

Abstract
It is now easier to discover thousands of protein sequences in a new microbial genome than it is to biochemically characterize the specific activity of a single protein of unknown function. The molecular functions of protein sequences have typically been predicted using homology-based computational methods, which rely on the principle that homologous proteins share a similar function. However, some protein families include groups of proteins with different molecular functions. A phylogenetic approach for predicting molecular function (sometimes called “phylogenomics”) is an effective means to predict protein molecular function. These methods incorporate functional evidence from all members of a family that have functional characterizations using the evolutionary history of the protein family to make robust predictions for the uncharacterized proteins. However, they are often difficult to apply on a genome-wide scale because of the time-consuming step of reconstructing the phylogenies of each protein to be annotated. Our automated approach for function annotation using phylogeny, the SIFTER (Statistical Inference of Function Through Evolutionary Relationships) methodology, uses a statistical graphical model to compute the probabilities of molecular functions for unannotated proteins. Our benchmark tests showed that SIFTER provides accurate functional predictions on various protein families, outperforming other available methods.
1. Introduction
As sequencing technologies develop, sequence data is accruing at a fast rate, and the potential for medical applications of genomic data to human biology is just beginning to be realized. Sequencing also heralds unprecedented opportunities for understanding human-associated microbiota, whose genetic diversity is perhaps 100 times that of the human genome [1]. However, despite this large body of new sequence information, functional annotation remains a major challenge. Molecular functions of proteins in the human genome continue to be discovered, in large part by homology to those experimentally characterized in model organisms.
Typically, protein function annotation involves finding homologs of a protein sequence, followed by database queries and computational techniques to predict function from the annotated homologs. These methods rely on the principle that proteins from a common ancestor may share a similar function. However, most protein families have sets of proteins with different functions and therefore traditional bioinformatics approaches are unable to reliably assign the appropriate function to unannotated proteins. Currently, protein function databases have a large proportion of erroneously annotated proteins, where the incorrect annotations were either derived using an imprecise computational technique or inferred using another incorrect annotation [2–4].
We have proposed integrating available functional data using the evolutionary relationships of a protein family, and we implemented this method in the program SIFTER. This phylogenetic approach to molecular function annotation, sometimes termed “phylogenomics” [5–8], uses an explicit phylogenetic tree to make functional predictions. The basic principle of phylogenetic function annotation is that function will tend to evolve in parallel with sequence [9], and that function is more likely to change after a duplication than after a speciation event [10–12]. Thus, the traditional evolutionary based approach involves building a phylogenetic tree from homologous protein sequences, identifying the most likely location of duplication events, and propagating known functions within each clade descendant from a duplication event. This evolutionary approach is a more sophisticated method of determining protein function than using sequence similarity (BLAST), or unstructured homology data (COGs) because it is unclear how to transfer function annotation in an evolutionarily consistent way when homology is represented by clusters rather than by a phylogeny. Accelerated rates of evolution will result in closely related sequences having less significant sequence similarity scores; this means that BLAST will systematically find proteins that appear more distant in an evolutionary tree as being most similar [13]. By contrast, a phylogenetic tree reflects the evolutionary path of a set of homologous proteins (figure 1). By using phylogenetic trees, we can directly apply the assumptions about how function evolves in order to enable a consistent, meaningful method of transferring sparse and noisy functional evidence.
Figure 1.
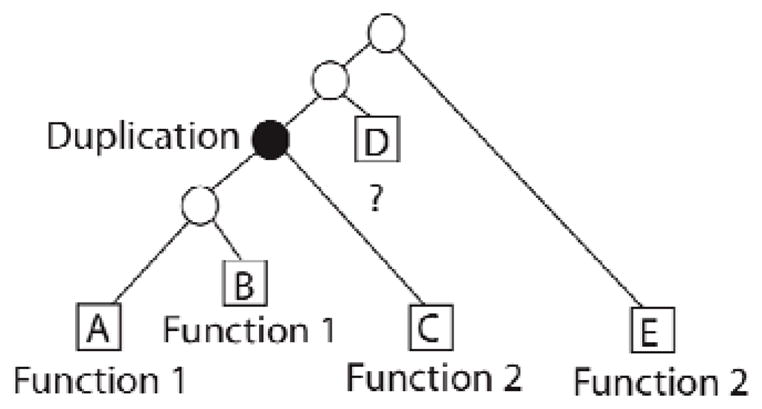
Function evolution is generally parsimonious; BLAST makes systematic errors in predicting function. Protein D is unannotated, while proteins A and B share a function, as do C and E. Parsimonious reconstruction dictates that D shares a function with C and E; a BLAST search gives the highest score to protein B, yielding erroneous annotations.
The use of phylogenetic function prediction to annotate individual proteins has proliferated [e.g., 6, 14–16] and provided some of the most reliable function annotations [17]. Manual phylogenetic function prediction studies on a genome-wide level are rare, because of the time-consuming step of reconstructing and then analyzing phylogenies for each of the unannotated proteins, but they do occur [18].
2. The SIFTER approach
The SIFTER methodology [19] is based on evolutionary principles [6], using a statistical representation. Currently, SIFTER takes as input a reconciled phylogeny and a set of annotations for some of the proteins in the protein family. Given a query sequence, the appropriate Pfam [20] protein family can be used to build the reconciled phylogeny. The Pfam alignment of the query sequence and its homologs is used as input to a phylogenetic reconstruction program, such as PAUP* with maximum likelihood [21]. The phylogeny is reconciled [22] against a known species phylogeny using, for example, the Forester [23] software to determine which nodes of the tree represent protein duplication events and which represent speciation events. (This approach will treat horizontal transfer effectively like a duplication, which is appropriate in the sense that we would like to assume that function is more likely to change after a horizontal transfer event.) We include prior information about function by computing the probability of each of the candidate functions given the available evidence for the proteins in the tree with functional evidence from the GOA database [24]. The candidate molecular functions are represented as a boolean vector, where initially the probability associated with each candidate function is a function of the set of annotations for that protein and their corresponding evidence types (e.g., experimental, electronic). Finally, from this reconciled phylogeny with sparse observations, SIFTER computes the posterior probability of each molecular function for all proteins in the family using a simple statistical model of protein function evolution.
The model of protein function evolution in SIFTER allows every candidate molecular function to mutate to every other candidate function for the family, and the likelihood of a mutation is related to estimated mutability of each pair of molecular functions, branch length, and whether an internal tree node represents a speciation or a duplication event. Whereas the branch length is a (fixed) value that is input to the method through the reconciled tree, we are free to estimate the two parameters controlling function mutability and general rate of mutation along a branch. The mutability parameters in effect relate sequence change to functional change for every pair of candidate functions if you consider a constant rate of amino acid substitution along each branch. In other words, if one function has a high rate of mutability to another function, then on average it takes fewer mutations to change the first function into the second. Two different parameters reflect the relative rates of functional change after a speciation event versus after a duplication event.
The phylogenetic tree is the structure for inferring molecular function, with the phylogenetic characters replaced by the molecular function random variables. Using message passing [25–27] it is possible to propagate this information throughout the tree to infer the posterior probability of each candidate function for all nodes. We chose a probabilistic approach to protein function prediction because it is well suited to the nature of the evidence. As in other areas of computational biology [see e.g. 28], a probabilistic framework has the major advantage that it allows multiple, noisy sources of evidence to be used for a single prediction, by weighting and combining this evidence appropriately. This is a fundamental feature of SIFTER—it computes posterior probabilities for each possible function of a query protein by combining evidence from related proteins in a coherent, evolutionarily motivated way through the phylogeny. Finally, a key feature of the protein function prediction problem is the sparsity of available experimental annotations within any particular protein family. The probabilistic approach takes sparsity into account in a natural way, as posterior probabilities are lower when the supporting evidence is weak or conflicting.
The SIFTER algorithm makes predictions using the evolutionary structure of a protein family and all available functional information. It provides traceable evidence, making it straightforward to understand the posterior probability of any leaf node by looking at posterior probability of the hidden nodes throughout the phylogeny, and it provides probabilistic results for each possible function.
3. Performance of SIFTER
We tested the performance of SIFTER on two different protein families: AMP/adenosine deaminases, and aminotransferases [29]. The sequences and alignments for each family were downloaded from the Pfam database [20] and the function annotations were from the GOA database [24] and a manual literature search. Each family posed unique challenges to function prediction.
The performance of SIFTER was estimated with leave-one-out cross validation experiments, where the available annotation of each protein was removed from the training set before a SIFTER run for the protein family was performed [29], then checking whether the maximum posterior probability for the predicted protein agreed with the held-out annotation. We ran cross validation for each of the two protein families with experimental annotations, and also with a combination of experimental and electronic annotations. SIFTER’s performance was compared with three other function prediction algorithms: BLAST [30], GOtcha [31] and Orthostrapper [32].
Our first dataset was composed of the Pfam AMP/adenosine deaminase family, which contained 251 proteins. This family has 33 proteins with experimental annotations, which came from the GOA database, a manual literature search, and our own characterization experiment. The challenge in assigning functions for proteins in this family is that a subset of proteins has multiple functions, where the additional function is active in a second protein domain. Thus, in our analysis we conclude that a prediction was correct if one of the two functions of a protein was assigned correctly. Cross validation on experimental annotations yielded 93.9% accuracy, while the accuracy for cross validation on experimental and electronic annotations was 96.3% [29]. In comparison, cross validation on experimental data yielded 66.7% accuracy for BLAST and GOtcha (GOtcha-exp), while Orthostrapper achieved 78.8% accuracy. GOtcha achieved 87.9% accuracy with both experimental and electronic annotation [29]. In Figure 2, we show the ROC analysis of this family, where SIFTER outperforms all the other methods on the deaminase family. This analysis looks at the percentage of false positive predictions relative to the percentage of true positive predictions as the cutoff for the posterior probability for function prediction goes from 0 to 1 (in SIFTER). This method of comparison was especially suitable for the deaminase family due to the multiple functions some of the family members have.
Figure 2.
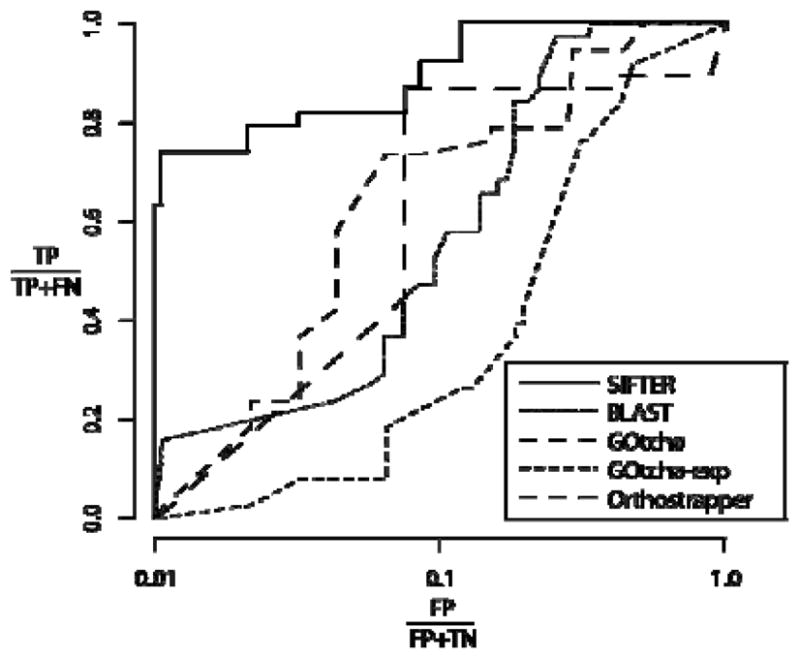
ROC figure comparing the five tested methods on the deaminase family using experimental annotations [29] (For GOtcha, both experimental and electronic annotation, GOtcha-exp means experimental annotation only). The X-axis is in log scale, and the key to axis labels is: TP=true positives, FP=false positives, TN=true negatives, and FN=false negatives.
The aminotransferase dataset was a difficult test case for our function prediction method due to homoplasy within the family. It appears that the preference for one of the substrates might have appeared multiple times independently during evolution in this family. Despite this, SIFTER maintained good prediction accuracy (75%; 9 of 12) when only the experimental data was used in cross validation [29]. The inclusion of electronic annotations increased SIFTER prediction accuracy to 92.6% (50 of 54). BLAST, GOtcha and GOtcha-exp achieved 66.7% accuracy (8 of 12) in the cross-validation experiments with experimental data, while Orthostrapper was unable to annotate any of the proteins [29].
4. Conclusions
The development of SIFTER is an ongoing project and we now have a new version of the program available (manuscript currently submitted). The new version includes a more general model of protein function evolution and a fast method for calculating the posterior probabilities; these improvements make SIFTER applicable on large and functionally diverse protein families and on genome-scale function annotation. Furthermore, we are validating SIFTER predictions experimentally using the extremely diverse Nudix family of hydrolases as a test bed.
References
- 1.Gill SR, Pop M, Deboy RT, Eckburg PB, et al. Science. 2006;312:1355–9. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner SE. Trends Genet. 1999;15:132–3. doi: 10.1016/s0168-9525(99)01706-0. [DOI] [PubMed] [Google Scholar]
- 3.Galperin MY, Koonin EV. In Silico Biol. 1998;1:55–67. [PubMed] [Google Scholar]
- 4.Jones CE, Brown AL, Baumann U. BMC Bioinformatics. 2007;8:170. doi: 10.1186/1471-2105-8-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisen JA, Hanawalt PC. Mutat Res. 1999;435:171–213. doi: 10.1016/s0921-8777(99)00050-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisen JA. Genome Res. 1998;8:163–7. doi: 10.1101/gr.8.3.163. [DOI] [PubMed] [Google Scholar]
- 7.Sjölander K. Bioinformatics. 2004;20:170–9. doi: 10.1093/bioinformatics/bth021. [DOI] [PubMed] [Google Scholar]
- 8.Brown D, Sjölander K. PLoS Comput Biol. 2006;2:e77. doi: 10.1371/journal.pcbi.0020077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atchley WR, Fitch WM. Proc Natl Acad Sci U S A. 1997;94:5172–6. doi: 10.1073/pnas.94.10.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohno S. Evolution by gene duplication. New York: Springer-Verlag; 1970. [Google Scholar]
- 11.Fitch WM. Syst Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 12.Lynch M, Conery JS. Science. 2000;290:1151–5. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 13.Koski LB, Golding GB. Journal of Molecular Evolution. 2001;52:540–2. doi: 10.1007/s002390010184. [DOI] [PubMed] [Google Scholar]
- 14.Theodorides K, De Riva A, Gomez-Zurita J, Foster PG, Vogler AP. Insect Mol Biol. 2002;11:467–75. doi: 10.1046/j.1365-2583.2002.00355.x. [DOI] [PubMed] [Google Scholar]
- 15.Danchin A. Curr Issues Mol Biol. 2003;5:37–42. [PubMed] [Google Scholar]
- 16.Gadelle D, Filee J, Buhler C, Forterre P. Bioessays. 2003;25:232–42. doi: 10.1002/bies.10245. [DOI] [PubMed] [Google Scholar]
- 17.Eisen JA, Fraser CM. Science. 2003;300:1706–7. doi: 10.1126/science.1086292. [DOI] [PubMed] [Google Scholar]
- 18.Tettelin H, Masignani V, Cieslewicz MJ, Eisen JA, et al. Proc Natl Acad Sci U S A. 2002;99:12391–6. doi: 10.1073/pnas.182380799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelhardt BE, Jordan MI, Muratore KE, Brenner SE. PLoS Comput Biol. 2005;1:e45. doi: 10.1371/journal.pcbi.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bateman A, Coin L, Durbin R, Finn RD, et al. Nucleic Acids Res. 2004;32:D138–41. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swofford DL. Paup*: Phylogenetic analysis using parsimony (*and other methods) Sunderland, Massachusetts: Sinauer Associates; 2001. [Google Scholar]
- 22.Perriere G, Duret L, Gouy M. Genome Res. 2000;10:379–85. doi: 10.1101/gr.10.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zmasek CM, Eddy SR. Bioinformatics. 2001;17:821–8. doi: 10.1093/bioinformatics/17.9.821. [DOI] [PubMed] [Google Scholar]
- 24.Camon E, Magrane M, Barrell D, Lee V, et al. Nucleic Acids Res. 2004;32:D262–6. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elston RC, Stewart J. Hum Hered. 1971;21:523–42. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- 26.Felsenstein J. J Mol Evol. 1981;17:368–76. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 27.Hilden J. Clin Genet. 1970;1:319–48. [Google Scholar]
- 28.Durbin R, Eddy S, Krogh A, Mitchison G. Biological sequence analysis. Cambridge University Press; 1998. [Google Scholar]
- 29.Engelhardt BE, Jordan MI, Brenner SE. Proc 23rd Intl Conf Machine Learning. 2006:038.1–8. [Google Scholar]
- 30.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 31.Martin DM, Berriman M, Barton GJ. BMC Bioinformatics. 2004;5:178. doi: 10.1186/1471-2105-5-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Storm CE, Sonnhammer EL. Bioinformatics. 2002;18:92–9. doi: 10.1093/bioinformatics/18.1.92. [DOI] [PubMed] [Google Scholar]