated them with DMSO, EGC or EGCG for 2h or 24h. After 2h, contacts remained intact and even after 24h DMSO and EGC had no effect (Fig. 4d). NM4E contacts resisted perturbation by EGCG (Fig. 4d). By contrast, the intermolecular contacts of NM4 were partially susceptible to EGCG (Fig. 4d), whereas those of NM25 were more disrupted (Fig. 4d). EGCG also remodeled the intramolecular contacts of NM25 (Supplementary Fig. 7c). Thus, the various contacts of distinct strains displayed differential sensitivity to EGCG.
EGCG selectively cures weak [PSI+]
Selectivity was maintained in vivo. EGCG cured weak [PSI+] more effectively than strong [PSI+] (Fig. 5a). Moreover, EGCG did not cure strong [PSI+] induced by NM4E (Fig. 5a). Thus, [PSI+] exists as a spectrum of variants with differential sensitivity to EGCG.
Figure 5. EGCG eliminates select Sup35 prion strains.
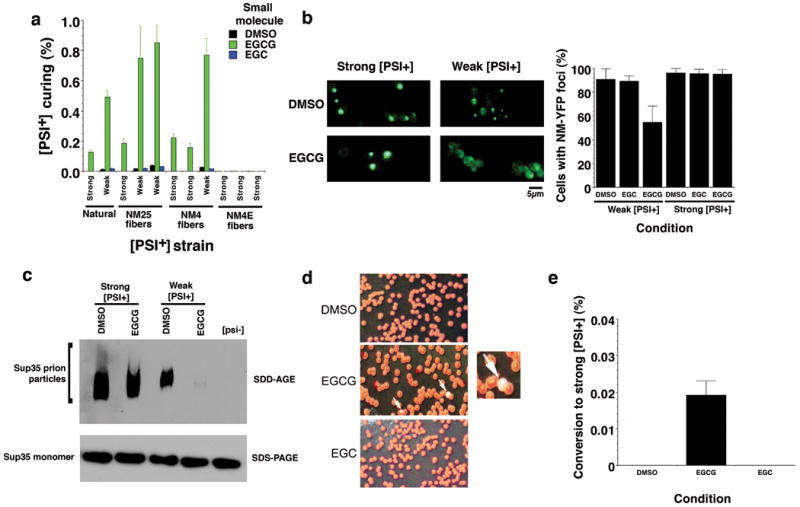
(a) We assessed EGCG curing of natural [PSI+] strain variants and several new [PSI+] strain variants generated by infecting [psi-] Δpdr5 cells with NM25 (two weak variants, one strong), NM4 (two strong variants, one weak) or NM4E (all strong). The indicated [PSI+] Δpdr5 strain variants were treated with DMSO (1%), EGCG or EGC (125μM) for 72h in liquid culture. Cells were plated on 25% YPD and the proportion of red [psi-] colonies was determined. Values represent means±SD (n=4).
(b) NM-YFP was expressed for 5h in weak or strong [PSI+] Δpdr5 cells. Expression was then shut off in the presence of DMSO (1%), EGCG or EGC (250μM) for 2h, and cells were then imaged (left). Note the more diffuse fluorescence in weak [PSI+] cells treated with EGCG. The proportion of cells with NM-YFP foci was then determined (right). Values represent means±SD (n=16).
(c) The indicated [PSI+] Δpdr5 strain variants were treated with either DMSO (1%) or EGCG (125μM) for 48h in liquid culture and then processed for SDD-AGE or SDS-PAGE followed by immunoblot to detect Sup35 prion particles or Sup35 monomers.
(d, e) Weak [PSI+] Δpdr5 were treated with DMSO (1%), EGCG or EGC (125μM) for 48h in liquid culture. Cells were plated on 25% YPD (d). White arrows denote strong [PSI+] colonies that formed in the presence of EGCG, and one example is shown at higher magnification. The proportion of strong [PSI+] colonies was determined (e). Values represent means±SD (n=9).
To test whether EGCG remodeled Sup35 prions in vivo, we employed NM-YFP, which forms fluorescent foci in [PSI+] cells, but is diffuse in [psi-] cells43. We preformed NM-YFP foci in weak and strong [PSI+] and then treated with EGCG. EGCG selectively eliminated NM-YFP foci from weak [PSI+] strains, indicating that prions were cured, whereas DMSO had no effect (Fig. 5b). Yet, prions might persist but be too small to detect by fluorescence. Hence, we employed semi-denaturing detergent-agarose gel electrophoresis (SDD-AGE), which detects prion particles44 (750kDa and bigger). EGCG selectively eliminated Sup35 prion particles in weak but not strong [PSI+], without affecting Sup35 levels (Fig. 5c). We excluded that EGCG effects arise from Hsp104 inhibition (Supplementary Fig. 8a) or by eliciting a heat shock or unfolded protein response (Supplementary Fig. 8b-d). Furthermore, the growth rates of [psi-] and [PSI+] Δpdr5 cells were equal and unaffected by these EGCG concentrations (data not shown). Thus, together with our in vitro findings (Figs. 2 and 4), these data strongly suggest that EGCG cures [PSI+] by directly remodeling Sup35 prions.
EGCG promotes switching from weak to strong [PSI+]
Once established, [PSI+] variants do not generally switch from weak to strong or vice versa36,45. Remarkably, EGCG cured weak [PSI+] variants and simultaneously increased switching from weak to strong [PSI+], whereas DMSO or EGC did not (Fig. 5d, e). Some weak [PSI+] variants may have harbored low levels of EGCG-resistant Sup35 prions that encode strong [PSI+]. Upon exposure to EGCG, these would amplify and cause switching to strong [PSI+]. Thus, a single small molecule may be insufficient to eliminate all amyloid forms of a specific protein and can select for resistant amyloid polymorphs.
Combinations of DAPH-12 and EGCG antagonize prion diversity
We reasoned that coapplication of another small molecule that antagonizes Sup35 prionogenesis might counter EGCG-resistant strains. We selected DAPH-12 (Fig. 1e), which directly inhibits and reverses Sup35 prionogenesis at 25°C and cures strong [PSI+]11. DAPH-12 neither inhibited Hsp104 (Supplementary Fig. 8a) nor elicited a heat-shock response11 (Supplementary Fig. 8c). Yet, how DAPH-12 affects different strains is unclear. In contrast to EGCG, DAPH-12 inhibited spontaneous NM fibrillization at 4°C and 25°C (Fig. 6a). At higher concentrations, DAPH-12 inhibited fibrillization seeded by NM4 and NM25 (Supplementary Fig. 9a). However, inhibition at 4°C was less pronounced than at 25°C (Fig. 6a and Supplementary Fig. 9a). This was due to NM accessing different strains because DAPH-12 was less effective in inhibiting NM mutants that were BMB-crosslinked in the Head compared to the Tail (Fig. 1c), regardless of assembly temperature (Fig. 6b). DAPH-12 disrupted early folding events in molten NM oligomers detected by position-specific acrylodan fluorescence (Supplementary Fig. 9b). Unlike EGCG, DAPH-12 inhibited acrylodan fluorescence increases at N- and C-terminal portions of N, but was less effective at 4°C (Supplementary Fig. 9b). Furthermore, distinct patterns of position-specific pyrene excimer fluorescence, indicative of intermolecular prion contacts, emerged after assembly in the presence of DAPH-12 compared to EGCG, particularly at 4°C (Fig. 6c). Thus, DAPH-12 and EGCG antagonized NM fibrillization in a distinct manner. Infection of [psi-] cells revealed that prions encoding both weak and strong [PSI+] were antagonized by DAPH-12 (Fig. 6d). However, infectious conformations encoding weak [PSI+] were more extensively inhibited than those encoding strong [PSI+].
Figure 6. Combinations of DAPH-12 and EGCG prevent formation of multiple prion strains.

(a) Spontaneous, agitated NM (5μM) fibrillization after 6h at 25°C or 4°C in the presence of DMSO (1%), DAPH-12, EGCG or EGCG plus DAPH-12 (0.5:0.5). The total concentration of small molecule was kept constant at 0.125μM, 0.25μM, 5μM or 10μM. Fibrillization was measured by CR binding and 100% reflects assembly in the absence of DMSO, DAPH-12 or EGCG. Values represent means±SD (n=3).
(b) NM cysteine variants were crosslinked under denaturing conditions with a flexible 11Å BMB crosslink at position 25 or 96. The indicated NM protein (5μM) was then assembled with agitation at 25°C or 4°C for 6h in the presence of DMSO (1%), DAPH-12 (10μM), EGCG (10μM) or DAPH-12 plus EGCG (5μM of each). Fibrillization was measured by ThT fluorescence. Values represent means±SD (n=3).
(c) Proximity analysis assessed by excimer fluorescence. NM proteins (5μM) carrying pyrene labels at the indicated single sites were incubated for 6h with agitation at 25°C or 4°C in the presence of DMSO (1%), DAPH-12 (10μM), EGCG (10μM) or DAPH-12 plus EGCG (5μM of each). The ratio of excimer fluorescence to non-excimer fluorescence (I465nm/I375nm) is plotted.
(d) NM (5μM) was incubated with agitation at 25°C or 4°C for 6h in the presence of DMSO (1%), DAPH-12 (10μM), EGCG (10μM) or DAPH-12 plus EGCG (5μM of each). Reactions were dialyzed to remove unbound small molecule, concentrated, sonicated and transformed into [psi-] cells. The proportion of [psi-], weak [PSI+] or strong [PSI+] transformants was then determined. Values represent means±SD (n=3).
Next, we tested DAPH-12 and EGCG combinations. We kept the total concentration of small molecule constant, and compared the efficacy of an equimolar combination (0.5:0.5) to either small molecule (1:0 or 0:1) alone. Combinations of DAPH-12 and EGCG more effectively inhibited spontaneous and seeded NM fibrillization (Fig. 6a, b and Supplementary Fig. 9a), prionogenic folding of N (Supplementary Fig. 9b) and intermolecular contact formation (Fig. 6c) at 4°C and 25°C than either small molecule alone. At lower concentrations, DAPH-12 and EGCG synergized to inhibit both spontaneous and seeded assembly (Fig. 6a and Supplementary Fig. 9a). These effects were specific for the DAPH-12 and EGCG pair. Pairing DAPH-12 or EGCG with the inactive analogs EGC or DAPH-611 (Fig. 1e; 6) did not improve inhibition (Supplementary Fig. 9c). DAPH-12 prevented formation of NM4E strain (Fig. 3d), which formed at 4°C in the presence of EGCG (Fig. 6a, c, d). Indeed, the DAPH-12 and EGCG combination restricted formation of distinct infectious conformers more effectively than either small molecule alone (Fig. 6d).
DAPH-12 remodeled preformed NM4 and NM25 fibers (Fig. 7a). NM25 fibers were converted to small oligomers by DAPH-12 (Fig. 7b), whereas some fibers persisted after DAPH-12 treatment of NM4 (Fig. 7b). DAPH-12 disrupted intermolecular prion contacts of both strains (Fig. 7c). Yet in contrast to EGCG, DAPH-12 did not disrupt the intramolecular contacts of NM2511 (Supplementary Fig. 9d). The oligomers generated by DAPH-12 were not recognized by conformation-specific antibodies31,41 that recognize amyloidogenic oligomers or mature fibers (Supplementary Fig. 9e), and had diminished seeding activity (Supplementary Fig. 9f). Thus, DAPH-12 converted NM fibers to non-templating species in a manner distinct to EGCG.
Figure 7. Combinations of DAPH-12 and EGCG remodel multiple prion strains.
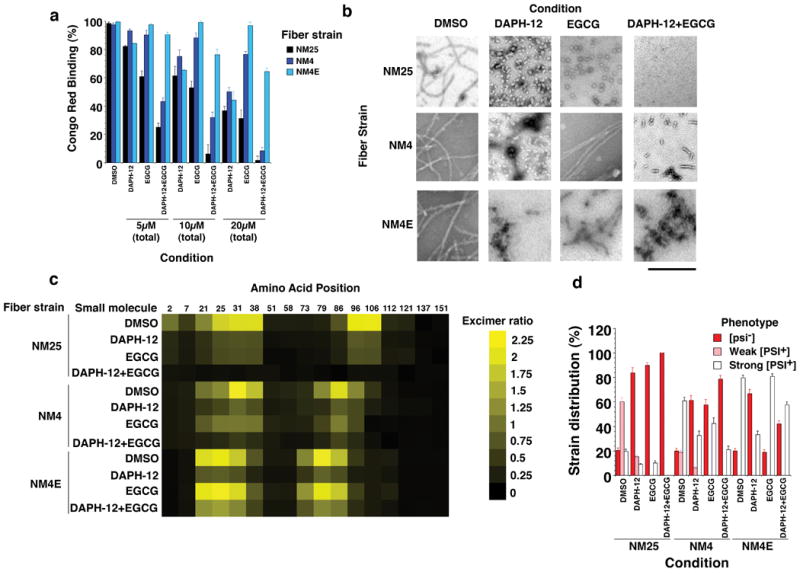
(a) NM25, NM4 or NM4E fibers (2.5μM monomer) were incubated for 24h at 25°C with DMSO (1%), DAPH-12, EGCG or DAPH-12 plus EGCG (0.5:0.5). The total concentration of small molecule was kept constant at 5μM, 10μM or 20μM. Fiber integrity was then determined by CR binding. Values represent means±SD (n=3).
(b) NM25, NM4 or NM4E fibers (2.5μM monomer) were incubated for 24h at 25°C with DMSO (1%), DAPH-12 (20μM), EGCG (20μM) or DAPH-12 plus EGCG (10μM of each). Reactions were then processed for EM. Bar, 0.5μm.
(c) NM proteins (5μM) carrying pyrene labels at single sites were assembled into fibers by incubation for 16h with agitation at either 4°C, 25°C or 4°C in the presence of EGCG. Assembled fibers (2.5μM monomer) were then treated with DMSO (1%), DAPH-12 (20μM), EGCG (20μM) or DAPH-12 plus EGCG (10μM of each) for 24h. The ratio of excimer fluorescence to non-excimer fluorescence (I465nm/I375nm) is plotted.
(d) NM25, NM4 or NM4E fibers (2.5μM monomer) were incubated with DMSO (1%), DAPH-12 (20μM), EGCG (20μM) or DAPH-12 plus EGCG (10μM of each) for 24h. Reactions were then dialyzed to remove unbound small molecule, concentrated, sonicated and transformed into [psi-] cells. The proportion of [psi-], weak [PSI+] or strong [PSI+] transformants was then determined. Values represent means±SD (n=3).
DAPH-12 also partially remodeled EGCG-resistant NM4E fibers (Fig. 7a-c). NM4E and NM4 fibers retained more seeding activity than NM25 fibers after DAPH-12 treatment in vitro and in vivo (Fig. 7d and Supplementary Fig. 9f). Strains encoding weak [PSI+] were more effectively remodeled by DAPH-12 than those encoding strong [PSI+] (Fig. 7d). Moreover, DAPH-12 cured weak [PSI+] more effectively than strong [PSI+] (Fig. 8a). Thus, like EGCG, DAPH-12 preferentially antagonized prion forms that encoded weak [PSI+]. Yet, in contrast to EGCG, DAPH-12 did not disrupt intramolecular contacts (Supplementary Fig. 9d).
Figure 8. EGCG and DAPH-12 synergize to cure various [PSI+] variants.

(a) The indicated [PSI+] Δpdr5 strain variants were treated with DMSO (1%), DAPH-12 (100μM), EGCG (100μM) or EGCG plus DAPH-12 (50μM of each or 100μM of each) for 72h in liquid culture. Cells were plated and the proportion of red [psi-] colonies was determined. Values represent means±SD (n=4).
(b, c) Weak [PSI+] Δpdr5 cells (b) or strong [PSI+] Δpdr5 cells (c) were treated with DMSO (1%), DAPH-12 (100μM), EGCG (100μM) or EGCG plus DAPH-12 (50μM of each or 100μM of each) for 0-96h in liquid culture. At the indicated times, cells were plated and the proportion of red [psi-] colonies was determined. Values represent means±SD (n=3).
(d) Weak [PSI+] Δpdr5 were treated with DMSO (1%), DAPH-12 (100μM), EGCG (100μM) or DAPH-12 plus EGCG (100μM of each) for 72h in liquid culture. Cells were plated and the proportion of strong [PSI+] colonies was determined. Values represent means±SD (n=5).
DAPH-12 and EGCG synergized in remodeling NM4 and NM25 and were more effective than either small molecule alone (Fig. 7a-c and Supplementary Fig. 9f). DAPH-12 plus EGCG completely resolved NM25 fibers (Fig. 7b), whereas NM4 fibers were converted to oligomeric and short fibrillar species (Fig. 7b). Infectivity studies confirmed that DAPH-12 plus EGCG more effectively eliminated NM4 and NM25 than either small molecule alone (Fig. 7d). By contrast, DAPH-12 and EGCG combinations were no more effective than DAPH-12 in remodeling NM4E (Fig. 7a-d and Supplementary Fig. 9f). DAPH-12 and EGCG combinations were more effective in curing the majority of weak and strong [PSI+] strains (Fig. 8a). The only exception was strong [PSI+] conferred by NM4E infection (Fig. 8a). Curing kinetics also revealed DAPH-12 and EGCG synergy (Fig. 8b, c). Finally, DAPH-12 prevented switching from weak to strong [PSI+] elicited by EGCG (Fig. 8d). We conclude that EGCG and DAPH-12 synergize to cure multiple [PSI+] strains.
Discussion
Drug resistance is a severe obstacle in the fight against numerous diseases. Here, we suggest that this problem likely extends to amyloid and prion disorders. Until now, it has been unclear how small molecules directly affect different pure prion strains. By employing Sup35, we discovered that a pure prion protein accesses multiple infectious strains with differential sensitivity to individual small molecules that directly disrupt amyloid structure. We demonstrated how small molecules can directly and differentially affect the spatial arrangement of individual amino acids that comprise the intra- and intermolecular contacts of different prion structures. Moreover, we described the first example of how the combination of two small molecules, that antagonize prionogenesis by distinct mechanisms, synergize to directly inhibit and reverse the formation of diverse prion strains.
Resistance manifested in different ways depending on whether the small molecule was present before or after prionogenesis. If EGCG was present prior to prionogenesis, Sup35's prion domain could (at 4°C) form infectious amyloids, which configured intermolecular contacts that do not ordinarily assemble and resist disruption by EGCG. Thus, small molecules altered the folding landscapes that underpin the repertoire of accessible amyloid forms. Both NM4 and NM4E conferred strong [PSI+]. Thus, the repertoire of fiber conformations encoding strong [PSI+] is more nuanced than previously suspected. Intriguingly, EGCG affected which strain was deployed. The ability to form entirely new strains and escape small molecule inhibition illustrates the plasticity of prionogenesis and the problem it poses to drug development. Small molecule scaffolds that facilitate new amyloid polymorphs should be identified and perhaps avoided. Equally problematic are prion structures maintained by different intermolecular contacts that display differential resistance to small molecules. For example, NM4 was more resistant to EGCG or DAPH-12 than NM25.
Importantly, DAPH-12 and EGCG synergized in directly inhibiting and reversing Sup35 prionogenesis in a manner that antagonized a broader spectrum of prion forms. This synergy was likely due to mechanistic differences in DAPH-12 and EGCG action. For example, if present prior to prionogenesis at 25°C, both DAPH-12 and EGCG precluded the intermolecular contacts required to nucleate assembly. They prevented NM from accessing an obligate oligomeric intermediate required for de novo fibrillization11. Formation of this intermediate requires reorganization of NM monomers within molten oligomers30,31 (Fig. 1b, steps 2 and 3). These events can be tracked using acrylodan-labeled NM, which has revealed that portions of N (residues ∼21-106 at 25°C and residues ∼21-96 at 4°C) become solvent inaccessible within oligomers during the lag phase prior to fibrillization11,26. DAPH-12 prevents N- and C-terminal portions of N from becoming solvent inaccessible11, whereas EGCG only prevented C-terminal portions. Thus, DAPH-12 and EGCG altered the folding of Sup35's prionogenic region in different ways.
These mechanistic differences likely determined the different efficacies of DAPH-12 and EGCG against distinct prion strains. For example, EGCG did not inhibit formation of strains that nucleated from the Head, whereas DAPH-12 partially inhibited their formation (Fig. 6b). Both small molecules strongly antagonized strains that nucleated from the Tail (Fig. 6b). When combined, these distinct modes of action antagonize multiple strains. DAPH-12 and EGCG combinations were more efficacious in inhibiting seeded fibrillization by NM4 and NM25. This was especially evident for seeding by NM4, which was more resistant to either small molecule alone. Thus, DAPH-12 and EGCG combinations antagonized intermolecular contact formation even in the presence of preformed templates.
Both EGCG and DAPH-12 slowly remodeled preformed prions to non-templating forms. Again, EGCG and DAPH-12 displayed subtly different modes of action, which likely contributed to their differing efficacies against different strains. EGCG disrupted both inter- and intramolecular contacts, whereas DAPH-12 disrupted only intermolecular contacts. Acting alone, either small molecule converted NM25 to distinct oligomeric forms. Yet, in combination, their subtly different activities synergized to remodel both NM4 and NM25. This was particularly striking for NM25 fibers, which were completely resolved (Fig. 7b). These direct prion-remodeling activities of DAPH-12 and EGCG exerted greater effects on NM25 than on NM4, which helps explain why weak [PSI+] is cured more effectively than strong [PSI+]. DAPH-12 also inhibited and reversed NM4E formation, and prevented switching from weak to strong [PSI+] elicited by EGCG.
Our findings have implications for developing successful small molecule treatments for amyloid disorders. Like [PSI+], neurodegenerative amyloidoses likely reflect a continuum of phenotypes caused by an underlying continuum of amyloid strains and misfolded forms, rather than a single pure form. Treatment with a small molecule that targets only one strain can only be effective if a disease state is caused by a purely susceptible strain. However, if mixtures of susceptible and resistant strains cause other disease states, then the resistant strain, even if present at low levels, might amplify. We draw analogy from the ability of EGCG to cure weak [PSI+], but simultaneously cause the appearance of strong [PSI+]. At the other extreme, disease states caused by purely resistant strains would be refractory to a small molecule that antagonizes other strains. Here, we draw analogy from EGCG being unable to cure strong [PSI+] strains conferred by NM4E. Such small molecules might fail in broad clinical trials if administered alone. Thus, small-molecule therapies must mitigate the vicissitudes of amyloidogenesis.
Ultimately, neurodegenerative amyloidoses may require combination therapies involving small molecule cocktails that target all strain permutations. Lessons learned from HIV and cancer treatments, which have been hampered by amplification of rare drug-resistant forms but eventually remedied with small molecule cocktails46,47, might have important applications for neurodegenerative amyloidoses. Our observations that DAPH-12 and EGCG synergized to block and reverse prionogenesis of multiple strains provide proof-of-principle that small molecule combinations can directly counter prion diversity. This is the first example of a synergistic small molecule combination that directly eradicates diverse prion strain structures.
Whether the DAPH-12 and EGCG pair will synergize against other amyloidogenic proteins is unclear. However, we suspect that synergistic small-molecule combinations can be found for each specific amyloidogenic protein. Even with Sup35, DAPH-12 and EGCG failed to synergize in curing strong [PSI+] conferred by NM4E. Yet, we suspect that other small molecule combinations might. Using [PSI+], we can screen for EGCG or DAPH-12 analogs with enhanced specificity or activity when used alone or in combination. Lead compounds can then be assessed against disease-associated proteins, such as Aβ42, which are antagonized by the parental EGCG and DAPH-12 scaffolds6,11. Thus, new small molecule combinations with optimized activity or specificity might be identified rapidly to antagonize entire strain spectra. Small molecule combinations that synergistically remodel diverse strains will provide a powerful strategy to compliment small molecule combinations that modulate proteostasis and synergistically ameliorate protein-misfolding disorders24,48.
Materials and Methods
Small molecules
EGCG, EGC, gallic acid and DAPH-6 (Sigma-Aldrich) were dissolved in DMSO. DAPH-12 was synthesized and characterized as described49,50.
Proteins
Aβ42 was obtained from Biosource. NM, NM cysteine mutants, Sup35 and Hsp104 were purified as described11,26,31,33. NM Δ21-38, NM Δ83-110 and NM Δ21-38, Δ83-110 constructs were generated by Quikchange mutagenesis (Stratagene), and proteins were purified as for NM. Single cysteine NM mutants were labeled with either pyrene maleimide or acrylodan (Molecular Probes) or crosslinked with BMB (Pierce) under denaturing conditions as described26. See Supplementary Methods for description of NM EGCG binding.
Fiber Assembly and Disassembly
Aβ42 fiber assembly was as described11. NM fiber assembly and disassembly was performed in the presence of the indicated concentrations of DMSO, DAPH-12, EGCG or EGC as described11,26,31. NM (5μM) fibrillization was in assembly buffer (AB): 40mM Hepes-KOH (pH 7.4), 150mM KCl, 20mM MgCl2 and 1mM DTT. Unseeded reactions were agitated at 1400rpm in a thermomixer (Eppendorf) for the indicated time at 4°C or 25°C. Seeded assembly was unagitated and was performed for 4h at the temperature (4°C or 25°C) used to make the fibers that acted as seed. Unbound small molecules were removed from seed preparations by dialysis for 1h at 25°C against AB using Slide-A-lyzer MINI dialysis unit, 10K MWCO (Pierce). SDS-PAGE followed by Coomassie staining revealed that ∼95% of the protein was recovered after dialysis. We confirmed that ∼95% of the EGCG was removed by dialysis using nitroblue tetrazolium staining (see Supplementary Methods). Sup35 fiber assembly was in AB supplemented with 10% glycerol and 1mM GTP as described33, except that reactions were agitated at 700rpm in a thermomixer (Eppendorf). For fiber disassembly, NM or Sup35 fibers (2.5μM monomer) were incubated without agitation at 25°C for the indicated time. Fiber assembly or disassembly was determined by CR binding, ThT fluorescence, EM, sedimentation analysis or SDS-resistance as described11,31. See also Supplementary Methods.
Acrylodan and Pyrene Fluorescence
Acrylodan and pyrene fluorescence were as described11,26. Samples were diluted 10-fold in AB before measurement to prevent potential spectroscopic interference. Very similar results were obtained if unbound small molecules were rapidly removed immediately before fluorescence measurements by Bio-spin 6 (Bio-Rad) gel filtration (for acrylodan measurements) or dialysis (for pyrene measurements).
Protein transformation
Yeast cells from a W303-derived strain (MATα leu2-3, -112 his3-11 trp1-1 ura3-1 ade1-14 can1-100 [pin-] [psi-] [ure-o]) that contained an ADE1 nonsense mutation suppressible by [PSI+] were transformed with NM conformers and a URA3 plasmid. Unbound small molecules were removed from seed preparations as described above. NM conformers were then resuspended in the transformation mix15,26. The proportion of Ura+ transformants that acquired 27 was determined. Very similar results were obtained using another yeast strain, 74-D694.
Peptide arrays
The N peptides: 5-25 (NQGNNQQNYQQYSQNGNQQQG), 20-38 (GNQQQGNNRYQ GYQAYNAQ), 41-71 (PAGGYYQNYQGYSGYQQGGYQQYNPDAGYQQ) and 88-106 (YQQQFNPQGGRGNYKNFNY) were synthesized by the MIT biopolymers laboratory. Peptides were dissolved in 8M urea, 20mM TrisHCl (pH 7.4) to yield 10mM stock solutions, which were spotted onto nitrocellulose, dried and blocked for 24h with 3% BSA. Arrays were then incubated with NM-his (1μM) in AB plus 3% BSA for 16h at 4°C or 25°C in the presence of DMSO (1%) or EGCG (20μM). Arrays were then washed extensively with 2% SDS. NM-his was detected using an anti-his monoclonal antibody (GE Healthcare) followed by an HRP–coupled secondary antibody (Sigma). The amount of NM-his bound was then determined by quantitative densitometry in comparison to known quantities of NM-his spotted on nitrocellulose. Similar results were obtained using peptides covalently coupled to glass slides27.
[PSI+] induction
Yeast cells from a W303-derived strain (MATα leu2-3, -112 his3-11 trp1-1 ura3-1 ade1-14 can1-100 [PIN+] [psi-] [ure-o] Δpdr5∷CAN Leu2∷GAL-NM-YFP pRS305) were grown in liquid medium containing raffinose (SRaff-Leu) as the sole carbon source to an optical density of 1 at 600nm. Cultures were then diluted to an optical density of 0.1 at 600nm with media containing galactose as the sole carbon source (SGal-Leu) to induce NM-YFP expression in the presence of the indicated concentrations of EGCG, EGC or DMSO. After 12h at 30°C, ∼103 cells were plated onto 25% YPD or SD-Ade and incubated for 3-7 days at 30°C. [PSI+] induction was scored as the number of white (strong [PSI+]) and pink (weak [PSI+]) colonies divided by the total number of colonies. Alternatively, the number of colonies on SD-Ade plates was divided by the number of colonies on the corresponding 25% YPD plates. Both assays produced virtually identical results. Alternatively, yeast cells were analyzed by microscopy using a Leica DMR microscope and Leica IM50 Image manager software. Pictures were taken at 40× magnification and the proportion of cells with one or more fluorescent focus determined. Six different microscopic fields with at least 80 cells in each field were scored for each condition.
[PSI+] curing
Liquid cultures of a W303-derived yeast strain (MATα leu2-3, -112 his3-11 trp1-1 ura3-1 ade1-14 can1-100 [PIN+] [PSI+] [ure-o] Δpdr5∷CAN) were grown at 30°C in YPD containing the indicated concentrations of EGCG, DAPH-12, EGC or DMSO for 0-24h. Cultures were maintained in mid-log growth phase by dilution with YPD and grown for another 24-72h. Subsequently, ∼8 × 103 cells were plated on 25% YPD for each condition, and grown at 30°C for 3 days. [PSI+] curing was scored as the proportion of red ade- [psi-] colonies.
For fluorescence studies, yeast cells from a W303-derived strain (MATα leu2-3, -112 his3-11 trp1-1 ura3-1 ade1-14 can1-100 [PIN+] [PSI+] [ure-o] Δpdr5∷CAN Leu2∷GAL-NM-YFP pRS305) were grown in SRaff-Leu at 30°C to an optical density of 1 at 600nm. Cultures were then diluted into SGAL-Leu containing the indicated concentrations of EGCG, EGC or DMSO. After 8h of NM-YFP expression, cells were analyzed by microscopy as above. Alternatively, NM-YFP was expressed for 5h. Expression was then shut down by adding 2% glucose in the presence of DMSO (1%), EGCG or EGC (250μM) for 2h, and cells were then imaged. The proportion of cells with one or more NM-YFP focus was then determined.
Supplementary Material
Acknowledgments
We thank E. Hennessy (Massachusetts Institute of Technology), S. Buchwald (Massachusetts Institute of Technology), R. Krishnan (Whitehead Institute for Biomedical Research), S. Lindquist (Whitehead Institute for Biomedical Research), J. Weissman (University of California San Francisco), R. Wetzel (University of Pittsburgh School of Medicine) and C. Glabe (University of California Irvine) for generous provision of reagents; J. Chan for preliminary in vivo experiments; and M. Lemmon, A. Gitler, S.B. Cullinan, N. Bonini and S.W. Englander for comments on the manuscript. This work was supported by US National Institutes of Health (NIH) training grant 2T32GM008275-21 (E.A.S), an NIH Director’s New Innovator Award (DP2OD002177), an Ellison Medical Foundation New Scholar in Aging Award, an American Heart Association Scientist Development Grant, and University of Pennsylvania Institute on Aging and Alzheimer’s Disease Core Center pilots (J.S.).
Footnotes
Competing interests statement: The authors declare no competing financial interests.
Author contributions: B.R., M.D., H.W., C.C., N.P.L., E.A.S. and J.S. designed experiments, contributed key reagents, performed experiments and interpreted data. M.N.K., E.J.H. and S.L.B. contributed key reagents. M.D. and J.S. wrote the manuscript.
References
- 1.Nelson R, Eisenberg D. Structural models of amyloid-like fibrils. Adv Protein Chem. 2006;73:235–82. doi: 10.1016/S0065-3233(06)73008-X. [DOI] [PubMed] [Google Scholar]
- 2.Skovronsky DM, Lee VMY, Trojanowski JQ. Neurodegenerative Diseases: New Concepts of Pathogenesis and Their Therapeutic Implications. Annu Rev Pathol Mech Dis. 2006;1:151–170. doi: 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts BE, Shorter J. Escaping amyloid fate. Nat Struct Mol Biol. 2008;15:544–6. doi: 10.1038/nsmb0608-544. [DOI] [PubMed] [Google Scholar]
- 5.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–9. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 6.Ehrnhoefer DE, et al. Redirecting aggregation pathways: small molecule-mediated conversion of amyloidogenic polypeptides in unstructured, off-pathway oligomers. Nature Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 7.Ehrnhoefer DE, et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models. Hum Mol Genet. 2006;15:2743–51. doi: 10.1093/hmg/ddl210. [DOI] [PubMed] [Google Scholar]
- 8.Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science. 2004;306:865–9. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 9.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–6. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Zhu M, Rajamani S, Uversky VN, Fink AL. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem Biol. 2004;11:1513–21. doi: 10.1016/j.chembiol.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, et al. Direct and selective elimination of specific prions and amyloids by 4,5-dianilinophthalimide and analogs. Proc Natl Acad Sci U S A. 2008;105:7159–64. doi: 10.1073/pnas.0801934105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legname G, et al. Synthetic mammalian prions. Science. 2004;305:673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 13.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 14.Safar J, et al. Eight prion strains have PrPSc molecules with different conformations. Nat Med. 1998;4:1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–8. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 16.Tessier PM, Lindquist S. Unraveling infectious structures, strain variants and species barriers for the yeast prion [PSI+] Nat Struct Mol Biol. 2009;16:598–605. doi: 10.1038/nsmb.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tribouillard D, et al. Antiprion drugs as chemical tools to uncover mechanisms of prion propagation. Prion. 2007;1:48–52. doi: 10.4161/pri.1.1.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguzzi A. Staining, straining and restraining prions. Nat Neurosci. 2008;11:1239–40. doi: 10.1038/nn1108-1239. [DOI] [PubMed] [Google Scholar]
- 19.Castilla J, et al. Crossing the species barrier by PrPSc replication in vitro generates unique infectious prions. Cell. 2008;134:757–68. doi: 10.1016/j.cell.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kocisko DA, et al. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77:10288–94. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trevitt CR, Collinge J. A systematic review of prion therapeutics in experimental models. Brain. 2006;129:2241–65. doi: 10.1093/brain/awl150. [DOI] [PubMed] [Google Scholar]
- 22.Kocisko DA, et al. Comparison of protease-resistant prion protein inhibitors in cell cultures infected with two strains of mouse and sheep scrapie. Neurosci Lett. 2005;388:106–11. doi: 10.1016/j.neulet.2005.06.053. [DOI] [PubMed] [Google Scholar]
- 23.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 24.Mu TW, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–81. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–50. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 26.Krishnan R, Lindquist SL. Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature. 2005;435:765–72. doi: 10.1038/nature03679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tessier PM, Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature. 2007;447:556–61. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheibel T, Bloom J, Lindquist SL. The elongation of yeast prion fibers involves separable steps of association and conversion. Proc Natl Acad Sci U S A. 2004;101:2287–92. doi: 10.1073/pnas.0308754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheibel T, Lindquist SL. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat Struct Biol. 2001;8:958–62. doi: 10.1038/nsb1101-958. [DOI] [PubMed] [Google Scholar]
- 30.Serio TR, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–21. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 31.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–7. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 32.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci U S A. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shorter J, Lindquist S. Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol Cell. 2006;23:425–38. doi: 10.1016/j.molcel.2006.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheibel T, Kowal AS, Bloom JD, Lindquist SL. Bidirectional amyloid fiber growth for a yeast prion determinant. Curr Biol. 2001;11:366–9. doi: 10.1016/s0960-9822(01)00099-9. [DOI] [PubMed] [Google Scholar]
- 35.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–7. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 36.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI+] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375–86. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–9. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 38.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 39.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc Natl Acad Sci U S A. 2006;103:19754–9. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diaz-Avalos R, King CY, Wall J, Simon M, Caspar DL. Strain-specific morphologies of yeast prion amyloid fibrils. Proc Natl Acad Sci U S A. 2005;102:10165–70. doi: 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 42.O'Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci U S A. 2002;99:1485–90. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patino MM, Liu JJ, Glover JR, Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–6. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 44.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49636–43. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 45.Kochneva-Pervukhova NV, et al. [PSI+] prion generation in yeast: characterization of the ‘strain’ difference. Yeast. 2001;18:489–97. doi: 10.1002/yea.700. [DOI] [PubMed] [Google Scholar]
- 46.Staszewski S, et al. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. N Engl J Med. 1999;341:1865–73. doi: 10.1056/NEJM199912163412501. [DOI] [PubMed] [Google Scholar]
- 47.Burgess MR, Skaggs BJ, Shah NP, Lee FY, Sawyers CL. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc Natl Acad Sci U S A. 2005;102:3395–400. doi: 10.1073/pnas.0409770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spilman P, et al. A gamma-secretase inhibitor and quinacrine reduce prions and prevent dendritic degeneration in murine brains. Proc Natl Acad Sci U S A. 2008;105:10595–600. doi: 10.1073/pnas.0803671105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hennessy EJ, Buchwald SL. Synthesis of 4,5-dianilinophthalimide and related analogues for potential treatment of Alzheimer's disease via palladium-catalyzed amination. J Org Chem. 2005;70:7371–5. doi: 10.1021/jo051096o. [DOI] [PubMed] [Google Scholar]
- 50.Trinks U, et al. Dianilinophthalimides: potent and selective, ATP-competitive inhibitors of the EGF-receptor protein tyrosine kinase. J Med Chem. 1994;37:1015–27. doi: 10.1021/jm00033a019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.