

Abstract
Synthesis of the complex (BDI)Nb(NtBu)Cl2py (BDI = HC[C(Me)N(2,6-iPr2-C6H3)]2) was achieved in high yield following the treatment of Nb(NtBu)Cl3py2 with Li(BDI)(OEt2). Substitution of the chlorides for fluorides was effected by introducing 2.0 equiv of Me3SnF in toluene, providing the pyridine-coordinated difluoride complex (BDI)Nb(NtBu)F2py in modest yield. The pyridine ligands from both halide compounds were removed by treatment of the pyridine adducts with B(C6F5)3, affording the Lewis base-free complexes (BDI)Nb(NtBu)X2 (X = Cl, F). Additionally, the Lewis base-free dichlorides of the tBu-imido and Ar-imido (Ar = 2,6-iPr2-C6H3) complexes were obtained following treatment of Nb(NR)Cl3(dme) (R = tBu, Ar) with Li(BDI)(OEt2). The pyridine-coordinated dichloride was alkylated and arylated to form the dimethyl complex (BDI)Nb(NtBu)Me2 (described previously, see text) and the mono(p-tolyl) complex (BDI)Nb(NtBu)Cl(p-tol), the latter of which was methylated with MeMgBr to yield the mixed alkyl/aryl complex (BDI)Nb(NtBu)Me(p-tol) in good yield. A rare example of a Group 5 bis(tBu-imido) species was synthesized in good yield via treatment of (BDI)Nb(NtBu)Cl2py with 2.0 equiv of LiNHtBu to form (BDI)Nb(NtBu)2py. Exchange of the coordinated pyridine ligand for either pyridine-d5 or dmap (p-(dimethylamino)pyridine) was shown to occur through a dissociative mechanism, allowing for removal of the coordinated Lewis base by treatment with B(C6F5)3. The resulting average C2v-symmetric tetracoordinate bis(imido) complex (BDI)Nb(NtBu)2 was characterized in solution by NMR spectroscopy and observed to undergo clean thermal decomposition to yield (BDI#)Nb(NtBu)(NHtBu) (BDI# = H2C=C(NAr)CH=C(NAr)Me) over several hours at room temperature. Treatment of the four-coordinate bis(imido) with tBuNCO resulted in clean [2 + 2] cycloaddition to yield an oxaazametallacyclobutane complex, which was further observed to extrude tBuN=C=NtBu over 12 h at room temperature. The molecular structures of (BDI)Nb(NtBu)Cl2py, (BDI)Nb(NAr)Cl2, (BDI)Nb(NtBu)Me2, (BDI)Nb(NtBu)Cl(p-tol), (BDI)Nb(NtBu)2py, and (BDI)Nb(NtBu)2(dmap) were determined crystallographically. Finally, DFT (BP86) geometry optimization calculations on a model complex of the thermally unstable four-coordinate bis(imido) species allowed for identification of the orbital interactions leading to activation of the imido groups through mixing with the BDI frontier orbitals.
Introduction
The reactivity of early-metal imido functionalities depends in part on the relative energies of the metal xd (x = 3–5) and nitrogen 2p orbitals. As the energies of the xd orbitals decrease from left to right on the periodic table, the covalency of M=NR bonds should increase, leading to an overall stabilization of the M–N π-interaction. Studies on the chemistry of these complexes has led to an empirical trend to this effect, from Group 4 monoimido systems capable of a wide range of π- and σ-bond activation reactions1–7 to Group 6 imidos that often act as spectator ligands.8,9 Accordingly, Group 5 monoimido complexes have shown modest imido group reactivity, but such species are known to lead to unique products,10–13 prompting interest in developing methods for increasing the reactivity of the M=NR (M = V, Nb, Ta) bond.
One interesting concept for activating imido (and other multiply bonded) ligands is to electronically saturate the metal’s dπ orbitals (commonly termed π-loading) by introducing multiple π-donating ligands onto the metal center.14 When implemented with the appropriate symmetry and relative energy considerations, π-loading should have the effect of either polarizing or entirely localizing some imido π-electron density on the nitrogen atom,15–22 thereby decreasing the stabilization from the metal center and activating the π-electrons of the imido bond. This effect has been most successfully implemented with respect to early-metal imido chemistry through combinations of the imido ligand with the orbitally related cyclopentadienyl (Cp) ligands.1,7,23–30
A related method for achieving the π-loading effect is to form bis(imido) complexes.6,31,32 The bent bis(imido) motif can be compared to bent-metallocene systems due to the similarity between the donor orbitals of the cyclopentadienyl and imido ligands (both 1σ,2π, Fig. 1). This similarity indicates that the remaining unoccupied frontier orbitals for bent bis(imido) complexes would mimic those of the bent-metallocenes, with one b2 and two a1 orbitals located in the plane bisecting the angle between the two imido groups.33 These frontier orbitals (1a1, 2b2, and 2a1) lack any out-of-plane π-bonding component, suggesting that the incorporation of a π-donor at these positions would create competition between the imido π-donor orbitals and those of the non-imido ligands for the occupied metal-based π-symmetry orbitals through mixing with the 1b2 and 1b1 orbitals (Fig. 1).
Figure 1.

Left-center: An illustration of the isolobal relationship between the frontier orbitals of Cp2M and (RN)2M complexes. The given orbital occupation corresponds to a d0 electron configuration. The charges p and q are related by p = q + 2 for isoelectronic complexes of elements from the same group. Center-right: Molecular orbital interaction scheme depicting the symmetry and relative energy considerations for forming π-loaded four-coordinate bis(imido) complexes by using a π-donating bidentate ligand.
Nevertheless, in contrast to the Group 6 metals for which bis(imido) dihalide starting materials are readily available, the bis(imido) moiety is rare for Group 5 metals, suggesting the need for specialized auxiliary ligands. The diaryl-β-diketiminato ligand class (Ar2-BDI, HC[C(Me)NAr]2) has many desirable characteristics, being monoanionic, bi-dentate ligands with six-electron π-systems capable of mixing with π-symmetric orbitals on a metal center.34 Many examples are now known in which the use of the Ar2-BDI ligands have allowed for the stabilization of rare and generally reactive first-row transition metal fragments, including early-metal alkylidene and alkylidyne complexes,35–37 three-coordinate iron species,38 and late-metal nitrene complexes,39–41 suggesting that the Ar2-BDI ligand may be suitable for stabilizing the low-coordinate bis(imido)niobium fragment as well.
In comparison to the wealth of 3d transition metal chemistry supported by Ar2-BDI ligands, relatively few reports exist describing the application of the Ar2-BDI ligand class toward the heavier congeners of the middle transition metals (Groups 5 – 8). Thus, while a number of Group 4 d0 complexes are known,34,42–46 only the neutral and cationic d0 Mo complexes (Ar2-BDI)(X)Mo(NR)(Y) (Y = CHCMe2R, NR; see Fig. 2) have been reported for the 4/5d mid-transition metals.47–49 Several late metal Ar2-BDI systems are also known (Groups 9 – 10), but these tend toward either higher formal oxidation states or lower oxidation state metal centers bound by strong π-acids.34,50–58
Figure 2.

a) A representation of the delocalized structure of a Ar2-BDI ligand. b) A representation of the molybdenum imido alkylidene complexes supported by the Ar2-BDI ligands.47,48 c) A representation of the molybdenum bis(imido) complexes supported by the Ar2-BDI ligands.49
Two competing factors appear to have significantly limited research into 4d and 5d middle transition metal Ar2-BDI complexes. The first is the propensity of the Ar2-BDI ligand to undergo reductive cleavage of the ligand N–Cimine bond to yield terminal metal imido complexes bound by a five-membered monoazabutadiene ligand.46,59,60 This transformation is the predominant reactivity pattern observed for (Ar2-BDI)MX3 (M = Zr, Hf; X = monovalent, σ-donor ligand) systems when the metal is reduced to the d2 electronic state (Scheme 1). This instability seemingly excludes common entries into Group 5 and related chemistry via the d2 metal halides, which would mimic the route taken by Budzelaar61 in forming the (Ar2-BDI)MCl2 (M = Ti, V) complexes.
Scheme 1.

This inaccessibility of common dn≥2 starting materials for 4/5d early-metal Ar2-BDI complexes indicates that the use of high oxidation state starting materials may be necessary for the introduction of a Ar2-BDI ligand onto the heavier middle-transition metals. Empirically, however, it appears that Ar2-BDI complexes with substituted aryl rings tend toward four- and five-coordinate complexes,34 which indicates that the steric imposition of the flanking aryl groups may lead to unstable complexes on formation of (Ar2-BDI)MX4 species (M = Nb, Ta, Mo, W; X = halide) should the common pentahalides be used as starting materials. As an example of the circumvention of these steric requirements, the only known niobium β-diketiminato complex is that of the niobium tetrahalide (PhN=CPh–CMe=CPh–NH)NbCl4.62 While the product was not structurally characterized to confirm the bidentate coordination mode of the ligand, it is notable that the β-diketiminato nitrogens were minimally substituted (Ph, H).
Continuing our interest in the synthesis of early-metal low-coordinate complexes,63–66 we have sought Group 5 species supported by the bulky diaryl-β-diketiminato ligand HC[C(Me)N(2,6-iPr2-C6H3)]2 (hereafter referred to as BDI). Our solution to the electronic and steric requirements of ligand introduction extends from the synthetic protocols taken by Schrock and Mösch-Zanetti (see Fig. 2), who both made use of d0 molybdenum bis(divalent) ligand species as the direct synthetic precursors to their Ar2-BDI complexes. Their success would indicate that by concentrating two valencies at one coordination site, the steric requirements for forming stable d0 BDI metal complexes can be satisfied while avoiding low-valent starting materials. For our purposes, use of the imido group as the divalent ligand has the advantages of i) lending stability to the Nb(V) metal center through its 1σ,2π-bonding character,67 ii) allowing study of the effects of a mono-imido group on the BDI ligand, and iii) providing facile entry into four- and five-coordinate bis(imido)niobium complexes. The following text describes our efforts to develop a range of suitable starting materials for exploratory BDI-supported imidoniobium chemistry. We will conclude by discussing our preliminary studies of five- and four-coordinate bis(imido) complexes, along with theoretical support for the claim of π-loading resulting from mixing of the BDI π-system with the Nb=NR π-bonds.
Results and Discussion
Synthesis of dihalide complexes
The reaction of Nb(NtBu)Cl3py2 (py = pyridine) with Li(BDI)·OEt2 (BDI = HC(C(Me)NAr)2, Ar = 2,6-iPr-C6H3) in THF resulted in the formation of a deep red solution from which (BDI)Nb(NtBu)Cl2py (1, Scheme 2) was obtained in high yield as a red crystalline material. The room temperature 1H NMR spectrum displays significant line broadening due to pyridine dissociation, as confirmed by the rapid exchange of coordinated pyridine for pyridine-d5; NMR spectra obtained at 243K reveal average Cs-symmetry (Tc = 293K, ΔG‡ = 14.1(1) kcal/mol) with one singlet for each backbone methyl group, consistent with a geometry that places the chlorides trans to one another. The signals in the 1H and 13C{1H} NMR spectra appear in regions similar to those observed for related early transition metal BDI complexes.42,68,69
Scheme 2.

The chemical shift of the tBu group in the 1H NMR spectrum (0.64 ppm) is notable, however, as the signal is shifted ca. 1 ppm upfield from the starting material. This effect is due to the proximity of the imido group to one of the BDI aryl rings, as the ring current of aryl groups is well-known to provide a shielding effect for nuclei situated centrally above the ring. The 1H NMR chemical shift of the tBu-imido group has thus been a useful indicator of the molecular geometry of these species in solution.
The molecular structure of 1 was determined crystallographically and is represented in Figure 3. As indicated by the low-temperature 1H NMR spectrum, the structure of 1 is that of a distorted octahedron, with the two chlorides mutually trans, and the imido group and the pyridine oriented trans to the nitrogens of the BDI ligand. This geometry places one of the tBu methyl groups directly above the π-system of one of the BDI aryl rings (d(CMe-CAr) = 3.648 – 3.820 Å), consistent with the upfield chemical shift of the tBu group in the 1H NMR spectrum. The displacement of the chlorides toward the pyridine (Cl(l)-Nb(1)-Cl(2) = 160.68(2)°) suggests a weakened Nb-py binding interaction (Nb(1)-N(4) = 2.376(4) Å) and is suggestive of the trigonal bipyramidal geometry of the related Lewis base-free (BDI)Nb(NtBu)Me2 pentacoordinate complex (see below).
Figure 3.


Molecular structure of 1 as determined by a single crystal X-ray diffraction study. Hydrogen atoms omitted for clarity; thermal ellipsoids set at the 50% probability level. Selected bond lengths are given in Å. Selected bond angles (°): Nb(1)-N(1)-C(1) 163.59(19), Cl(1)-Nb(1)-Cl(2) 160.68(2), N(1)-Nb(1)-N(3) 171.92(8), N(2)-Nb(1)-N(4) 172.98(7).
On closer inspection of the crystal structure of 1, some features of the π-bonding within the BDI NCCCN metallacycle important to our later discussion of how the BDI π-system influences Nb=NtBu bonding become apparent. The Nb(1)-N(2) bond length of 2.100(2) Å is close to the range expected for a Nb amide,70 indicating π-donation from N(2) to Nb(1). In contrast, the long Nb(1)-N(3) bond length of 2.420(2) Å reflects the strong trans-influence of the imido ligand and indicates that any N(3)(pπ) to Nb(dπ) donation is minimal. This asymmetric binding of the BDI nitrogen atoms to the metal center is further reflected in the bond lengths within the backbone of the BDI ligand, as the nitrogen trans to the imido group has a shorter N(3)-C(20)imine bond length than that of the N(2)-C(18)imine bond of the nitrogen trans to the pyridine ligand (1.317(3) vs. 1.369(3) Å, respectively). Still, the long N(2)-C(18) bond is shorter than expected for an alkyl amine (1.469 Å) and close to that of N-C(O) bond lengths of N,N-dialkyl ureas (1.363 Å).71 A related trend is observed for the N(3) atom, as the N(3)-C(20) distance reflects the bond’s greater imine character with the interaction found to be intermediate between that of an isolated imine (1.279 Å) and the N-C bond lengths within pyridine (1.337 Å).71 This bond length alternation propagates through the C-C bonds, with a shorter C(18)-C(19) bond length (1.372(4) Å) than the C(19)-C(20) distance (1.421(4) Å). Thus, while the niobacycle formed by the BDI NCCCN ligand backbone has the electronic and orbital structure to form an aromatic metallacycle,72 the bond lengths within the six-membered ring more closely represent resonance structure A (Figure 4) than the resonance-averaged structure C that is commonly found on early-metal systems.
Figure 4.

Resonance structures used for describing the ground state of β-diketiminato niobium imido complexes.
To improve the range of available starting materials for further synthetic chemistry, we sought the synthesis of other halide complexes, both with and without coordinated donor ligands. The difluoride complex (BDI)Nb(NtBu)F2py (2) was synthesized from 1 by treating the dichloride with 2.0 equiv of the mild fluorinating reagent Me3SnF in toluene at room temperature (Scheme 3).
Scheme 3.

The 1H NMR spectrum of 2 indicates average Cs-symmetry at room temperature. The sharp resonances observed at 298 K are consistent with tighter binding of pyridine (Tc = 348 K, ΔG‡ = 16.3(1) kcal/mol) to a metal center that is both less sterically encumbered and more electrophilic than 1 due to the smaller size and higher electronegativity of fluorine. The formation of a Cs-symmetric complex with a mirror plane relating the two fluorides is further supported by a single new 19F NMR resonance at 68.77 ppm – a region common for Group 5 d0 fluoride complexes73 – and by an upfield shifted tBu resonance (0.41 ppm) indicative of an imido group situated cis to one BDI nitrogen and trans to the other, placing the imido group centrally above one of the flanking aryl rings.
Forming 14/16 e− base-free dihalide species was accomplished by two routes, either by pyridine abstraction from complexes 1 or 2 or by ligand metallation on the readily attainable Nb(NR)Cl3(dme) (R = tBu, Ar) starting materials. Thus, addition of the known pyridine abstracting reagent B(C6F5)3 to benzene solutions of 1 or 2 yielded the base-free dihalide complexes (BDI)Nb(NtBu)X2 (3, X = Cl; 4, X = F; Scheme 4, route a).74 In each case, the color the solution lightened slightly on addition of the borane, and the formation of py·B(C6F5)3 was clearly observed by 1H and 19F NMR. Complex 3 and py·B(C6F5)3 were separated by repeated fractional crystallization from pentane, yielding a material that was found to be analytically identical to that obtained from the reaction of Nb(NtBu)Cl3(dme) with (see below). Li(BDI)·OEt2
Scheme 4.

Compound 4 was characterized in solution by NMR spectroscopy and found to have averaged C2v symmetry at room temperature. The 1H NMR resonances were broad at this temperature, indicating that the high symmetry may be attributable to fluxional processes in solution. While low temperature NMR experiments reveal an averaged Cs-symmetric complex, NMR spectra collected at elevated temperatures begin to re-broaden from the room temperature spectra, indicating that more than one fluxional process is responsible for the averaged symmetry observed by NMR spectroscopy. The formation of a fluoride-bridged dimer upon pyridine abstraction could account for this behavior, as the low temperature would slow down any bridging fluoride exchange mechanisms, while the high temperature spectra would start to approximate that expected for the mononuclear complex (Scheme 5), which would likely have average C2v symmetry in solution based on related compounds (see below).
Scheme 5.
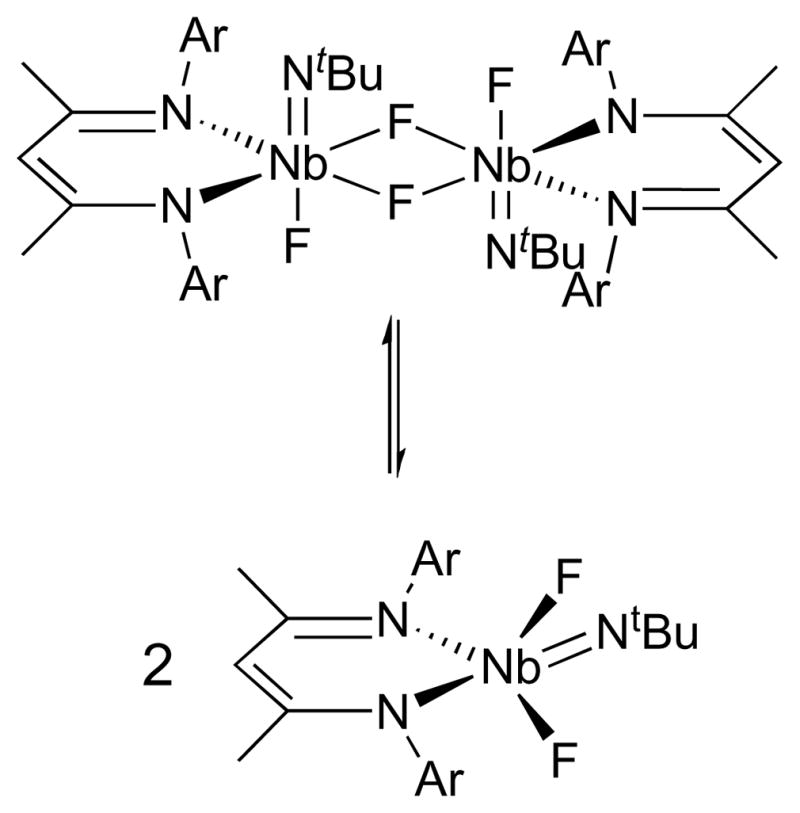
As mentioned above, the reaction of Nb(NtBu)Cl3(dme) with Li(BDI)·OEt2 in toluene resulted in a cherry red solution from which (BDI)Nb(NtBu)Cl2 (3; Scheme 4, route b) was crystallized in modest yields. The room temperature 1H NMR spectrum of 3 is again broadened due to fluxional motion in solution on the NMR timescale, but 1H and 13C{1H} NMR spectra collected at 223 K indicate overall Cs symmetry in solution. This solution state symmetry along with the downfield shift of the tBu resonance from 0.67 ppm for 1 to 1.54 ppm for 3 suggest an overall square pyramidal geometry with an apical imido group. The shift in the 1H NMR resonance for the tBu group is in accord with a change in position of the tBu group with respect to the nearest aryl ring of the BDI ligand. The chemical shift of the imido group for 3 lies in the range expected for early-metal tBu imido groups that are not flanked by aryl rings. Vapor diffusion osmometry measurements on 3 in benzene at room temperature further support the formulation of 3 as a monomer, ruling out the possibility of a dimer with bridging chlorides.
A related complex that is believed to have a geometric structure analogous to that of 3 was synthesized from the arylimido precursor Nb(NAr)Cl3(dme) (Ar = 2,6-iPr2-C6H3). Reaction of Li(BDI)·OEt2 with Nb(NAr)Cl3(dme) in THF led to isolation of the purple, base-free complex (BDI)Nb(NAr)Cl2 (5; Scheme 4, route b) in 57% yield after crystallization from Et2O. The room temperature 1H and 13C{1H} NMR spectra of 5 revealed that the geometry of the complex was fluxionally static on the NMR timescale, and the overall Cs symmetry observed in solution was supported by the crystallographic data collected for 5 (Figure 5), wherein an approximate molecular mirror plane includes the metal center, the α-carbon common to both BDI imine groups, and the nitrogen and arene portions of the imido group.
Figure 5.

The molecular structure of 5 as determined by a single crystal X-ray diffraction study. The hydrogen atoms were omitted for clarity; the thermal ellipsoids were set at the 50% probability level. Selected bond lengths are given in Å. Selected bond angles (°): Nb(1)-N(1)-C(1) 173.27(16), N(1)-Nb(1)-N(2) 103.93(7), N(1)-Nb(1)-N(3) 98.99(7), N(1)-Nb(1)-Cl(2) 107.81(6), N(1)-Nb(1)-Cl(1) 104.00(6), N(2)-Nb(1)-Cl(2) 148.05(5), N(3)-Nb(1)-Cl(1) 156.86(5).
As a likely mimic for the coordination geometry of 3, complex 5 reflects the versatility of the binding orientation of the imido group with respect to the BDI ligand. The ability of the ligand to adapt to the electronic situation at the metal center is indicated by the bond distances within the ligand NCCCN backbone.
In comparison to 1, which had alternating long and short bond distances within the BDI metallacycle resulting from the trans influence of the imido group, complex 5 has an apical imido group, with chlorides occupying both coordination sites trans to the BDI nitrogens. This leads to roughly equivalent Nb-NBDI bond lengths (Nb(1)-N(2) 2.1699(17) Å, Nb(1)-N(3) 2.1879(18) Å) and statistically equivalent N-Cimine (N(2)-C(26) 1.332(3) Å, N(3)-C(28) 1.336(3) Å) and Cimine-Cα (C(26)-C(27) 1.392(3) Å, C(27)-C(28) 1.395(3) Å) bond lengths. The implication of these findings is that the asymmetric binding motif found for 1 is a consequence of competing π-donation from ligands about the basal plane of the metal and not a general feature of the BDI ligand binding to a Nb(V) center.
Synthesis of alkyl and aryl Nb(V) complexes
Due to the relative ease of reduction to Nb(IV), substitution reactions at Nb(V) centers involving hard nucleophiles are often hampered by reduction reactions.75 This trend may be responsible for the fact that only a handful of Nb(V) aryl complexes are known,76–84 only one of which has been structurally characterized.80a Owing to the stabilizing nature of both the BDI ligand and imido groups toward high oxidation states, we sought niobium complexes with common alkyl and aryl ligands, in an effort to probe the electronic factors controlling the geometric parameters within the BDI ligand. A synthetic procedure for the dimethyl complex (BDI)Nb(NtBu)Me2 has been previously communicated, but the compound was not discussed in detail.85 A full analysis of this complex will be presented for comparative purposes.
Synthesis and characterization of (BDI)Nb(NtBu)Me2
Dimethylation of 1 could be performed cleanly using the Grignard reagent. Addition of a solution containing 2.0 equiv of MeMgBr in Et2O to a stirred slurry of 1 in Et2O at −72 °C caused the solution to turn from dark red to orange, then pale yellow on warming to room temperature. The isolated product (BDI)Nb(NtBu)Me2 (6, Scheme 6) crystallized from pentane as large yellow blocks in 70–80% yields. Complex 6 is thermally unstable both in the solid state (days) and in the solution (hours) at room temperature, decomposing into a brown unidentified material. Storage of 6 at −35 °C allowed the complex to persist for several months without significant decomposition.
Scheme 6.

A 1H NMR spectrum of 6 taken in THF-d8 at room temperature displays broad peaks associated with a dynamic molecular geometry on the NMR time scale; cooling the sample to 233K reveals a molecular geometry with overall Cs-symmetry, in which the BDI ligand bridges axial and equatorial positions and the Nb-Me groups occupy the remaining equatorial positions. This geometry is proposed from several key features in the 1H NMR spectrum of 6, including the appearance of i) two resonances for the backbone methyl groups (1.73 and 2.00 ppm), ii) a single resonance for the two Nb-Me groups at 0.41 ppm, and iii) a downfield shifted tBu group (0.88 ppm) due to its close proximity to an aryl group. These features rule out the possibility of a square pyramidal complex with basal methyl groups, in contrast to the pentacoordinate dihalide complexes 3 and 5, which place the imido group in the apical position of a square-based pyramid. The 13C{1H} NMR spectrum taken at 213K exhibits a single broad resonance at 47.5 ppm, attributable to the two Nb-Me groups with unresolved coupling to the 93Nb (I = 9/2, 100%) nucleus.
The fluxional motion observed in the room temperature NMR spectrum does not likely involve an ionic mechanism for interchange of the methyl groups as the energy barrier for the fluxional process did not change appreciably when the complex was dissolved in benzene or toluene as opposed to THF. While a Berry pseudorotation mechanism involving interchange of the axial and equatorial substituents in the pentacoordinate complex seems likely, a mechanism involving dissociation of a neutral nitrogen donor of the BDI ligand86 to give a four-coordinate complex capable of methyl group exchange has not been ruled out. A lengthening of the Nb(1)-N(3) bond – as seen in 1 – due to the trans-influence of the imido ligand could again be operative in 6, whereby the imido group’s trans-influence could lead to a trans-effect, facilitating the dissociation of the BDI nitrogen trans to the imido group.
The structure of 6 in the solid-state was found to be distorted trigonal bipyramidal87 (τ = 0.71), with an axial imido group, equatorial methyl groups, and the BDI ligand spanning axial and equatorial positions (Figure 6), matching the geometry proposed from the NMR data. As seen for 1, the Nb-NBDI distance for the nitrogen trans to the imido group is significantly longer than the Nb-NBDI distance for the equatorial nitrogen (2.3578(13) vs. 2.1354(13) Å), attributable to the trans-influence of the imido group (Nb(1)-N(1) 1.7773(13) Å). This bond alternation proceeds around the metallacycle, although not to the degree observed for 1. Also, the difference between the Nb(1)-CMe bond lengths (Nb(1)-C(1) 2.1776(17) Å; Nb(1)-C(2) 2.1874(17) Å) is consistent with the distorted nature of the trigonal bipyramid, placing the C(1) methyl group in a more apical position on the continuum between an idealized trigonal bipyramid and an idealized square-based pyramid.
Figure 6.

The molecular structure of 6 as determined by a single crystal X-ray diffraction study. The hydrogen atoms were omitted for clarity; the thermal ellipsoids were set at the 50 % probability level. Selected bond lengths are given in Å. Selected bond angles (°): N1-Nb1-N2 99.71(5), N1-Nb1-C1 94.83(6), N2-Nb1-C1 113.11(6), N1-Nb1-C2 89.58(6), N2-Nb1-C2 129.55(6), C1-Nb1-C2 115.33(7), N1-Nb1-N3 172.01(5), N2-Nb1-N3 83.33(5), C1-Nb1-N3 90.71(6), C2-Nb1-N3 82.83(6), C3-N1-Nb1 168.96(12).
Attempts at forming a monomethylniobium chloride product by using < 2.0 equiv of MeMgBr resulted only in mixtures of 1 and the dimethyl complex 6, indicating either that i) disproportionation processes are occurring rapidly in solution or that ii) the rate of addition of the second methyl group to the methyl chloride complex is much faster than the rate of addition of the first methyl group to the dichloride complex. Notably, attempts at forming other alkyl complexes resulted in intractable mixtures of products, presumably due to sterically-induced reactivity.
Synthesis and characterization of (BDI)Nb(NtBu)X(p-tol) (X = Cl, Me)
Aryl ligands should be more stable toward sterically induced decomposition reactions due to their lack of α-hydrogens and their smaller volume compared to common non-methyl alkyl ligands. The addition of 1.0 equiv of p-tol2Mg (p-tol = 4-MeC6H4) to 1 cleanly afforded (BDI)Nb(NtBu)Cl(p-tol) (7) as an orange solid (Scheme 7). The complex exhibits C1-symmetry by 1H NMR spectroscopy, with a p-tolyl methyl resonance at 2.28 ppm. The use of aryl Grignard reagents failed to give clean conversion to monoaryl products, and over-arylation was only observed when a greater excess of p-tol2Mg was allowed to react with 1 for longer periods of time. Although complex 7 is indefinitely stable in the solid state, it decomposes in solution over a few days into unidentified products.
Scheme 7.

Alkylation of 7 with MeMgBr cleanly formed the mixed aryl/alkyl complex (BDI)Nb(NtBu)Me(p-tol) (8, Scheme 7). This compound is stable at room temperature and, judging by the 298K 1H NMR spectrum, non-fluxional on the NMR time-scale at room temperature. The spectrum indicates molecular C1-symmetry, with eight HCMe2 doublets, four (overlapping) HCMe2 septuplets, and two backbone methyl singlets. We were intrigued by the low symmetry of these complexes in solution, given the fluxional nature of related pentacoordinate complex (BDI)Nb(NtBu)Me2. To determine the effect the aryl group had on the overall molecular structure, we obtained crystallographic information on one of the aryl complexes.
Single crystals of the p-tolyl chloride complex 7 were grown from pentane; the molecular structure is represented in Figure 7. The complex adopts a square pyramidal geometry (τ = 0.13), with an apical p-tolyl group. This geometry is surprising given the preference of square pyramidal complexes with one multiply-bonded ligand for placing the multiply bonded-ligand in the apical coordination site,21 akin to the common 4-legged piano stool complexes CpML4. The geometric preference of 7 may be solely indicative of a steric preference for placing the larger tBu group in a basal position, away from the bulky iPr substituents of the BDI ligand. Still, this complex provides a close structural analogy to 5, allowing for comparison of the effect the imido group on the bonding within the NCCCN framework. The trans-influence of the imido group can again be seen in the lengthening of the Nb(1)-N(3) bond length to 2.332(4) Å compared to the Nb(1)-N(2) bond length of 2.114(4) Å for the bond trans to the chloride.
Figure 7.

The molecular structure of 7 as determined by a single crystal X-ray diffraction study. The hydrogen atoms were omitted for clarity; the thermal ellipsoids were set at the 50 % probability level. Selected bond lengths are given in Å. Selected bond angles (°): N(1)-Nb(1)-N(2) 96.12(15), N(1)-Nb(1)-C(5) 101.31(19), N(2)-Nb(1)-C(5) 100.23(16), N(1)-Nb(1)-N(3) 151.03(17), N(2)-Nb(1)-N(3) 82.56(13), C(5)-Nb(1)-N(3) 107.42(16), N(1)-Nb(1)-Cl(1) 89.71(13), N(2)-Nb(1)-Cl(1) 159.03(11), C(5)-Nb(1)-Cl(1) 98.34(13), N(3)-Nb(1)-Cl(1) 82.64(9), Nb(1)-C(5)-C(8) 171.92.
The BDI NCCCN bond lengths alternate as predicted by resonance structure A in Scheme 3, although not to the degree seen for 1. The Nb(1)-C(5)Ar bond length of 2.145(5) Å is slightly longer than that observed previously for the other known Nb(V) aryl complex (2.122 Å).80 The aryl group is also canted slightly with respect to the niobium center (Nb(1)-C(5) ipso-C(8)para = 172°), and the metal lies 0.223 Å from the mean plane of the aryl group. This likely reflects crystal-packing forces, as little π-interaction would be expected between the aryl group and the metal center, and an intermolecular interaction between the p-tolyl methyl group and a near-by tBu group (2.516 Å) can be observed in the compound’s crystal-packing diagram.
The clean conversion of the halides into the aryl and alkyl products indicate that the combination of the BDI ligand with the imido group provides thermodynamic stability for the complexes containing a formal Nb(V) center, allowing for substitution reactions while resisting reduction to dn>0 complexes. We were further interested in testing the ability of this ligand framework to stabilize a low-coordinate 4d metal center, similar to the stabilization imparted to low-coordinate 3d metal systems.1–7 Such stabilization may then lead to increased reactivity compared with that exhibited by more coordinately-saturated systems.
Synthesis and characterization of pentacoordinate bis(tert-butylimido) complexes
(BDI)Nb(NtBu)2py
The BDI ligand is well positioned to stabilize low-coordinate fragments, given the size and orientation of the flanking aryl groups. Initial attempts at substitution of the chlorides of 1 with LiNHtBu in ethereal solvents resulted in mixtures of products, all displaying similar solubility properties. However, on stirring a slurry of LiNHtBu and 1 in pentane for 12 h at room temperature, the solution turned bright yellow and was found to contain a single complex that could be isolated by crystallization from Et2O. The resonances in the room temperature 1H NMR spectrum of the product were broadened by fluxional processes, but a spectrum taken in THF-d8 at 238 K revealed a C1-symmetric structure, with two tBu methyl resonances, one at 1.39 ppm and the other at 0.27 ppm. The appearance of two tBu peaks could reflect the presence of either the desired bis(tert-butylimido) complex (BDI)Nb(NtBu)2py or the product from a single amide substitution at 1, (BDI) Nb(NtBu)(NHtBu)Cl. Observation of primary amido N-H peaks in 1H NMR spectra can be problematic due to their characteristically broad signals in ill-defined chemical shift regions.88 However, a flame test for halide was negative, and analytical combustion data were consistent with the molecular formula of the bis(imido) complex, allowing us to identify the material as (BDI)Nb(NtBu)2py (9, Scheme 8). The fluxional process occurring in solution was found to have ΔG‡ = 13.2(2) kcal/mol, based on the separation of the two tBu resonances at 223 K and a Tc = 298 K. Addition of an excess of pyridine-d5 to a solution of 9 in THF-d8 at 223 K caused rapid ligand exchange, as evidenced by the appearance of one equivalent of free C5H5N.
Scheme 8.

In contrast to Group 6 chemistry, in which a number of bis(tBu-imido) complexes are known, complex 9 represents just the second isolated example of a neutral, monomeric bis(alkylimido) Group 5 complex89 and the third example of an isolated Nb bis(imido) complex with any imido group substitution.75,90 The reaction leading to this product likely proceeds through the aforementioned amido chloride complex, (BDI)Nb(NtBu)(NHtBu)Cl, which can be either directly dehydrohalogenated by the second equivalent of LiNHtBu, forming 9, or amidated to form the bis(amido) complex (BDI) Nb(NtBu)(NHtBu)2; an α-hydrogen abstraction reaction would then yield the bis(imido) complex and H2NtBu. Notably, attempts at forming the monoamido complex (BDI)Nb(NtBu)(NHtBu)Cl by reaction of 1.0 equiv of LiNHtBu with 1 in pentane yielded a 1:1 mixture of 1 and 9. Related reactions designed to generate imido complexes have been shown to proceed through an α-H abstraction mechanism;90 the main factor limiting the potential for this mechanism in the present study is the steric requirement of placing two tBu-amido groups on the (BDI)Nb(NtBu) fragment, a situation reminiscent of our unsuccessful attempts at preparing dialkyl complexes with substituted methide groups.
The proposed molecular structure of 9 was confirmed by X-ray crystallography (Figure 8). The compound crystallized in the orthorhombic space group Pbca, and the molecular geometry is distorted square pyramidal (τ = 0.23).
Figure 8.

The molecular structure of 9 as determined by a single crystal X-ray diffraction study. The hydrogen atoms were omitted for clarity; the thermal ellipsoids were set at the 50 % probability level. Selected bond lengths are given in Å. Selected bond angles (°): N(1)-Nb(1)-N(2) 113.10(11), N(1)-Nb(1)-N(3) 104.52(10), N(2)-Nb(1)-N(3) 96.08(10), N(1)-Nb(1)-N(5) 98.71(10), N(2)-Nb(1)-N(5) 83.60(9), N(3)-Nb(1)-N(5) 154.75(9), N(1)-Nb(1)-N(4) 104.67(9), N(2)-Nb(1)-N(4) 140.91(9), N(3)-Nb(1)-N(4) 83.55(8), N(5)-Nb(1)-N(4) 81.09(8), C(1)-N(1)-Nb(1) 165.5(2), C(5)-N(2)-Nb(1) 166.7(2).
The two tBu imido groups, while in different chemical environments, have remarkably similar metric parameters (Nb(1)-N(1)apical = 1.800(2) Å; Nb(1)-N(2)basal = 1.809(2) Å; Nb(1)-N(1) apical-C(1) = 165.5(2)°; Nb(1)-N(2) basal-C(5) = 166.6(2)°). The Nb=NtBu bond distances in 9 are shorter than those found for the only other neutral, structurally-characterized Nb bis(imido) complex Nb(NAr)2Cl(PMe3)2 (Ar = 2,6-iPr2C6H3) (Nb-N = 1.819(5), 1.827(5) Å),75 consistent with the effect of localizing the π-bonding between the nitrogen atoms and the niobium center, but longer than the Nb=NtBu distances reported for the monoimido compounds 1, 6, and 7, due to the anticipated π-loading effect. The Nb=NtBu bond distances in 9 are close to those found for Ta(NAr)2Clpy2 (1.809(6) and 1.812(6) Å)90 and Ta(NSitBu3)2Mepy2 (1.810(13) and 1.819(13) Å),31 but longer than the average Mo=NtBu bond distance of the 25 crystallographically-characterized compounds containing the Mo(NtBu)2 fragment found in the Cambridge Structural Database by 0.075 Å (Mo=NtBu bond distances range from 1.671 to 1.772 Å; davg(Mo=NtBu) = 1.732 Å), in accord with the larger atomic radius of d0 Nb versus d0 Mo. The angles about both BDI nitrogens in 9 sum to >359°, indicating that they are participating in significant π-bonding. In comparison with complex 1, however, the bond lengths within the NCCCN ligand framework are identical for the local mirror plane reflecting the two halves of the BDI ligand, indicating that the averaged resonance form C (Scheme 3) provides the best description of the electronic structure within the BDI backbone of 9. This symmetry implies that the BDI ligand is not participating in π-bonding with the metal center and that the asymmetry in Nb-NBDI bond lengths is the result of a σ-based trans-influence.
(BDI)Nb(NtBu)2(dmap)
In an attempt to perturb the π-bonding within the complex and ascertain any effect that further π-bonding may have on the structure, we synthesized the related dmap (4-(dimethylamino)pyridine) adduct (BDI)Nb(NtBu)2(dmap) (10, Scheme 9) by addition of dmap to a solution of 9 in toluene, followed by pyridine removal via distillation as the toluene azeotrope.
Scheme 9.

Complex 10 crystallizes from Et2O in the space group P21/n; it was found to have a square pyramidal geometry (τ = 0.28), with one apical and one basal imido group (Figure 9), similar to that observed for 9. The imido group bond lengths within 10 are equivalent within experimental error (Nb(1)-N(1)apical = 1.797(3) Å; Nb(1)-N(2)basal = 1.795(3) Å), and the difference between the two Nb-N-C bond angles for the imido groups is insignificant considering the flat potential energy surface for moderately-bent alkyl imido groups91 (Nb(1)-N(1) apical-C(1) = 165.3(3)°; Nb(1)-N(2) basal-C(5) = 166.7(3)°). Of the bonds to the metal and within the NCCCN framework, the only differences between 9 and 10 that are statistically significant are the Nb(1)-N(2)basal, Nb(1)-N(3), and C(22)-C(23) distances, with the Nb(1)-N(3) of 10 being longer by 0.012 Å and the Nb(1)-N(2)basal and C(22)-C(23) of 10 being shorter by 0.014 Å and 0.016 Å, respectively.
Figure 9.

The molecular structure of 10 as determined by a single crystal X-ray diffraction study. The hydrogen atoms were omitted for clarity; the thermal ellipsoids were set at the 50 % probability level. Selected bond lengths are given in Å. Selected bond angles (°): N(1)-Nb(1)-N(2) 112.80(13), N(1)-Nb(1)-N(3) 103.75(12), N(2)-Nb(1)-N(3) 95.85(12), N(1)-Nb(1)-N(5) 96.98(12), N(2)-Nb(1)-N(5) 83.94(11), N(3)-Nb(1)-N(5) 157.48(10), N(1)-Nb(1)-N(4) 105.38(11), N(2)-Nb(1)-N(4) 140.94(12), N(3)-Nb(1)-N(4) 82.52(10), N(5)-Nb(1)-N(4) 83.60(10), C(1)-N(1)-Nb(1) 165.3(3), C(5)-N(2)-Nb(1) 166.7(3).
As seen for 9, the product was fluxional on the NMR timescale at room temperature, but a spectrum of 10 taken at 223 K revealed a C1-symmetric complex analogous to that observed for 9. Again, two imido group signals can be clearly distinguished (δ = 0.32, 1.36 ppm), with one shifted upfield due to its proximity to the flanking aryl group. The methyl signals for the p-NMe2 group were still unresolved at this temperature, as indicated by a broad singlet at 2.88 ppm integrating to 6H. Cooling the sample further revealed isolated singlets for the p-NMe2 group at 193 K, indicating an activation energy for methyl group interchange of less than 10.8 kcal/mol.
The 13C{1H} NMR spectrum of 10 at 223 K reveals the expected set of four signals for the Cα and Cβ carbons of the imido groups; with these the Δδαβ values (Δδαβ = δ(Cα) − δ(Cβ)) associated with each imido nitrogen can be determined. The Δδαβ value has been shown to qualitatively represent the degree of bonding between tert-butylimido nitrogens and the metal centers to which they are bound.92a The difference is representative of the degree of shielding of the imido nitrogen, where larger Δδαβ values are indicative of electron-poor or electronegative metal centers, and smaller Δδαβ values are representative of localization of significant electron density on the imido nitrogen. The Δδαβ values for complexes 9 and 10 are generally lower in magnitude than the related monoimido complexes (Table 1). These low values for 9 and 10 indicate that π-loading of the metal center has decreased the degree of imido bonding to the metal, creating more electron-rich imido nitrogens.92b
Table 1.
A comparison of the Δδαβ values for compounds 1–4, 6–11.
| Compound | Δδαβ | |
|---|---|---|
| (BDI)Nb(NtBu)Cl2py | 1 | 39.9 |
| (BDI)Nb(NtBu)F2py | 2 | 36.5 |
| (BDI)Nb(NtBu)Cl2 | 3 | 44.0 |
| (BDI)Nb(NtBu)F2 | 4 | 40.1 |
| (BDI)Nb(NtBu)Me2 | 6 | 35.2a, 35.0b |
| (BDI)Nb(NtBu)Cl(p-tol) | 7 | 39.0 |
| (BDI)Nb(NtBu)Me(p-tol) | 8 | 35.8 |
| (BDI)Nb(NtBu)2py | 9 | 30.2c, 31.7d |
| (BDI)Nb(NtBu)2(dmap) | 10 | 29.7c, 30.9d |
| (BDI)Nb(NtBu)2 | 11 | 31.3 |
Δδαβ in THF-d8 at 213 K.
Δδαβ in toluene-d8 at 213 K.
For the apical imido group, as determined by 1JCH and 2JCH NMR 1H–13C correlation experiments (see experimental section).
For the basal imido, determined as for c.
Analysis of the solution-state behavior of the bis(imido) complexes 9 and 10
Rate law data on the reaction to form 10 and on the mechanism of pyridine group exchange on 9 could not be obtained by NMR measurements due to the rapid rate of the reactions over the temperature ranges investigated (−80/+20 °C), but circumstantial evidence points to a dissociative mechanism (Scheme 10) for these reactions, whereby pyridyl ligand dissociation forms a four coordinate intermediate that can then associate a second pyridyl ligand. This hypothesis is supported by the following observations: 1.) In a qualitative sense, complexes 9 and 10 are electronically-saturated such that ligand dissociation would generate a 16 e− complex. To maintain the 18-electron configuration of the starting material, addition of a second pyridyl ligand would force either dissociation of one of the BDI nitrogens or localization of a pair of electrons on one or both of the imido groups. 2.) Addition of an excess of either dmap or pyridine to solutions of 9 and 10 does not cause a change in the NMR spectral resonances attributable to the metal complexes when compared with those of the isolated complexes, even though the added ligand rapidly exchanges with the coordinated ligand. This implies either that the fluxional behavior is intramolecular or that pyridyl group exchange is occurring via a dissociative mechanism. 3.) NMR spectra collected at elevated temperatures cause the peaks to coalesce into a pattern indicative of a C2v-symmetric metal complex with free pyridine, strongly suggesting that the fluxional behavior in solution is due to pyridine dissociation, not intramolecular rearrangement. 4.) Addition of a suitable Lewis acid results in pyridine abstraction from 9 (see below).
Scheme 10.

It is interesting to compare complexes 9 and 10 with the related Group 6 NN, -(dipyrrolyl-α-methyl)-N-methylamine (dpma) bis(imido) complexes studied by Odom and co-workers.91 The authors made the observation that the combination of a tridentate, dianionic ligand with two monodentate substituents on a transition metal often leads to complexes with static molecular geometries in solution on the NMR time scale. The M(NtBu)2(dpma) (M = Cr, Mo, W) compounds they synthesized displayed this behavior, retaining non-equivalent imido groups from −80 to 80 °C. This contrasts with the majority of transition metal bis(imido) complexes that exhibit equivalent imido groups in solution regardless of dissimilarities in the solid state.75,93–95 The Odom compounds all have SP geometries (τ = 0.19 (W), 0.22 (Mo) and 0.28 (Cr)), with the apical position occupied by an imido group, very nearly approximating the geometry of (BDI)Nb(NtBu)2py, which has one apical imido group, one basal imido group, and a set of basal nitrogen ligands that form a combination of anionic and neutral donors. By analogy with the M(NtBu)2(dpma) compounds and by considering that the low temperature spectra of 9 and 10 clearly resolve the two imido groups, it seems likely that the fluxional processes observed in the NMR spectra are a result of pyridyl-ligand dissociation.
Synthesis, characterization and reactions of a four-coordinate bis(imido) complex
The lability of the pyridine ligand was important because it indicated that a four-coordinate bis(imido) complex may be accessible via utilization of a Lewis acid capable of preferentially binding pyridine in solutions of 9 or 10. Indeed, addition of 1.0 equiv of B(C6F5)3 to a solution of 9 in benzene quickly and quantitatively formed py·B(C6F5)3 and (BDI)Nb(NtBu)2 (11, Scheme 11) as judged by NMR spectroscopy. The NMR spectra of the transition metal product indicate that it has averaged C2v symmetry in solution, with the 1H spectrum revealing a single, sharp iPr methine septuplet at 3.42 ppm, two iPr methyl doublets integrating to 12 H each, and a single tBu resonance at 1.33 ppm integrating to 18 H. The 13C{1H} NMR spectrum supports the proposed C2v symmetric formulation and reveals Δδαβ = 31.3 ppm, a value that lies between the Δδαβ values for the two nonequivalent imido groups in 9 and at slightly lower magnitude than those for 10. The self-consistency in these values between the structurally-characterized bis(imido) complexes 9 and 10 and solution-state characterized bis(imido) 11 supports the formulation of 11 as a four-coordinate monomer instead of an imido-bridged dimer, the latter of which would have lower Δδαβ values for amido-like bridging imido groups.88
Scheme 11.

While 11 was stable enough to be spectroscopically characterized in solution, the complex undergoes clean thermal decomposition over several hours at room temperature to form (BDI#)Nb(NtBu)(NHtBu) (12, BDI# = H2C=C(NAr)CH=C(NAr)Me) via a C-H activation process (Scheme 11). Complex 12 exhibits 1H NMR signals characteristic of the new ligand motif, with two pseudo-singlets at 3.29 and 3.74 ppm shifted upfield due to their proximity to an aryl ring and attributable to the newly formed terminal methylene group. The overall C1-symmetry is evidenced by the spectroscopic observation of four nonequivalent iPr groups, each having two diastereotopic methyl groups. A broad singlet at 5.98 ppm integrates to 1H and lies in the range expected for primary alkyl amido ligands on Nb.88 Similar transformations have been observed before on related BDI systems; 34,42,96 however, the nuclearity of this reaction has not been determined for the present system due to inconsistent reaction rates. The decomposition reaction observed for 11 limits investigation into its reactivity with organic substrates; still, one reaction that proceeded cleanly before the C-H activation event could occur was observed with tBuNCO. On addition of 2.0 equiv of this substrate to a solution of 11 in C6D6, a single new product grew in cleanly over a period of 3 h and 1.0 equiv of tBuNCO was left unreacted. The product was identified by NMR spectroscopy as the oxaazaniobacyclobutane (BDI)Nb(NtBu)(κ2-O,N-OC(NtBu)NtBu) (13, Scheme 12), formed by a [2+2] reaction of the isocyanate with one of the imido groups on the metal.
Scheme 12.

The 1H NMR spectrum displays averaged Cs symmetry with three distinct tBu resonances (1.28, 1.34, and 1.47 ppm), and the 13C{1H} NMR spectrum indicates three nonequivalent tBu-Cα resonances at 52.0, 57.3, and 69.8 ppm. The upfield shifted Cα resonances are in regions similar to those found for organic imines and metal amides. After 14 h at room temperature, 13 decomposes into tBuNCNtBu (1.18 ppm, C6D6; Scheme 12) and unidentified metal species.
Computational studies on the electronic structure of 11
In contrast to earlier investigations which have found little variation to the bonding within the NCCCN metallacycle of the BDI ligand, the complexes discussed above reveal that the π-system of the BDI ligand can de-symmetrize as needed to accommodate the electronic requirements of the metal center. For C1- and Cs- (with the mirror plane containing the NCCCN ring) symmetric complexes, this variability of the bond lengths within the ligand metallacycle is indicative of the degree of mixing occurring between the high-lying BDI orbitals and the frontier orbitals on the metal center. The asymmetry of the metallacycle bond-lengths within complex 1 illustrates how the difference in π-bonding ability of the imido ligand compared to the chloride and pyridine ligands leads to non-equivalent C–N and C–C metallacycle bond lengths. The asymmetry further indicates that the BDI π-system is close enough in energy to the Nb=NR π-bonds to affect and be affected by the metal-imido bonding.
Thus, to evaluate the possibility that the BDI ligand is causing π-loading in the bis(imido) complex 11, we performed DFT calculations (BP86, see experimental section for details) on a truncated form of the metal complex. The geometry optimized structure of the model complex BDI′Nb(NMe)2 (11a, Fig. 10, BDI′ = HC[C(Me)N(2,6-Me2-C6H3)]2) reveals overall C2v symmetry in accord with the solution-state symmetry observed by NMR for complex 11. The geometry of 11a is best approximated as a tetrahedron, reminiscent of the bent-metallocene complexes Cp2MX2, with an Nimido–Nb–Nimido bonding angle of 110.8°, and an NBDI–Nb–NBDI bonding angle of 93.8°. The Nb=NMe bond lengths are both calculated to be 1.829 Å, indicating weakening of the Nb–Nimido interaction compared to those in the monoimido complexes described above and in accord with the trend observed for the bis(imido) complexes 9 and 10. The imido groups are bent about the nitrogens to ca. 155°, a common feature of alkyl imido groups on early transition metals, and bonding within the NCCCN ring of the BDI ligand is symmetric with respect to the molecular mirror plane containing the two imido groups and the metal center.
Figure 10.

Geometry optimized (BP86) coordinates of 11a. The hydrogen atoms were omitted for clarity. Selected bond lengths are given in Å. Selected bond angles: N(2)-Nb(1)-N(7) 110.8°, N(12)-Nb(1)-N(13) 93.8°, Nb(1)-N(2)-C(3) 157.3°, Nb(1)-Nb(7)-C(8) 153.9°, N(imido)-Nb(1)-N(BDI′) varies between 112.7° and 112.9°.
We next examined the frontier orbitals of 11a as compared to those for the molecular fragments BDI′− and Nb(NMe)2+. The fragment analysis was performed using the coordinates generated by the geometry optimization on 11a, resulting in the expected sets of orbitals based on previously published computational studies. Most important among these for the present study are the b1-symmetric frontier orbitals of both the BDI′− and Nb(NMe)2+ fragments. We were particularly interested to determine whether the highest occupied molecular orbital (HOMO) of the BDI′ anion would lie at an appropriate energy to effectively mix with the b1-symmetric HOMO of the Nb(NMe)2+ fragment, and if it does, whether the resulting anti-bonding combination would have more BDI′ or Nb(NMe)2 character.
The anticipated π-loading effect can be clearly discerned from the respective contribution of the BDI′ and imido ligands to the HOMO-2 (116) and HOMO (118) orbitals (Scheme 13). These two orbitals lie 15.3 kcal/mol apart in energy and account for the respective bonding and anti-bonding combinations of the b1-symmetric HOMOs of the BDI′− and Nb(NMe2)+ fragments. Orbital 116 is comprised of 9.5% metal character of which 5.5% comes from the 4d orbitals (5.4% dxz) and 4.1% from the 5p set (3.9% px). The imido nitrogens account for 19.6% of this molecular orbital, and the p-orbitals of the NCCCN atoms of the BDI′ ligand metallacycle contribute 52.2% to the total. By comparison, orbital 118, the molecular HOMO, is composed of 5.4% metal character, 46.3% imido nitrogen p-character, and 32.1% NCCCN metallacycle p-character, indicating that mixing of the BDI π-system with the imido π-bonding orbitals leads to an imido-based HOMO at higher energy than would be found in the absence of the π-bonding component from the BDI′ ligand.
Scheme 13.

Interestingly, the calculations also revealed that the HOMO-1 orbital results from a similar π-loading effect. The 5b2 σ-donor orbital of the BDI′ ligand forms an anti-bonding combination with the Nb(NMe)2+ HOMO-1 fragment orbital, itself derived from the bonding combination of the Nb-dyz orbital with the imido-nitrogen py orbitals. The corresponding bonding component to the molecular HOMO-1 orbital is found at HOMO-8. The relative contributions of the BDI′ and imido ligands to these two orbitals follow the trend described above for the HOMO and HOMO-2 orbitals, in that the bonding combination between the BDI′ ligand and the metal center is largely BDI′-based and the anti-bonding combination has a greater imido component, indicating that the Nb=NR π-bond has been effectively raised in energy compared to a theoretical imido-based molecular orbital that lacks significant non-imido ligand character. A more complete breakdown of the frontier orbitals of 11a, including the relative contribution of each fragment to the molecular orbitals, can be found in the supporting information.
Summary and Conclusions
The introduction of the BDI ligand onto niobium was accomplished via salt metathesis with an imidoniobium precursor. We believe that the imido group is particularly useful for forming this product as it both reduces the required coordination number about the metal and supports the d0 oxidation state through its strong donor capability. A number of halide, alkyl, and aryl complexes were characterized, that serve both as starting points for future investigations and as bases of comparison for the electronic properties of the metal center and the nature of BDI-metal bonding interactions.
A rare example of a Group 5 metal bis(imido) complex was prepared by introducing a second imido group via salt metathesis. The pentacoordinate complex 9 was found to lose the coordinated pyridine ligand, likely through a dissociative mechanism, allowing for pyridyl ligand abstraction by Lewis acids to generate the four-coordinate bis(imido) complex (BDI)Nb(NtBu)2 in solution. If left unsaturated, this species underwent a rearrangement to form a cyclo-bis(anilide) niobium complex, in which a hydrogen had been transferred to one of the imido nitrogens. This reactivity supports the conjecture that forming heavily π-loaded systems creates metal centers with more highly polarized M-Lπ bonds. The imido group in this case activates a C–H bond, albeit presumably via an intramolecular reaction. Cycloaddition of tBuNCO was found to be competitive with the rearrangement process; the metallacycle could be observed spectroscopically and was found to yield tBuNCNtBu on thermal decomposition.
Finally, a computational investigation into the electronic structure of the four-coordinate bis(imido) complex 11a revealed two distinct interactions between the BDI′ ligand and the Nb=NMe π-bonds near the molecular frontier orbitals. The HOMO and HOMO-1 orbitals were found to be largely imido-based and to result from anti-bonding combinations with the BDI′ ligand, thus illustrating the intended π-loading effect, which results from the proper balance of orbital occupation, symmetry, and relative energy between the imido- and supporting ligand-based orbitals.
Experimental Section
General Considerations
Unless otherwise noted, all reactions were performed using standard Schlenk line techniques or in an MBraun inert atmosphere box under an atmosphere of nitrogen (<1 ppm O2/H2O). Glassware, cannulae, and Celite were stored in an oven at ca. 425 K. Pentane, hexane, Et2O, THF, toluene, benzene, and DME were purified by passage through a column of activated alumina and degassed prior to use.97 Pyridine and tBuNH2 were distilled from CaH2. Deuterated solvents were vacuum-transferred from sodium/benzophenone and degassed with three freeze-pump-thaw cycles. NMR spectra were recorded on Bruker AV-300, AVQ-400 and DRX-500 spectrometers. 1H and 13C{1H} chemical shifts are given relative to residual solvent peaks. Proton and carbon NMR assignments were routinely confirmed by 1H-1H (COSY) or 1H-13C (HSQC and HMBC) experiments. Vapor diffusion osmometry was performed according to published procedures, using sublimed ferrocene as a standard.98 Infrared (IR) samples were prepared as Nujol mulls and were taken between KBr disks. NbCl5 was purified by sublimation prior to use. Li(BDI)·OEt2,61 Nb(NtBu)Cl3py2,23 Me3SnF,99 B(C6F5)3,100 [Nb(NtBu)(H2NtBu)Cl3]2,101 and (p-tol)2Mg,102 were prepared using the literature procedures. LiNHtBu was prepared by adding 1.0 equiv nBuLi to a solution of H2NtBu in pentane; the precipitate was collected by filtration, washed thoroughly with pentane and dried under vacuum. All other reagents were acquired from commercial sources and used as received. Elemental analyses were determined at the College of Chemistry, University of California, Berkeley. The X-ray structural determinations were performed at either CHEXRAY, University of California, Berkeley (1, 5, 7, 9 and 10) using either Bruker SMART 1000 or SMART APEX diffractometers or at the Advanced Light Source, Beamline 11.3.1, Lawrence Berkeley National Lab, Berkeley, CA (6). Full crystallographic details for 6 can be found elsewhere.85
Representative procedure for X-ray crystallography for compounds 1, 5, 7, 9, & 10
A crystal of appropriate size was coated in Paratone-N oil and mounted on a Kaptan© loop. The loop was transferred to a diffractometer equipped with a CCD area detector,103 centered in the beam, and cooled by a nitrogen flow low-temperature apparatus that had been previously calibrated by a thermocouple placed at the same position as the crystal. Preliminary orientation matrices and cell constants were determined by collection of 60 30 s frames, followed by spot integration and least-squares refinement. An arbitrary hemisphere of data was collected, and the raw data were integrated using SAINT.104 Cell dimensions reported were calculated from all reflections with I > 10 σ. The data were corrected for Lorentz and polarization effects; no correction for crystal decay was applied. Data were analyzed for agreement and possible absorption using XPREP.105 An empirical absorption correction based on comparison of redundant and equivalent reflections was applied using SADABS.106 Structures were solved by direct methods with the aid of successive difference Fourier maps and were refined on F2 using the SHELXTL 5.0 software package. Thermal parameters for all non-hydrogen atoms were refined anisotropically. ORTEP diagrams were created using the ORTEP-3 software package.107
(BDI)Nb(NtBu)Cl2py (1)
THF (50 mL) was added to a solid mixture of Nb(NtBu)Cl3py2 (5.0 g, 11.7 mmol) and Li(BDI)·OEt2 (5.81 g, 11.7 mmol) in a 250 mL Schlenk flask at room temperature. The mixture immediately took on an orange-red color. The solution was stirred at room temperature for 12 h, resulting in a deep red solution with a fine precipitate. The volatile materials were removed under vacuum, and the residue was extracted with 200 mL of hot hexane. The hexane extracts were filtered through a small pad of Celite. The filtrate was reheated to reflux, then filtered again and left to cool to room temperature. The red crystalline product that formed was collected and dried under vacuum. Yield: 5.8 g, 68 %. Higher yields of material of comparable purity may be obtained by performing a Soxhlet extraction with pentane or by extracting the crude product with benzene, filtering, evacuating to dryness and washing thoroughly with room temperature hexane (50 mL). Yield: 6.8–7.7 g, 80–90 %. 1H NMR (300 MHz, C6D6, 298 K): δ 8.57 (d, 2H, py), 7.15 (m, 6H, Ar), 6.61 (t,1H, py), 6.28 (t, 2H, py), 5.34 (s, 1H, HC(C(Me)NAr)2), 3.77 (br s, 4H, CHMe2), 1.72 (br s, 12 H, HC(C(Me)NAr)2, CHMe2), 1.17 (br s, 18H, CHMe2), 0.68 ppm (s, 9H, tBu); 1H NMR (500 MHz, C7D8, 243 K): δ 8.45 (d, 2H, py), 7.19 – 7.10 (m, 6H, Ar), 6.56 (t, 1H, py), 6.19 (t, 2H, py), 5.27 (s, 1H, HC(C(Me)NAr)2), 3.75 (sept, 2H, CHMe2), 3.63 (sept, 2H, CHMe2), 1.79 (d, 6H, CHMe2), 1.75 (s, 3H, HC(C(Me)NAr)2), 1.62 (s, 3H, HC(C(Me)NAr)2), 1.26 (d, 6H, CHMe2), 1.05 (d, 6H, CHMe2), 0.96 (d, 6H, CHMe2), 0.64 ppm (s, 9H, tBu); 13C{1H} NMR (125 MHz, C7D8, 243 K): δ 170.5 (HC(C(Me)NAr)2), 158.6 (HC(C(Me)NAr)2), 153.7 (Ar), 153.5 (Ar), 145.7 (Ar), 143.4 (Ar), 142.5 (Ar), 128.4 (Ar), 126.7 (Ar), 125.8 (Ar), 124.6 (Ar), 123.6 (Ar), 122.8 (Ar), 105.7 (HC(C(Me)NAr)2), 68.8 (Cα, tBu), 28.9 (Cβ, tBu), 28.5 (CHMe2), 28.2 (CHMe2), 28.0 (HC(C(Me)NAr)2), 25.9 (CHMe2), 25.8 (HC(C(Me)NAr)2), 25.7 (CHMe2), 25.1 (CHMe2), 23.7 ppm (CHMe2); Anal. Calcd for C38H55Cl2N4Nb: C 62.38, H 7.58, N 7.66; found: C 62.57, H 7.67, N 7.68.
(BDI)Nb(NtBu)F2py (2)
Toluene (15 mL) was added to a solid mixture of 1 (300 mg, 0.41 mmol) and Me3SnF (150 mg, 0.82 mmol) at room temperature. The solution was stirred vigorously for 24 h, during which time the color of the solution gradually changed from dark red to yellow-orange as the insoluble Me3SnF reacted. The solution was then filtered and concentrated to ca. 2 mL at which point a microcrystalline material began to precipitate. The flask was stored at −40 °C for 48 h, causing the precipitation of a yellow-orange crystalline material. The solid was collected by filtration and dried under vacuum. Yield: 113 mg, 40 %. 1H NMR (500 MHz, C7D8, 298K): δ 8.40 (d, 2H, py), 7.13 (s, 3H, Ar), 6.92 (m, 1H, Ar), 6.85 (d, 2H, Ar), 6.73 (m, 1H, py), 6.40 (m, 2H, py), 5.11 (s, 1H, HC(C(Me)NAr)2), 3.80 (sept, 2H, CHMe2), 3.43 (sept, 2H, CHMe2), 1.74 (d, 6H, CHMe2), 1.67 (s, 3H, HC(C(Me)NAr)2), 1.66 (s, 3H, HC(C(Me)NAr)2), 1.29 (d, 6H, CHMe2), 1.08 (d, 6H, CHMe2), 1.03 (d, 6H, CHMe2), 0.41 (s, 9H, tBu). 13C{1H} NMR (125 MHz, C7D8, 298 K): δ 168.4 (HC(C(Me)NAr)2), 159.0 (HC(C(Me)NAr)2), 154.4 (Ar), 151.0, 146.6 (Ar), 143.3 (Ar), 143.1 (Ar), 126.4 (Ar), 124.8 (Ar), 123.8 (Ar), 123.6 (Ar), 101.6 (HC(C(Me)NAr)2), 66.1 (Cα, tBu), 29.6 (Cβ, tBu), 28.7 (CHMe2), 28.2 (CHMe2), 26.6 (HC(C(Me)NAr)2), 26.2 (HC(C(Me)NAr)2), 26.1 (CHMe2), 25.6 (CHMe2), 25.2 (CHMe2), 24.5 (CHMe2). 19F NMR (376.5 MHz, C7D8, 298 K): δ 68.77 (s, 2F, NbF2). Anal. Calcd for C38H55F2N4Nb: C 65.32, H 7.93, N 8.02; found: C 63.14, H 7.88, N 7.88.
Nb(NtBu)Cl3(dme)
A solution of [Nb(N tBu)(H2NtBu)Cl3]2 (1.33 g, 1.94 mmol) in 40 mL of DME was cooled to −72 °C, and a solution of HCl in Et2O (1.94 mL, 3.87 mmol) was added by syringe. The flask was allowed to warm to room temperature. A white precipitate formed while the solution was stirred for 12 h. The resulting yellow solution was filtered and the volatile material was removed under vacuum. The resulting oily residue was extracted with toluene, and the extracts were combined and evacuated to dryness. The residue was triturated with Et2O (3 × 10 mL) to give a yellow powder. Yield: 1.16 g, 83 %. The spectral data agree with those reported in the literature.108
(BDI)Nb(NtBu)Cl2 (3) from Nb(NtBu)Cl3(dme)
A 100 mL flask containing solid Nb(NtBu)Cl3(dme) (723 mg, 2.01 mmol) and Li(BDI)·Et2O (1.0 g, 2.01 mmol) was cooled to −72 °C and 20 mL toluene was added slowly. The solution was allowed to warm to room temperature by which point the color turned dark red. Stirring was continued overnight, then the volatile materials were removed under vacuum. The residue was extracted with hot hexane (2 × 25 mL), and the extracts were combined and concentrated. Crystallization of the residue from hot hexane (+65/−35°C) yielded the product as red blocks. The solid was collected by filtration and dried under vacuum. Yield: 740 mg, 57 %. 1H NMR (500 MHz, C6D6, 298K): δ 7.1 (br s, 6H, Ar), 5.25 (s, 1H, HC(C(Me)NAr)2), 3.79 (br s, 2H, CHMe2), 2.92 (br s, 2H, CHMe2), 1.6 (br s, 6H, HC(C(Me)NAr)2), 1.54 (s, 9H, tBu), 1.31 (br s, 16H, CHMe2), 0.98 ppm (br s, 6H, CHMe2). 1H NMR (500 MHz, C7D8, 223 K): δ 7.2–6.9 (m, 6H, Ar), 5.14 (s, 1H, HC(C(Me)NAr)2), 3.76 (sept, 2H, CHMe2), 2.79 (sept, 2H, CHMe2), 1.63 (d, 6H, CHMe2), 1.42 (s, 6H, HC(C(Me)NAr)2), 1.31 (s, 9H, tBu), 1.27 (d, 6H, CHMe2), 1.23 (d, 6H, CHMe2), 0.98 (d, 6H, CHMe2). 13C{1H} NMR (125 MHz, C7D8, 223 K): δ 168.5 (HC(C(Me)NAr)2), 151.9 (Ar), 145.8 (Ar), 141.2 (Ar), 127.3 (Ar), 125.5 (Ar), 123.7 (Ar), 105.1 (HC(C(Me)NAr)2), 75.3 (Cα, tBu), 31.3 (Cβ, tBu), 30.3, 29.2, 28.1, 26.1, 24.7, 24.5, 24.5. Anal. Calcd for C33H50Cl2N3Nb: C, 60.74; H, 7.72; N, 6.44. Found: C, 61.03, H 7.86; N, 6.22.
(BDI)Nb(NtBu)Cl2 (3) from 1
To a solution of 1 (200 mg, 0.27 mmol) in 10 mL C6H6 was added a solution of B(C6F5)3 (140 mg, 0.27 mmol) in 5 mL C6H6. The color quickly changed from dark red to light red. The solution was stirred and after 30 min, the volatile materials were removed under vacuum, leaving a bright red oily solid. Pentane (5 mL) was added, and the suspension was agitated to dissolve any remaining benzene. The volatile materials were again removed under vacuum, the residue was extracted with pentane (2 × 10 mL), and the combined extracts were filtered. Storing the filtrate at −35 °C produced a mixture of bright red and white crystalline material. This material was redissolved in a minimal amount of pentane, filtered and stored at −35 °C. This process was repeated several times until the crystalline material was homogeneous in appearance and analyzed as expected (see above). Yield: 29 mg, 16 %.
(BDI)Nb(NtBu)F2 (4) from 1
To a solution of 2 (30 mg, 0.43 mmol) in 0.3 mL C6D6 was added a solution of B(C6F5)3 (22 mg, 0.043 mmol) in 0.3 mL C6D6. The solution color changed slightly from dark orange to a lighter orange. The product was analyzed by NMR spectroscopy without purification. The 19F NMR data match those for py·B(C6F5)3. 1H NMR (500 MHz, C7D8, 303 K): δ 7.10 (m, 6H, Ar), 5.35 (s, 1H, HC(C(Me)NAr)2), 3.35 (br s, 4H, CHMe2), 1.61 (s, 6H, HC(C(Me)NAr)2), 1.33 (d, 12H, CHMe2), 1.17 (s, 9H, tBu), 1.13 (d, 12H, CHMe2). 13C{1H} NMR (125 MHz, C7D7): δ 170.1 (HC(C(Me)NAr)2), 146.7 (Ar), 142.7 (Ar), 127.8 (Ar), 125.0 (Ar), 105.6 (HC(C(Me)NAr), 72.2 (NtBu, Cα), 32.1 (tBu, Cβ), 28.9 (CHMe2), 25.8 (CHMe2), 25.3 (HC(C(Me)NAr)2), 25.1 (CHMe2). 19F NMR (376.5 MHz, C6D6, 298 K): δ 83.96 (br s, Δν1/2 = 460 Hz).
(BDI)Nb(NAr)Cl2 (5)
A solution of Li(BDI)·OEt2 (644 mg, 1.29 mmol) in THF (10 mL) was added to a solution of Nb(NAr)Cl3(dme) (600 mg, 1.29 mmol) in THF (10 mL) at room temperature. The flask was sealed and the solution was stirred at room temperature for 12 h, after which time the color had turned deep purple. The volatile materials were removed under vacuum, and the product was extracted with Et2O (2 × 15 mL). The filtered solution was concentrated until a microcrystalline precipitate began to form, at which point the solution was filtered again and stored at −40 °C. A dark purple crystalline material was collected and dried under vacuum. Yield: 554 mg, 57 %. X-ray quality crystals were grown by storing a concentrated Et2O solution at 0 °C for several days. 1H NMR (500 MHz, C6D6, 298K): δ 7.13 (br m, 2H, BDI Ar), 7.06 (br m, 4H, BDIAr), 7.01 (br d, 2H, imido Ar), 6.91 (br t, 1H, imido Ar), 5.42 (s, 1H, HC(C(Me)NAr)2), 4.55 (sept, 1H, imido CHMe2), 4.01 (sept, 1H, imido CHMe2) 3.34 (sept, 2H, BDI CHMe2), 3.09 (sept, 2H, BDI CHMe2), 1.65 (s, 6H, HC(C(Me)NAr)2), 1.45 (d, 6H, BDI CHMe2), 1.31 (d, 6H, imido CHMe2), 1.21 (d, 12H, overlapping imido and BDI CHMe2), 1.11 (d, 6H, BDI CHMe2), 1.07 ppm (d, 6H, imido CHMe2). 13C{1H} NMR (125 MHz, C6D6, 298K): δ 168.28, 153.45, 147.93, 142.05, 141.63, 141.60, 128.68, 127.81, 127.73, 125.49, 124.49, 124.08, 122.64, 104.51, 30.45, 29.13, 28.81, 28.44, 26.94, 25.62, 25.26, 25.21, 25.17, 24.65. Anal. Calcd for C41H58Cl2N3Nb: C, 65.07; H, 7.73; N, 5.55. Found: C, 65.15; H, 7.66; N, 5.38.
(BDI)Nb(NtBu)Me2 (6)
An alternative procedure for synthesizing 6 has been reported previously.85 The following presents a preferred synthetic method, along with more extensive NMR data than had previously been reported. To a slurry of 1 (2.13 g, 2.91 mmol) in cold Et2O (50 mL, −72 °C) was added MeMgBr (1.94 mL, 5.82 mmol) by syringe. The solution immediately took on an orange color. Upon allowing the solution to warm to room temperature, the solution rapidly became bright yellow. After 30 min. at room temperature, the volatile materials were removed in vacuo, and the residue was thoroughly extracted with pentane. The filtrate was concentrated and stored at −40 °C until crystalline material formed. The product was collected from two crops as bright yellow blocks. Yield: 1.41 g, 79 %. The product is thermally unstable at room temperature both in solution and in the solid state, decomposing into a brown, unidentified product over several hours in solution and several days in the solid state, but it may be stored at −40 °C for months without significant decomposition. 1H NMR (300 MHz, THF-d8, 293 K): δ 7.16 (s, 6H, Ar), 5.57 (s, 1H, HC(C(Me)NAr)2), 2.99 (br s, 4H, CHMe2), 1.86 (br s, 6H, HC(C(Me)NAr)2), 1.25 (br s, 18H, CHMe2), 1.16 (d, 6H, CHMe2), 0.91 (s, 9H, tBu), 0.44 (s, 6H, NbMe2). 1H NMR (500 MHz, C6D6, 293 K): δ 7.14 (br s, 6H, Ar), 5.26 (s, 1H, HC(C(Me)NAr)2), 3.19 (br s, 4H, CHMe2), 1.67 (br s, 6H), 1.36 (br s, 12H), 1.16 (br s, 12H), 1.10 (s, 9H, tBu), 0.78 (s, 6H, NbMe2). 1H NMR (500 MHz, THF-d8, 233 K): δ 7.21 (m, 6H, Ar), 5.63 (s, 1H, HC(C(Me)NAr)2), 3.15 (sept, 2H, CHMe2), 2.73 (sept, 2H, CHMe2), 2.00 (s, 3H, HC(C(Me)NAr)2), 1.73 (s, 3H, HC(C(Me)NAr)2), 1.41 (d, 6H, CHMe2), 1.19 (d, 6H, CHMe2), 1.13 (d, 6H, CHMe2), 1.08 (d, 6H, CHMe2), 0.88 (s, 9H, tBu), 0.41 (s, 6H, NbMe2). 13C{1H} NMR (125 MHz, THF-d8, 213K): δ 170.0 (HC(C(Me)NAr)2), 164.8 (HC(C(Me)NAr)2), 153.6, 149.5, 126.8, 126.5, 125.1, 124.9, 101.9 (HC(C(Me)NAr)2), 65.8 (Cα, tBu), 47.5 (NbMe2), 30.6 (Cβ, tBu), 29.7, 29.0, 27.5, 26.2, 25.4, 25.2, 24.9, 24.6. 1H NMR (500 MHz, C7D8, 213 K): δ 7.18 – 6.99 (m, 6H, Ar), 5.15 (s, 1H, HC(C(Me)NAr)2), 3.29 (sept, 2H, CHMe2), 2.82 (sept, 2H, CHMe2), 1.77 (s, 3H, HC(C(Me)NAr)2), 1.51 (d, 6H, CHMe2), 1.47 (s, 3H, HC(C(Me)NAr)2), 1.21 (t, 12H, CHMe2), 1.09 (m, 15H, tBu and CHMe2), 0.88 (s, 6H, NbMe2). 13C{1H} NMR (125 MHz, C7D8, 213K): δ 168.6 (HC(C(Me)NAr)2), 163.5 (HC(C(Me)NAr)2), 152.6, 148.6, 141.4, 139.8, 125.9, 124.3, 124.0, 100.9 (HC(C(Me)NAr)2), 65.2 (Cα, tBu), 47.6 (NbMe2), 30.2 (Cβ, tBu), 28.8, 28.1, 26.9, 25.4, 24.9, 24.8, 24.1, 24.0 (one aryl resonance could not be found). Anal. Calcd for C35H56N3Nb: C, 68.72; H, 9.23; N, 6.87. Found: C, 68.47, H 9.52; N, 6.98.
(BDI)Nb(NtBu)Cl(p-tol) (7)
To a stirred suspension of 1 (600 mg, 0.82 mmol) in 20 mL Et2O, cooled to −72 °C, was added a slurry of (p-tol)2Mg (169 mg, 0.82 mmol) in 10 mL Et2O. The color quickly turned yellow as the flask warmed to room temperature. After stirring the mixture for 30 min at room temperature, the volatile materials were removed in vacuo. The residue was extracted with pentane (2 × 20 mL), and the extracts were filtered and concentrated to ca. 10 mL. The flask was stored at −35 °C until a yellow-orange crystalline material formed. The material was collected and dried under vacuum. Yield: 345 mg, 59 %. 1H NMR (500 MHz, THF-d8, 298K): δ 7.81 (br s, 2H), 7.14 (s, 2H), 7.10 (d, 1H), 7.06 (m, 2H), 6.97 (m, 3H), 6.03 (s, 1H, HC(C(Me)NAr)2), 3.00 (sept, 1H, CHMe2), 2.69 (m, 3H, CHMe2), 2.28 (s, 3H, 4-Me-C6H4), 1.92 (s, 3H, HC(C(Me)NAr)2), 1.76 (s, 3H, HC(C(Me)NAr)2), 1.38 (d, 3H, CHMe2), 1.21 (d, 3H, CHMe2), 1.09 (dd, 6H, CHMe2), 0.98 (dd, 6H, CHMe2), 0.94 (s, 9H, tBu), 0.90 (d, 3H, CHMe2), 0.41 (d, 3H, CHMe2). 1H NMR (500 MHz, C6D6, 278 K): δ 8.05 (br s, 2H), 7.15 (m, 2H), 7.07 (m, 2H), 7.00 (m, 4H), 5.43 (s, 1H, HC(C(Me)NAr)2), 3.05 (sept, 1H, CHMe2), 2.89 (sept, 3H, CHMe2), 2.81 (m, 6H, CHMe2), 2.05 (s, 3H, 4-Me-C6H4), 1.63 (s, 3H, HC(C(Me)NAr)2), 1.45 (s, 3H, HC(C(Me)NAr)2), 1.38 (d, 3H, CHMe2), 1.37 (d, 3H, CHMe2), 1.09 (m, 18H, tBu and 3 × CHMe2), 1.02 (d, 3H, CHMe2), 1.01 (d, 3H, CHMe2), 0.77 (d, 3H, CHMe2). 13C{1H} NMR (125 MHz, C6D6, 298K): δ 171.0, 162.8, 154.9, 149.4, 141.7, 141.5, 141.2, 140.5, 140.42, 127.3, 126.9, 126.5, 125.8, 124.4, 123.9, 123.7, 105.4, 69.4, 30.4, 30.2, 29.8, 28.8, 28.4, 27.1, 26.1, 26.0, 25.1, 25.1, 24.6, 24.6, 24.5, 24.5, 24.4, 22.3. Anal. Calcd for C40H57ClN3Nb: C, 67.83; H, 8.11; N, 5.93. Found: C, 67.80, H 8.27; N, 5.94.
(BDI)Nb(NtBu)Me(p-tol) (8)
A solution of 7 (319 mg, 0.45 mmol) in 20 mL Et2O was cooled to −72 °C, and a solution of MeMgBr in Et2O (0.15 mL, 0.45 mmol) was added dropwise by syringe. The solution became bright yellow as the flask was allowed to warm to room temperature. After stirring at room temperature for 30 min, the volatile materials were removed under vacuum. The residue was extracted with pentane (3 × 15 mL) and filtered from a fine white precipitate until the solution was entirely clear. The filtrate was concentrated under vacuum until a yellow crystalline material began to precipitate. The solution was then warmed gently to re-dissolve the precipitated material and stored at −40°C, leading to the formation of yellow blocks which were collected by filtration and dried under vacuum. Yield: 133 mg, 43 %. 1H NMR (500 MHz, C6D6, 298K): δ 7.28 (br s), 7.17 (s), 7.09 (m), 7.03 (m), 5.56 (s, 1H, HC(C(Me)NAr)2), 2.91 (m, 4H, CHMe2), 2.16 (s, 3H, 4-MeC6H4), 1.82 (s, 3H, HC(C(Me)NAr)2), 1.59 (s, 3H, HC(C(Me)NAr)2), 1.35 (d, 3H, CHMe2), 1.28 (d, 3H, CHMe2), 1.16 (d, 3H, CHMe2), 1.14 (d, 3H, CHMe2), 1.13 (s, 3H, NbMe), 1.09 (s, 9H, tBu), 1.09 (d, 3H, CHMe2), 1.04 (d, 3H, CHMe2), 0.95 (d, 3H, CHMe2), 0.67 (d, 3H, CHMe2). 13C{1H} NMR (125 MHz, C6D6, 298K): δ 169.8 (HC(C(Me)NAr)2), 164.4 (HC(C(Me)NAr)2), 154.0, 149.6, 142.9, 142.2, 141.2, 140.7, 137.6, 128.7, 127.7, 126.5, 125.6, 124.9, 124.7, 123.7, 102.5 (HC(C(Me)NAr)2), 66.8 (Cα, tBu), 54.8 (NbMe), 31.0 (Cβ, tBu), 30.1, 29.0, 28.7, 28.1, 27.5, 26.1, 25.7, 25.5, 25.5, 24.9, 24.8, 24.6, 24.6, 23.6, 23.1, 22.1. Anal. Calcd for C41H60N3Nb: C, 71.59; H, 8.79; N, 6.11. Found: C, 71.78, H 8.59; N, 5.83.
(BDI)Nb(NtBu)2py (9)
To a solid mixture of 1 (700 mg, 0.96 mmol) and LiNHtBu (151 mg, 1.91 mmol) was added pentane (50 mL) at room temperature. The slurry was stirred vigorously for 12 h, after which time the solution had turned bright yellow and a white precipitate was present. The volatile materials were removed in vacuo, the product was extracted with Et2O (3 × 20 mL), and the combined extracts were filtered repeatedly until a clear solution was obtained. The yellow solution was then concentrated until crystalline material began to form. The flask was stored at −35 °C for 48 h. The yellow crystalline product was collected and dried in vacuo. Yield from two crops: 371 mg, 53 %. 1H NMR (500 MHz, C6D6, 298K): δ 8.33 (br s, 2H, py), 7.15-6.90 (br m, 6H, Ar), 6.66 (br s, 1H, py), 6.35 (br s, 2H, py), 5.18 (br s, 1H, HC(C(Me)NAr)2), 3.68 (br s, 4H, CHMe2), 1.65 (s, 6H, HC(C(Me)NAr)2), 1.35 (br s, 30 H, tBu and CHMe2), 1.25 (d, 12H, CHMe2). 1H NMR (500 MHz, THF-d8, 238K): δ 8.86 (br s, 1H), 7.54 (t, 2H), 7.31 (br s, 1H), 7.15-7.00 (m, 4H, Ar), 6.91 (t, 1H), 6.73 (br s, 1H), 6.65 (d, 1H), 5.19 (s, 1H, HC(C(Me)NAr)2), 4.04 (sept, 1H, CHMe2), 3.84 (sept, 1H, CHMe2), 3.50 (sept, 1H, CHMe2), 3.35 (sept, 1H, CHMe2), 1.68 (s, 3H, HC(C(Me)NAr)2), 1.65 (s, 3H, HC(C(Me)NAr)2), 1.50 (d, 3H, CHMe2), 1.44 (d, 3H, CHMe2), 1.42 (d, 3H, CHMe2), 1.39 (s, 9H, tBu), 1.23 (d, 3H, CHMe2), 1.19 (d, 6H, 2 × CHMe2), 1.08 (d, 3H, CHMe2), 0.67 (d, 3H, CHMe2), 0.28 (s, 9H, tBu). 13C NMR (125 MHz, THF-d8, 223K): δ 167.51, 166.38, 154.57, 154.46, 151.00, 150.29, 143.71, 142.09, 142.05, 141.64, 138.04, 126.06, 125.48, 124.85, 124.58, 124.47, 124.44, 124.42, 123.05, 64.50, 63.93, 34.35, 32.23, 29.49, 29.25, 28.09, 28.03, 27.97, 26.73, 25.98, 25.86, 25.01, 24.92, 24.55. Anal. Calcd for C42H64N5Nb: C, 68.92; H, 8.81; N, 9.57. Found: C, 68.82, H 8.81; N, 9.75.
(BDI)Nb(NtBu)2(dmap) (10)
Solid dmap (17 mg, 0.14 mmol) was added to a solution of 9 (100 mg, 0.14 mmol) in toluene (5 mL) at room temperature. The solution was stirred at room temperature until all the solids had dissolved (ca. 5 min), then the volatile materials were removed under vacuum. The solid was redissolved in toluene and evacuated to dryness two more times to remove the pyridine. The resulting yellow solid was dissolved in Et2O, filtered, and concentrated until a microcrystalline material began to form. The solution was then stored at −35 °C to yield the product as yellow blocks. Yield: 43 mg, 40 %. 1H NMR (500 MHz, THF-d8, 223 K): δ 8.35 (br d, 1H, Ar-dmap), 7.09 (t, 2H, Ar), 7.05-6.98 (m, 3H, Ar and Ar-dmap), 6.93 (t, 1H, Ar), 6.40 (d, 1H, Ar), 6.48 (br d, 1H, Ar-dmap), 5.89 (br d, 1H, Ar-dmap), 5.13 (s, 1H, HC(C(Me)NAr)2), 4.14 (sept, 1H, CHMe2), 3.87 (sept, 1H, CHMe2), 3.49 (sept, 1H, CHMe2), 3.32 (sept, 1H, CHMe2), 2.88 (br d, 6H, p-NMe2), 1.64 (s, 3H, HC(C(Me)NAr)2), 1.62 (s, 3H, HC(C(Me)NAr)2), 1.51 (d, 3H, CHMe2), 1.41 (d, 6H, 2 × CHMe2), 1.36 (s, 9H, tBu), 1.22 (d, 3H, CHMe2), 1.19 (d, 3H, CHMe2), 1.18 (d, 3H, CHMe2), 1.08 (d, 3H, CHMe2), 0.77 (d, 3H, CHMe2), 0.32 (s, 9H, tBu). 13C NMR (125 MHz, THF-d8, 223K): δ 167.1, 166.0, 155.1, 154.8, 154.2, 150.8, 144.1, 142.0, 141.8, 141.7, 138.5, 129.9, 129.2, 126.3, 125.6, 125.2, 124.8, 124.6, 124.3, 124.1, 104.4, 99.4, 64.1, 63.6, 39.1, 34.4, 32.7, 29.4, 29.2, 28.2, 28.0, 27.9, 26.8, 26.0, 25.0, 24.9. Anal. Calcd for C44H69N6Nb: C, 68.19; H, 8.97; N, 10.84. Found: C, 68.14, H 9.17; N, 10.66.
(BDI)Nb(NtBu)2 (11)
To a stirred solution of 9 (232 mg, 0.32 mmol) in C6H6 (25 mL) was added a solution of B(C6F5)3 (162 mg, 0.32 mmol) in C6H6 (5 mL) at room temperature. The resulting solution was stirred for 10 min, then the solvent was removed in vacuo, leaving a yellow residue. The residue was triturated with pentane (2 × 10 mL) until a solid was obtained; this was extracted with pentane and the combined extracts were filtered from a white solid to give a clear yellow filtrate, which was evaporated in vacuo, leaving a yellow solid. Yield: 178 mg, 85 %. NMR analysis of the isolated material revealed residual py·B(C6F5)3 along with varying proportions of 12. This contamination along with the thermal instability of the product prevented full characterization of 11. 1H NMR (400 MHz, C7D8, 298 K): δ 7.06 (m, 6H, Ar), 5.35 (s, 1H, HC(C(Me)NAr)2), 3.83 (sept, 4H, CHMe2), 1.79 (s, 6H, HC(C(Me)NAr)2), 1.38 (d, 12H, CHMe2), 1.32 (s, 18H, tBu), 1.17 (d, 12H, CHMe2). 13C{1H} NMR (100 MHz, C7D8): δ 170.29 (HC(C(Me)NAr)2), 144.94 (Ar), 141.77 (Ar), 126.78 (Ar), 124.16 (Ar), 100.13 (HC(C(Me)NAr), 64.65 (Nb=NtBu, Cα), 34.34 (tBu, Cβ), 28.04 (CHMe2), 25.66 (CHMe2), 24.74 (HC(C(Me)NAr)2), 24.28 (CHMe2).
(BDI#)Nb(NtBu)(NHtBu) (12)
To a stirred solution of 9 (232 mg, 0.32 mmol) in C6H6 (25 mL) was added a solution of B(C6F5)3 (162 mg, 0.32 mmol) in C6H6 (5 mL) at room temperature. The solution was stirred for 12 h at room temperature, then the solvent was removed in vacuo. The residue was triturated with pentane (2 × 15 mL) and then extracted into pentane (10 mL), leaving a white solid. Removal of the volatile materials from the yellow filtrate gave the product as a yellow powder. Yield: 182 mg, 87 %. 1H NMR (400 MHz, C6D6): δ 7.27 (m, 1H, Ar), 7.22 (m, 2H, Ar), 7.10 (m, 3H, Ar), 5.98 (br s, 1H, Nb(NHtBu), 5.28 (s, 1H, HC(C(Me)NAr)2), 3.85 (sept, 1H, CHMe2), 3.74 (m, 2H, CH2 and CHMe2), 5.54 (sept, 1H, CHMe2), 3.35 (sept, 1H, CHMe2), 3.29 (s, 1H, CH2), 1.54 (d, 3H, CHMe2), 1.50 (s, 3H, HC(C(Me)NAr)2), 1.49 (d, 3H, CHMe2), 1.43 (d, 3H, CHMe2), 1.43 (d, 3H, CHMe2), 1.41 (d, 3H, CHMe2), 1.38 (d, 3H, CHMe2), 1.38 (s, 9H, tBu), 1.24 (d, 3H, CHMe2), 1.19 (d, 3H, CHMe2), 0.89 (s, 9H, tBu). Satisfactory elemental analysis data could not be obtain due to contamination of the product with residual py·B(C6F5)3.
(BDI)Nb(NtBu)(κ2-O,N-OC(NtBu)NtBu) (13)
To a solution of 9 (20 mg, 0.029 mmol) in 0.5 mL C6D6 was added a solution of B(C6F5)3 (14 mg, 0.029 mmol) in 0.5 mL C6D6. After 5 min, tBuNCO (6.2 μL, 0.055 mmol) was added by syringe. The solution was transferred to an NMR tube, and the tube was sealed under vacuum. The solution was allowed to stand at room temperature for 3 h then analyzed by NMR spectroscopy without purification due to the thermal instability of the product. 1H NMR (500 MHz, C6D6): δ 7.20-7.05 (m, 6H, Ar), 5.36 (s, 1H, HC(C(Me)NAr)2), 3.57 (sept, 2H, CHMe2), 2.81 (sept, 2H, CHMe2), 1.70 (s, 6H, HC(C(Me)NAr)2), 1.47 (s, 9H, tBu), 1.38 (d, 6H, CHMe2), 1.34 (s, 9H, tBu), 1.33 (d, 6H, CHMe2), 1.28 (s, 9H, tBu), 1.20 (d, 6H, CHMe2), 1.05 (d, 6H, CHMe2). 13C NMR (125 MHz, C6D6): δ 170.92 (HC(C(Me)NAr)2), 156.83 (OCN), 149.50 (C6F5), 147.60 (C6F5), 146.69 (Ar), 144.41 (Ar), 143.80 (Ar), 143.02 (Ar), 142.04 (Ar), 139.00 (C6F5), 137.05 (C6F5), 127.57 (Ar), 125.30 (Ar), 124.65 (Ar), 104.71 (HC(C(Me)NAr), 69.82 (Nb=NtBu, Cα), 57.34 (cycle, tBu, Cα), 52.03 (cycle, tBu, Cα), 33.14 (tBu, Cβ), 32.59 (tBu, Cβ), 31.88 (tBu, Cβ), 29.66 (CHMe2), 28.06 (CHMe2), 26.06 (CHMe2), 25.85 (CHMe2), 25.71 (CHMe2), 25.56 (CHMe2), 24.75 (HC(C(Me)NAr)2).
Supplementary Material
Table 2.
Crystallographic data for complexes 1, 5, 7, 9 and 10.
| 1 | 5 | 7 | 9 | 10 | |
|---|---|---|---|---|---|
| formula | C38H55Cl2N4Nb | C41H58Cl2N3Nb | C40H57ClN3Nb | C42H64N5Nb | C44H69N6Nb |
| fw | 731.67 | 756.71 | 708.25 | 731.89 | 774.96 |
| space group | P21/c | Pbca | P212121 | Pbca | P21/n |
| a (Å) | 8.865(1) | 17.840(5) | 13.757(2) | 19.649(1) | 15.530(3) |
| b (Å) | 17.986(2) | 21.065(6) | 16.524(3) | 18.887(1) | 12.284(2) |
| c (Å) | 24.392(2) | 21.778(6) | 17.039(3) | 22.364(1) | 23.724(4) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 99.792(1) | 90 | 90 | 90 | 107.887(3) |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 3832.8(6) | 8184(4) | 3873.3(11) | 8299.5(9) | 4307.3(14) |
| Z | 4 | 8 | 4 | 8 | 4 |
| ρcalcd (g/cm3) | 1.268 | 1.228 | 1.215 | 1.171 | 1.195 |
| F000 | 1544 | 3200 | 1504 | 3136 | 1664 |
| μ (cm−1) | 0.48 | 0.46 | 0.41 | 0.32 | 0.31 |
| Tmin/Tmax | 0.917772 | 0.90033 | 0.870265 | 0.887106 | 0.921823 |
| no. rflns measured | 16890 | 37957 | 17262 | 33520 | 17130 |
| no. indep. rflns | 6495 | 8076 | 6488 | 5593 | 6328 |
| Rint | 0.0291 | 0.0398 | 0.0626 | 0.0517 | 0.0457 |
| no. obs. (I > 2.00σ(I)) | 5096 | 6046 | 5497 | 4337 | 4105 |
| no. variables | 406 | 426 | 407 | 433 | 460 |
| R1a, wR2b | 0.0367, 0.0872 | 0.0356, 0.0860 | 0.0473, 0.1137 | 0.0349, 0.0862 | 0.0406, 0.0910 |
| R1 (all data) | 0.0531 | 0.0560 | 0.0617 | 0.0521 | 0.0792 |
| goodness-of-fit on F2 | 1.206 | 1.117 | 0.969 | 1.382 | 1.071 |
| res. peak, hole (e−/Å3) | 1.014, −0.399 | 0.882, −0.287 | 1.261, −0.298 | 0.417, −0.308 | 0.717, −0.393 |
| CCDC ref. # | 675084 | 758024 | 758023 | 675086 | 758025 |
R1 = Σ(|Fo| − |Fc|)/Σ(|Fo|);
wR2 = [Σ{w(Fo2 − Fc2)2}/Σ{w(Fo2)2}]1/2
Acknowledgments
We thank the American Chemical Society Petroleum Research Fund (ACS-47249AC3), the National Science Foundation (CHE-0416309), and the National Institutes of Health (R01-GM025459-29) for funding.
Footnotes
Supporting Information Available. Complete crystallographic data for compounds 1, 5, 6, 7, 9, and 10 (CIF), and complete information on the DFT calculations performed on compound 11a. This information is available free of charge via the Internet at http://www.pubs.acs.org.
References
- 1.Duncan AP, Bergman RG. Chem Rec. 2002;2:431–445. doi: 10.1002/tcr.10039. [DOI] [PubMed] [Google Scholar]
- 2.Wigley D. Prog Inorg Chem. 1994;42:239–482. [Google Scholar]
- 3.Schaller CP, Cummins CC, Wolczanski PT. J Am Chem Soc. 1996;118:591–611. [Google Scholar]
- 4.Cummins CC, Schaller CP, Vanduyne GD, Wolczanski PT, Chan AWE, Hoffmann R. J Am Chem Soc. 1991;113:2985–2994. [Google Scholar]
- 5.DeWith J, Horton AD, Orpen AG. Organometallics. 1993;12:1493–1496. [Google Scholar]
- 6.DeWith J, Horton AD. Angew Chem Int Ed. 1993;32:903–905. [Google Scholar]
- 7.Blake RE, Antonelli DM, Henling LM, Schaefer WP, Hardcastle KI, Bercaw JE. Organometallics. 1998;17:718–725. [Google Scholar]
- 8.Schrock RR. J Mol Catal A: Chem. 2004;213:21–30. [Google Scholar]
- 9.Bolton PD, Mountford P. Adv Synth Catal. 2005;347:355–366. [Google Scholar]
- 10.Royo P, Sanchez-Nieves J. J Organomet Chem. 2000;597:61–68. [Google Scholar]
- 11.Burland MC, Pontz TW, Meyer TY. Organometallics. 2002;21:1933–1941. [Google Scholar]
- 12.Ong TG, Yap GPA, Richeson DS. J Chem Soc, Chem Commun. 2003:2612–2613. doi: 10.1039/b308710g. [DOI] [PubMed] [Google Scholar]
- 13.Anderson LL, Arnold J, Bergman RG. Org Lett. 2004;6:2519–2522. doi: 10.1021/ol0492851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao YW, Rodgers PM, Wigley DE, Alexander SJ, Rheingold AL. J Am Chem Soc. 1991;113:6326–6328. [Google Scholar]
- 15.Williams D, Schrock RR. Organometallics. 1993;12:1148–1160. [Google Scholar]
- 16.Williams D, Schofield M, Schrock RR, Davis W, Anhaus J. J Am Chem Soc. 1991;113:5480–5481. [Google Scholar]
- 17.Williams D, Schofield M, Anhaus J, Schrock RR. J Am Chem Soc. 1990;112:6728–6729. [Google Scholar]
- 18.Weinstock I, Schrock RR, Williams D, Crowe W. Organometallics. 1991;10:1–2. [Google Scholar]
- 19.Schofield M, Kee T, Anhaus J, Schrock RR, Johnson K, Davis W. Inorg Chem. 1991;30:3595–3604. [Google Scholar]
- 20.Lin ZY, Hall MB. Coord Chem Rev. 1993;123:149–167. [Google Scholar]
- 21.Cundari TR. Chem Rev. 2000;100:807–818. doi: 10.1021/cr980406i. [DOI] [PubMed] [Google Scholar]
- 22.Benson MT, Bryan JC, Burrell AK, Cundari TR. Inorg Chem. 1995;34:2348–2355. [Google Scholar]
- 23.Sundermeyer J, Putterlik J, Foth M, Field JS, Ramesar N. Chem Ber. 1994;127:1207–1212. [Google Scholar]
- 24.Sundermeyer J, Runge D. Angew Chem Int Ed. 1994;33:1255–1257. [Google Scholar]
- 25.Schorm A, Sundermeyer J. Eur J Inorg Chem. 2001:2947–2955. [Google Scholar]
- 26.Parkin G, Vanasselt A, Leahy DJ, Whinnery L, Hua NG, Quan RW, Henling LM, Schaefer WP, Santarsiero BD, Bercaw JE. Inorg Chem. 1992;31:82–85. [Google Scholar]
- 27.Walsh PJ, Hollander FJ, Bergman RG. J Am Chem Soc. 1988;110:8729–8731. [Google Scholar]
- 28.Walsh PJ, Baranger AM, Bergman RG. J Am Chem Soc. 1992;114:1708–1719. [Google Scholar]
- 29.Baranger AM, Walsh PJ, Bergman RG. J Am Chem Soc. 1993;115:2753–2763. [Google Scholar]
- 30.Hoyt HM, Michael FE, Bergman RG. J Am Chem Soc. 2004;126:1018–1019. doi: 10.1021/ja0385944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaller CP, Wolczanski PT. Inorg Chem. 1993;32:131–144. [Google Scholar]
- 32.Cundari TR. Organometallics. 1994;13:2987–2994. [Google Scholar]
- 33.Williams DS, Schofield MH, Schrock RR. Organometallics. 1993;12:4560–4571. [Google Scholar]
- 34.Bourget-Merle L, Lappert MF, Severn JR. Chem Rev. 2002;102:3031–3065. doi: 10.1021/cr010424r. [DOI] [PubMed] [Google Scholar]
- 35.Basuli F, Kilgore U, Hu X, Meyer K, Pink M, Huffman J, Mindiola D. Angew Chem Int Ed. 2004;43:3156–3159. doi: 10.1002/anie.200353210. [DOI] [PubMed] [Google Scholar]
- 36.Basuli F, Clark R, Bailey B, Brown D, Huffman J, Mindiola D. J Chem Soc, Chem Commun. 2005:2250. doi: 10.1039/b500350d. [DOI] [PubMed] [Google Scholar]
- 37.Mindiola D, Bailey B, Basuli F. Eur J Inorg Chem. 2006;2006:3135–3146. [Google Scholar]
- 38.Holland PL. Acc Chem Res. 2008;41:905–914. doi: 10.1021/ar700267b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kogut E, Wiencko HL, Zhang LB, Cordeau DE, Warren TH. J Am Chem Soc. 2005;127:11248–11249. doi: 10.1021/ja0533186. [DOI] [PubMed] [Google Scholar]
- 40.Dai XL, Kapoor P, Warren TH. J Am Chem Soc. 2004;126:4798–4799. doi: 10.1021/ja036308i. [DOI] [PubMed] [Google Scholar]
- 41.Badiei YM, Krishnaswamy A, Melzer MM, Warren TH. J Am Chem Soc. 2006;128:15056–15057. doi: 10.1021/ja065299l. [DOI] [PubMed] [Google Scholar]
- 42.Basuli F, Huffman JC, Mindiola DJ. Inorg Chem. 2003;42:8003–8010. doi: 10.1021/ic034853e. [DOI] [PubMed] [Google Scholar]
- 43.Franceschini PL, Morstein M, Berke H, Schmalle HW. Inorg Chem. 2003;42:7273–7282. doi: 10.1021/ic034328f. [DOI] [PubMed] [Google Scholar]
- 44.Nikiforov GB, Roesky HW, Magull J, Labahn T, Vidovic D, Noltemeyer M, Schmidt HG, Hosmane NS. Polyhedron. 2003;22:2669–2681. [Google Scholar]
- 45.Shaviv E, Botoshansky M, Eisen MS. J Organomet Chem. 2003;683:165–180. [Google Scholar]
- 46.Hamaki H, Takeda N, Tokitoh N. Organometallics. 2006;25:2457–2464. [Google Scholar]
- 47.Tonzetich ZJ, Jiang AJ, Schrock RR, Mueller P. Organometallics. 2007;26:3771–3783. [Google Scholar]
- 48.Tonzetich ZJ, Jiang AJ, Schrock RR, Muller P. Organometallics. 2006;25:4725–4727. [Google Scholar]
- 49.Lyashenko G, Herbst-Irmer R, Jancik V, Pal A, Mosch-Zanetti NC. Inorg Chem. 2008;47:113–120. doi: 10.1021/ic701534a. [DOI] [PubMed] [Google Scholar]
- 50.Fekl U, Goldberg K. J Am Chem Soc. 2002;124:6804–6805. doi: 10.1021/ja0258677. [DOI] [PubMed] [Google Scholar]
- 51.Fekl U, Kaminsky W, Goldberg KI. J Am Chem Soc. 2003;125:15286–15287. doi: 10.1021/ja037781z. [DOI] [PubMed] [Google Scholar]
- 52.Bernskoetter WH, Lobkovsky E, Chirik PJ. J Chem Soc, Chem Commun. 2004:764–765. doi: 10.1039/b315817a. [DOI] [PubMed] [Google Scholar]
- 53.Bernskoetter WH, Lobkovsky E, Chirik PJ. Organometallics. 2005;24:4367–4373. [Google Scholar]
- 54.Bernskoetter WH, Lobkovsky E, Chirik PJ. Organometallics. 2005;24:6250–6259. [Google Scholar]
- 55.Landolsi K, Richard P, Bouachir F. J Organomet Chem. 2005;690:513–518. [Google Scholar]
- 56.Masuda JD, Stephan DW. Can J Chem. 2005;83:324–327. [Google Scholar]
- 57.Fandos R, Walter MD, Kazhdan D, Andersen RA. Organometallics. 2006;25:3678–3687. [Google Scholar]
- 58.Tian X, Goddard R, Poerschke K. Organometallics. 2006;25:5854–5862. [Google Scholar]
- 59.Basuli F, Kilgore UJ, Brown D, Huffman JC, Mindiola DJ. Organometallics. 2004;23:6166–6175. [Google Scholar]
- 60.Basuli F, Huffman J, Mindiola D. Inorg Chim Acta. 2007;360:246–254. [Google Scholar]
- 61.Budzelaar PHM, van Oort AB, Orpen AG, Lw Eur J Inorg Chem. 1998:1485–1494. [Google Scholar]
- 62.Roesky HW, Mellerrehbein B, Noltemeyer M. Z Naturforsch, B: Chem Sci. 1991;46:1053–1058. [Google Scholar]
- 63.Dawson DY, Arnold J. Organometallics. 1994;16:1111–1113. [Google Scholar]
- 64.Brand H, Arnold J. Angew Chem Int Ed. 1997;33:95–97. [Google Scholar]
- 65.Gerlach C, Arnold J. Inorg Chem. 1996;35:5770–5780. [Google Scholar]
- 66.Anderson LL, Schmidt JAR, Arnold J, Bergman R. Organometallics. 2006;25:3394–3406. doi: 10.1021/om060081t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams DN, Mitchell JP, Poole AD, Siemeling U, Clegg W, Hockless DCR, Oneil PA, Gibson VC. J Chem Soc, Dalton Trans. 1992:739–751. [Google Scholar]
- 68.Basuli F, Bailey BC, Watson LA, Tomaszewski J, Huffman JC, Mindiola DJ. Organometallics. 2005;24:1886–1906. [Google Scholar]
- 69.Basuli F, Bailey BC, Huffman JC, Mindiola DJ. Organometallics. 2005;24:3321–3334. [Google Scholar]
- 70.Orpen AG, Brammer L, Allen FH, Kennard O, Watson DG, Taylor R. J Chem Soc, Dalton Trans. 1989:S1–S83. [Google Scholar]
- 71.Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor R. J Chem Soc, Perkin Trans 2. 1987:S1–S19. [Google Scholar]
- 72.Masui H. Coord Chem Rev. 2001;219:957–992. [Google Scholar]
- 73.Mullins SM, Hagadorn JR, Bergman RG, Arnold J. J Organomet Chem. 2000;607:227–232. [Google Scholar]
- 74.Sanchez-Nieves J, Frutos LM, Royo P, Castano O, Herdtweck E. Organometallics. 2005;24:2004–2007. [Google Scholar]
- 75.Bott SG, Hoffman DM, Rangarajan P. Inorg Chem. 1995;34:4305–4310. [Google Scholar]
- 76.Jamieson G, Lindsell WE. Inorg Chim Acta. 1978;28:113–118. [Google Scholar]
- 77.McLain SJ, Schrock RR, Sharp PR, Churchill MR, Youngs WJ. J Am Chem Soc. 1979;101:263–265. [Google Scholar]
- 78.Sharp PR, Astruc D, Schrock RR. J Organomet Chem. 1979;182:477–488. [Google Scholar]
- 79.Antinolo A, Deilarduya JM, Otero A, Royo P, Lanfredi AMM, Tiripicchio A. J Chem Soc, Dalton Trans. 1988:2685–2693. [Google Scholar]
- 80.(a) Chesnut RW, Yu JS, Fanwick PE, Rothwell IP, Huffman JC. Polyhedron. 1990;9:1051–1058. [Google Scholar]; (b) Lockwood MA, Potyen MC, Steffey BD, Fanwick PE, Rothwell IP. Polyhedron. 1995;14:3293–3313. [Google Scholar]
- 81.Cockcroft JK, Gibson VC, Howard JAK, Poole AD, Siemeling U, Wilson C. J Chem Soc, Chem Commun. 1992:1668–1670. [Google Scholar]
- 82.Etienne M, White PS, Templeton JL. Organometallics. 1993;12:4010–4015. [Google Scholar]
- 83.Djakovitch L, Herrmann WA. J Organomet Chem. 1997;546:399–405. [Google Scholar]
- 84.Chan MCW, Cole JM, Gibson VC, Howard JAK, Lehmann C, Poole AD, Siemeling U. J Chem Soc, Dalton Trans. 1998:103–111. [Google Scholar]
- 85.Tomson NC, Yan A, Arnold J, Bergman R. J Am Chem Soc. 2008;130:11262–11263. doi: 10.1021/ja803729k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.(a) Wright RJ, Power PP, Scott BL, Kiplinger JL. Organometallics. 2004;23:4801–4803. [Google Scholar]; (b) Clegg W, Coles SJ, Cope EK, Mair FS. Angew Chem, Int Ed. 1998;37:796–798. doi: 10.1002/(SICI)1521-3773(19980403)37:6<796::AID-ANIE796>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 87.Determined using the continuous symmetry parameter τ = (α − β)/60, where α and β are the largest and second largest angles about the metal center, respectively; Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. J Chem Soc; Dalton Trans. 1984:1349.
- 88.Humphries M, Green M, Leech M, Gibson V, Jolly M, Williams D, Elsegood M, Clegg W. J Chem Soc, Dalton Trans. 2000:4044–4051. [Google Scholar]
- 89.Kilgore U, Karty J, Pink M, Gao X, Mindiola D. Angew Chem Int Ed. 2009;48:2394–2397. doi: 10.1002/anie.200806022. [DOI] [PubMed] [Google Scholar]
- 90.Chao YW, Wexler PA, Wigley DE, Eh Inorg Chem. 1990;29:4592–4594. [Google Scholar]
- 91.Ciszewski J, Harrison J, Odom A. Inorg Chem. 2004;43:3605–3617. doi: 10.1021/ic049644o. [DOI] [PubMed] [Google Scholar]
- 92.Nugent WA, Mayer JM. Metal-Ligand Multiple Bonds. John Wiley & Sons; New York: 1988. [Google Scholar]; (b) While the the values of Δδαβ can provide useful qualitative data for comparing species with closely related ligand environments, care should be taken when comparing systems with significantly different electronic properties. e.g. The Δδαβ for the fluoride complexes 2 and 4 are lower than those for the chloride complexes 1 and 3, even though fluorine is more electronegative than chlorine. This discrepency possibly indicates better Nb d-orbital overlap with the fluoride 2p-orbitals than with the 3p-orbitals of the chlorides. Due to the complexity of these interactions, we have refrained from providing further analysis of the Δδαβ values beyond what is presented in the text.
- 93.Bradley DC, Hodge SR, Runnacles JD, Hughes M, Mason J, Richards RL. J Chem Soc, Dalton Trans. 1992:1663–1668. [Google Scholar]
- 94.Barrie P, Coffey TA, Forster GD, Hogarth G. J Chem Soc, Dalton Trans. 1999:4519–4528. [Google Scholar]
- 95.Strong JB, Yap GPA, Ostrander R, Liable-Sands LM, Rheingold AL, Thouvenot R, Gouzerh P, Maatta EA. J Am Chem Soc. 2000;122:639–649. [Google Scholar]
- 96.(a) Ding Y, Hao H, Roesky HW, Noltemeyer M, Schmidt HG. Organometallics. 2001;20:4806. [Google Scholar]; (b) Meltzer A, Inoue S, Praesang C, Driess M. J Am Chem Soc. 2010;132:3038–3046. doi: 10.1021/ja910305p. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 97.Alaimo PJ, Peters DW, Arnold J, Bergman RG. J Chem Educ. 2001;78:64–64. [Google Scholar]
- 98.Burger BJ, Bercaw JE. In: Experimental Organometallic Chemistry. Wayda AL, Darensbourg MY, editors. American Chemical Society; Washington, DC: 1987. pp. 79–98. [Google Scholar]
- 99.Vela J, Smith JM, Yu Y, Ketterer NA, Flaschenriem CJ, Lachicotte RJ, Holland PL. J Am Chem Soc. 2005;127:7857–7870. doi: 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]
- 100.Koppe K, Bilir V, Frohn H, Mercier HPA, Schrobilgen GJ. Inorg Chem. 2007;46:9425–9437. doi: 10.1021/ic7010138. [DOI] [PubMed] [Google Scholar]
- 101.Bates PA, Nielson AJ, Waters JM. Polyhedron. 1985;4:1391–1401. [Google Scholar]
- 102.Pajerski AD, Squiller EP, Parvez M, Whittle RR, Richey HG. Organometallics. 2005;24:809–814. [Google Scholar]
- 103.SMART: Area-Detector Software Package. Bruker Analytical X-ray Systems, Inc; Madison, WI: 2001–2003. [Google Scholar]
- 104.SAINT: SAX Area-Detector Integration Program, V6.40. Bruker Analytical X-ray Systems Inc; Madison, WI: 2003. [Google Scholar]
- 105.XPREP. Bruker Analytical X-ray Systems Inc; Madison, WI: 2003. [Google Scholar]
- 106.SADABS: Bruker-Nonius Area Detector Scaling and Absorption v. 2.05. Bruker Analytical X-ray Systems, Inc; Madison, WI: 2003. [Google Scholar]
- 107.Farrugia LJ. J Appl Crystallogr. 1997;30:565. [Google Scholar]
- 108.Korolev AV, Rheingold AL, Williams DS. Inorg Chem. 1997;36:2647–2655. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.