

Abstract
AIMS
To assess the impact of specialty society guidelines on the use of corticosteroids and cytotoxic agents in the initial management of patients with idiopathic pulmonary fibrosis.
METHODS
A retrospective cohort study of 941 patients with an incident diagnosis of IPF was conducted using a large medical records database. The primary outcome was a new prescription for corticosteroids with or without a cytotoxic agent within 30 days of diagnosis. The primary exposure was whether diagnosis occurred before or after the publication of treatment guidelines. Logistic regression was used to control for changes in population demographics and disease characteristics across time.
RESULTS
In total, 187 patients (19.9%) received a new corticosteroid prescription within 30 days of diagnosis. Fewer patients received corticosteroids after the publication of guidelines (22.2% vs. 17.7%; adjusted OR for steroid use after the publication of guidelines 0.65, 95% confidence interval 0.46, 0.92, P = 0.014). Among the 187 patients treated with corticosteroids, 22 (11.8%) also received a cytotoxic agent. The use of cytotoxic agents among users of corticosteroids increased significantly after the publication of guidelines (5.1% vs. 19.3%) with a fully adjusted OR = 4.71 (95% CI 1.56, 14.21, P = 0.006).
CONCLUSIONS
Since the publication of treatment guidelines, there has been a small reduction in the overall use of corticosteroids. Consistent with these guidelines, the use of cytotoxic agents among those prescribed corticosteroids has increased significantly; however, the use of these agents remains uncommon.
Keywords: corticosteroids, idiopathic pulmonary fibrosis, pharmacoepidemiology, treatment guidelines
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Idiopathic pulmonary fibrosis (IPF) is a progressive, fatal disease with no known aetiology and no proven treatment. Despite the absence of efficacy data, many physicians treat IPF with corticosteroids either as monotherapy or in combination with a cytotoxic agent.
Specialty society guidelines published in 1999 and 2000 recognize that treatment may not be appropriate for all patients with IPF, but recommend that if treatment is to be initiated, a combination of corticosteroids with a cytotoxic agent is preferred over corticosteroids alone.
It is not known how the use of corticosteroids and cytotoxic agents in the treatment of IPF has changed over time and whether published guidelines have altered prescribing practices.
WHAT THIS STUDY ADDS
The results of this study demonstrate a modest but statistically significant reduction in the overall use of corticosteroids since the publication of treatment guidelines. At the same time, there was a more pronounced increase in the combined use of corticosteroids and cytotoxic agents consistent with recommendations.
Even with the increase in the use of combination therapy, corticosteroid monotherapy remained the most commonly prescribed regimen among treated patients.
Given the lack of established benefit and the risks associated with corticosteroid therapy, the reasons for the continued use of corticosteroid monotherapy in the majority of treated patients warrant further investigation.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common form of idiopathic interstitial pneumonia and is associated with significantly shortened survival [1]. No pharmacologic agents have been shown to be consistently effective in reversing the natural history of IPF or in reducing mortality. Corticosteroids have been used as a mainstay of therapy. However, to date no studies have established a consistent benefit of corticosteroid monotherapy in the management of IPF [2].
Several studies have examined the combination of other agents with prednisone. One small study suggested that combination therapy with azathioprine may be effective in decreasing age-adjusted mortality [3], while another suggested that the use of prednisone, azathioprine and N-acetylcysteine may delay the progression of pulmonary function abnormalities [4]. Both of these studies had important limitations, and other studies using alternative agents have been disappointing [5–8].
Despite the lack of convincing efficacy data to support any single therapeutic approach, in 1999 the British Thoracic Society (BTS) and in 2000 the American Thoracic Society (ATS)/European Respiratory Society (ERS) adopted similar guidelines for the treatment of IPF [9, 10]. These published guidelines recommend that the decision whether or not to initiate therapy should take into account individual patient characteristics such as disease severity, symptoms, age and other co-morbid conditions. Once the decision to initiate treatment has been made, both sets of guidelines recommend the use of a combination of prednisone and either azathioprine or cyclophosphamide with close follow-up for an objective response to therapy.
It is not clear how physicians interpret the existing literature and whether the adoption of specialty society guidelines has impacted on the use of pharmacologic agents in the treatment of IPF. The few existing studies examining the treatment of IPF in practice are either from a time period well before the publication of treatment guidelines or unable to examine patient level prescription data at the time of diagnosis [11–13]. While these studies suggest that treatment of IPF with corticosteroids either alone or in combination with another agent is common, none has examined whether there have been temporal shifts in management or whether the publication of treatment guidelines has modified physician behaviour.
This study was designed to quantify how patients with IPF are treated at the time of their incident diagnosis using patient-level prescription data and to determine whether the use of medications has changed with the publication of treatment guidelines. Portions of the results of this study have been presented previously in abstract form [14, 15].
Methods
We conducted a retrospective cohort study using data collected from The Health Improvement Network (THIN) Database, a medical records database containing more than 5 million primary care records from participating general practices in the United Kingdom (UK). This database has been used in several past pharmacoepidemiology studies [16–19] and contains demographic and diagnostic information for each patient, as well as complete prescription information derived directly from the software used to generate prescriptions. This study was reviewed by the Institutional Review Board at the University of Pennsylvania.
Study subjects
We identified patients with an incident diagnosis of IPF defined as a new entry for a diagnostic code for either ‘idiopathic fibrosing alveolitis’ (H563.00) or ‘cryptogenic fibrosing alveolitis’ (H563.12). These terms have been used in prior research in THIN [20], and the accuracy of diagnostic codes in identifying patients with IPF has been previously established in a similar database, the General Practitioner's Research Database, that uses the same computer software for data entry [21].
We excluded patients with fewer than 12 months of medical records in the database prior to their first diagnosis with IPF in order to reduce the potential for misclassifying prevalent cases as incident cases [22]. Patients with a previous diagnosis of an autoimmune disease that might also be treated with corticosteroids were also excluded. Additionally, in order to analyse changes in the initiation of therapy for IPF, patients who received corticosteroids or a cytotoxic agent in the 3 months prior to diagnosis were excluded. Finally we excluded patients who were under 40 years old at diagnosis and patients whose records failed to meet quality standards as maintained by THIN.
Study outcomes
The primary outcome was defined as a new prescription for an oral corticosteroid with or without a cytotoxic agent written within 30 days of the first diagnosis of IPF. Combination therapy was defined as a prescription for corticosteroids and a prescription for either azathioprine or cyclophosphamide both within 30 days of diagnosis. The use of N-acetylcysteine (NAC) was not assessed because the years included in the database had minimal overlap with the period of time since NAC was shown to have a potential role in the treatment of IPF [4].
Exposure definition
In order to evaluate the effect of published treatment guidelines on prescribing behaviour, we grouped patients according to the year in which their initial diagnosis of IPF was made. We a priori used a date of 1 January 2000 to differentiate those diagnoses made before and after the publication of treatment guidelines in the UK. While this date does not coincide exactly with the publication date of BTS guidelines, it allows for a delay in dissemination of recommendations to clinicians and falls between the publication of the BTS guidelines and those issued by the ATS/ERS. As a sensitivity analysis, we repeated the analysis using the actual date of publication of BTS guidelines (1 April 1999) to dichotomize our primary exposure.
Finally, in order to determine whether any observed changes in prescribing behaviour were part of a temporal trend or event unrelated to the publication of treatment guidelines, we altered the year used to divide the cohort into pre- and post-eras across the range 1997–2003. We then repeated the primary analyses using each of these years as a cut-point to determine which year resulted in the greatest pre–post difference.
Statistical analysis
All statistical analyses were conducted using STATA version 9 (StataCorp, College Station, TX, USA). The proportions of patients treated with corticosteroids dichotomized before and after 1 January 2000 were compared using a binomial test of proportions. Among those who received corticosteroids, the proportions who received a cytotoxic agent were also compared.
In order to control for potential changes over time in disease characteristics, co-morbidities and demographic variables which might influence the use of corticosteroids, we created multivariable logistic regression models for the use of corticosteroids and for the use of cytotoxic agents among corticosteroid users. Pre-specified potential confounders were tested individually in separate bivariable analyses with each outcome. Those variables that were associated with the outcome with a P value < 0.2 were included in the final multivariable regression models [23]. Additionally, because age at diagnosis and current smoking were observed to be significantly higher after the publication of guidelines, these variables were forced into the final multivariable models. A P value of 0.05 was used as a threshold to define statistical significance in all analyses.
Results
We identified a total of 1583 patients with a diagnosis of IPF between the years of 1989 and 2006. Of these, 941 met the specified inclusion criteria (Figure 1). The mean age in this cohort at the time of diagnosis was 71.7 years (SD = 10.3) and 62.3% of patients were male. There were several changes in the patient population over the period of observation. The mean age at the time of diagnosis in all subjects in the cohort increased from 70.1 years in the period before 1 January 2000 to 73.1 years in the period after that date (P ≤ 0.0001). The frequency of smoking also increased. Additionally, the use of oxygen at the time of diagnosis increased from 7.4% to 14.7% (P = 0.0004). Similarly, the proportion of patients who had a diagnosis of dyspnoea prior to their diagnosis with IPF increased from 31.2% to 42.9% (P = 0.0002). Patient and disease characteristics for the whole cohort and by era of diagnosis are summarized in Table 1.
Figure 1.
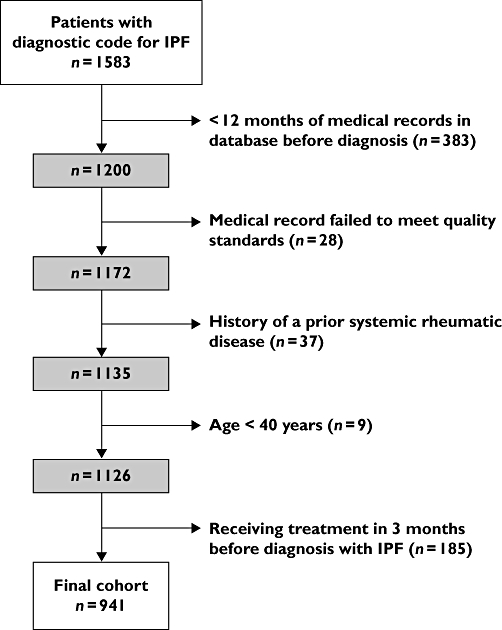
Exclusions applied to generate the final analysis cohort
Table 1.
Patient and disease characteristics in the total cohort and in groups diagnosed before and after publication of treatment guidelines
| Total | Before guidelines | After guidelines | P value* | |
|---|---|---|---|---|
| Number | 941 | 445 | 496 | N/A |
| Age (years) (SD) | 71.7 (10.3) | 70.1 (10.7) | 73.1 (9.8) | < 0.0001 |
| Gender | ||||
| Male | 586 (62.3%) | 276 (62.0%) | 310 (62.5%) | 0.88 |
| Female | 355 (37.7%) | 169 (38.0% | 186 (37.5%) | |
| Current smoking | 270 (28.7%) | 114 (25.6%) | 156 (31.5%) | 0.046 |
| Biopsy or Bronchoscopy | 84 (8.9%) | 46 (10.3%) | 38 (7.7%) | 0.15 |
| Oxygen use | 106 (11.3%) | 33 (7.4%) | 73 (14.7%) | 0.0004 |
| Dyspnoea | 352 (37.4%) | 139 (31.2%) | 213 (42.9%) | 0.0002 |
Comparison between groups before and after publication of guidelines.
In total, 187 patients (19.9%) received a new prescription for corticosteroids either alone or in combination with a cytotoxic agent at the time of diagnosis. Only 22 of these patients (11.8%) were treated with combination therapy. The proportion of patients who received corticosteroids decreased after the publication of guidelines (22.2% vs. 17.7%, OR = 0.75, 95% CI 0.55, 1.04, P = 0.084). Among those who received corticosteroids, the proportion who also received a prescription for a cytotoxic agent increased significantly from 5.1% to 19.3% (OR = 4.50, 95% CI 1.59, 12.78, P = 0.0025). These results are illustrated in Figure 2. Figure 3 shows the use of corticosteroids plotted alongside the use of oxygen and dyspnoea by year of diagnosis.
Figure 2.
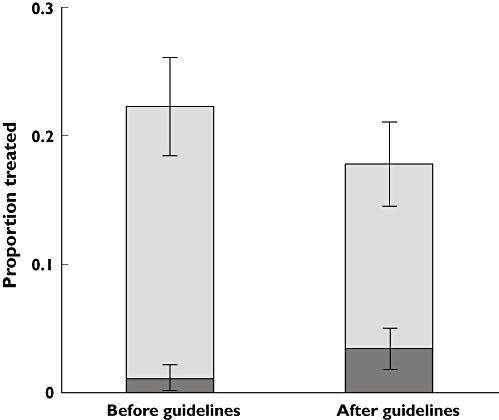
The contribution of corticosteroid monotherapy and combination therapy with a cytotoxic agent to the total use of corticosteroids before and after the publication of specialty society guidelines. Corticosteroid monotherapy ( ); corticosteroids prescribed in combination with a cytotoxic agent (
); corticosteroids prescribed in combination with a cytotoxic agent ( )
)
Figure 3.
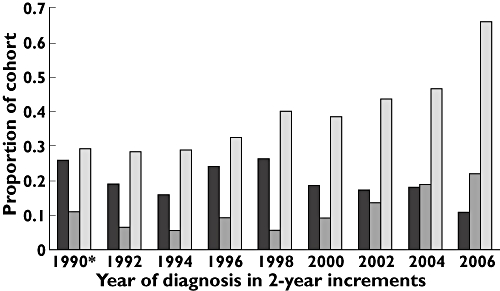
Oxygen use, dyspnoea, and the use of corticosteroids over time. Corticosteroid use ( ); oxygen use (
); oxygen use ( ); dyspnoea (
); dyspnoea ( )
)
We tested as potential confounders each of the factors included in Table 1 as well as co-morbid conditions such as diabetes and osteoporosis, and the number of patients diagnosed with IPF per year in the physician's practice. Also, because patients with IPF who do not have classic diagnostic features at the time of initial presentation might be more likely to receive corticosteroids, we included the presence of a diagnostic code for a different form of interstitial lung disease prior to the diagnosis of IPF as a potential confounder. Only oxygen use, a diagnostic code for dyspnoea, and a prior diagnosis with a different form of ILD were associated with a P value < 0.2 in bivariable analysis. These variables, as well as age in 10-year increments and current smoking status, were then included in a final multivariable regression model. After adjustment for these potential confounders (Table 2), the OR for receiving steroids in the period after 1 January 2000 compared with the period before that date was 0.65 (95% CI 0.46, 0.92, P = 0.014). Including past corticosteroid use (>90 days prior to diagnosis) in the regression model did not alter the results. Similarly, an analysis excluding those patients with a prior diagnosis of a different form of ILD was not substantially different from the adjusted model. A sensitivity analysis using 1 April 1999 as a closer approximation to the date of publication of BTS guidelines revealed similar results of both bivariable and multivariable analyses (results not shown).
Table 2.
Use of corticosteroids and use of cytotoxic agents among corticosteroid users after the publication of treatment guidelines
| Unadjusted OR (95% CI) | Adjusted OR (95% CI) | P value for adjusted OR | |
|---|---|---|---|
| All corticosteroid treatment | 0.75 (0.55, 1.04) | 0.65 (0.46, 0.92)* | 0.014 |
| Cytotoxic therapy among corticosteroid users | 4.50 (1.59, 12.78) | 4.71 (1.56, 14.21)† | 0.006 |
Adjusted for age, smoking status, past diagnosis with a different ILD, dyspnoea and use of oxygen.
Adjusted for age, smoking status, and use of oxygen.
We then tested the same variables to examine for potential confounding of the effect of diagnostic time period on the use of cytotoxic agents among those patients who were treated with corticosteroids. Only oxygen use at the time of diagnosis met criteria for inclusion as a potential confounder based on bivariable analyses. This was therefore included in a multivariable logistic regression model along with age and smoking status. The resulting fully adjusted OR for the use of cytotoxic agents after the publication of treatment guidelines recommending their use was 4.71 (95% CI 1.56, 14.21, P = 0.006) (see Table 2).
The results of dichotomizing the exposure by different years to test whether the year 2000 was a part of a pre-existing trend are presented in Table 3. In the case of both the overall use of corticosteroids and the use of combination therapy with corticosteroids and cytotoxic agents, dividing the exposure periods at the year 2000 resulted in the greatest difference in use of these treatment strategies.
Table 3.
Treatment with corticosteroids and combination therapy – results of dividing the cohort by different years
| Year | % treated before | % treated after | Difference % | Adjusted OR | 95% CI | P value |
|---|---|---|---|---|---|---|
| 1997 | ||||||
| CS | 19.9% | 19.9% | 0.0% | 0.89 | 0.60, 1.30 | 0.542 |
| CT | 5.6% | 14.3% | 8.7% | 2.83 | 0.76, 10.59 | 0.122 |
| 1998 | ||||||
| CS | 20.7% | 19.4% | −1.3% | 0.82 | 0.57, 1.18 | 0.293 |
| CT | 6.0% | 15.0% | 9.0% | 3.09 | 0.93, 10.30 | 0.066 |
| 1999 | ||||||
| CS | 22.4% | 18.1% | −4.3% | 0.66 | 0.47, 0.93 | 0.019 |
| CT | 5.9% | 16.7% | 10.8% | 3.18 | 1.02, 9.95 | 0.047 |
| 2000† | ||||||
| CS | 22.2% | 17.7% | −4.5% | 0.65 | 0.46, 0.92 | 0.014 |
| CT | 5.1% | 19.3% | 14.2% | 4.71 | 1.56, 14.21 | 0.006 |
| 2001 | ||||||
| CS | 21.3% | 18.1% | −3.2% | 0.70 | 0.49, 0.99 | 0.042 |
| CT | 8.1% | 17.1% | 9.0% | 2.22 | 0.84, 5.86 | 0.108 |
| 2002 | ||||||
| CS | 21.4% | 17.3% | −4.1% | 0.65 | 0.45, 0.94 | 0.023 |
| CT | 8.7% | 18.3% | 9.6% | 1.94 | 0.73, 5.16 | 0.182 |
| 2003 | ||||||
| CS | 20.9% | 17.3% | −3.6% | 0.66 | 0.44, 0.97 | 0.036 |
| CT | 9.9% | 17.4% | 7.5% | 1.65 | 0.59, 4.59 | 0.338 |
CS, all corticosteroid therapy; CT, corticosteroid users also receiving a cytotoxic agent.
A priori comparison before and after publication of guidelines.
Discussion
This study demonstrates that there has been a small decrease in the use of corticosteroids in the initial management of IPF since the publication of treatment guidelines. While the overall use of corticosteroids has decreased, the use of cytotoxic agents among those patients who receive corticosteroids has increased. Despite this increase, corticosteroid monotherapy was by far the more commonly prescribed treatment for IPF throughout the study period. The reasons for the apparent reluctance of physicians to prescribe cytotoxic agents warrant further study, especially in light of the most recent BTS guidelines that explicitly recommend against the use of high-dose corticosteroid monotherapy in favour of the combination of corticosteroids, azathioprine and N-acetylcysteine [24].
The overall use of corticosteroids in this cohort is less than that suggested by previous studies [11–13]. Johnston and colleagues found that 44% of patients seen by pulmonologists in the UK were offered treatment at the time of diagnosis [13]. However, these results were based on patients with an incident diagnosis made before 30 November 1992. Since that time, a growing body of literature has emerged suggesting the efficacy of corticosteroids in the treatment of IPF is limited and confirming the high risk of toxicity with treatment [2, 25]. More recently, Collard and colleagues conducted a survey in 2006 of academic physicians in the US and found that 52% of these physicians stated they ‘always’ or ‘frequently’ prescribed pharmacotherapy at the time of diagnosis with IPF [11]. This study, however, did not include patient level data reflecting actual prescribing practice. Additionally, because this study was limited to academic physicians in the US, it may not be representative of the population of IPF patients in other countries or practice settings. Finally, Hubbard and colleagues reported that 71% of patients with a diagnosis of IPF in the UK received corticosteroids at some time in their medical care. However, the timing of that treatment in the disease course was not reported [12].
The difference between the use of corticosteroids in this study and previous studies could also be related to our decision to limit the cohort to only those patients who were not on treatment at the time of diagnosis. We believe the decision to continue therapy is fundamentally different from the initiation of corticosteroid therapy and therefore we excluded these patients. Additionally, we used a strict definition of treatment initiation, limiting our outcomes to only those patients that received a prescription within 30 days of diagnosis.
Limitations of the database did not allow collection of pulmonary function test results or data on other validated markers of disease severity for the analysis of potential confounders. However, we did observe that the use of oxygen at the time of diagnosis and the presence of dyspnoea prior to the diagnosis of IPF both increased across the study period. It is possible that this reflects an increase in disease severity at presentation over time, although this has not been described elsewhere in the literature and is in contrast to the findings of Gribbin et al. who found that the median survival from the time of diagnosis has not changed [1]. It is also possible that the observed changes in these variables are not representative of an underlying change in the severity of IPF at presentation, but rather reflect temporal trends in recording symptoms such as dyspnoea in the electronic medical record or increased screening for hypoxaemia among patients with IPF. Similar attention to accurate data recording and health screening could explain the observed increase in cigarette smoking. Nonetheless, both dyspnoea and the use of oxygen met pre-specified criteria as potential confounders and were therefore included in the multivariable analysis.
This study has several important strengths. It is the first to examine prescription level data to determine the number of patients receiving corticosteroids and cytotoxic agents at the time of diagnosis, and the first to report changes in prescribing behaviour over time. We had a large number of patients covering multiple years both before and after the publication of guidelines allowing for a complete analysis of temporal trends in the treatment of IPF over nearly 15 years. Additionally, we were able to control for changes in the underlying population in order to determine whether trends were independent of other factors.
Despite these strengths, it is possible that the changes in treatment over time are related to an event other than the publication of specialty society guidelines. For instance, the classification of the idiopathic interstitial pneumonias underwent significant revisions in the years examined in this study [26, 27], and it is possible that the population of patients diagnosed with IPF in later years represents a more homogenous population of patients who would be expected to have less steroid-responsive disease. More stringent diagnostic criteria could also explain the observed increase in age at the time of diagnosis if younger patients with other forms of interstitial lung disease were previously classified as having IPF. This would not explain, however, the increased use of cytotoxic agents. Additionally, the observed differences in treatment were most pronounced both in absolute and relative terms when the study period was divided according to the year guidelines were published.
It is also possible that the observed differences in treatment simply reflect a temporal trend unrelated to any discrete event. When the use of corticosteroids is examined graphically by diagnostic year (see Figure 3), there is no clear trend before the year 2000 to suggest that guidelines were published during a pre-existing period of declining corticosteroid use. In fact, there appears to have been a trend of increasing use in the years immediately preceding 2000. Nevertheless, it is not possible to determine definitively using these data whether a trend toward reduced use after the year 2000 is directly attributable to published guidelines or other influences.
Although we cannot verify the clinical criteria used to confirm the diagnosis of IPF in THIN, our criteria are consistent with past work in this area, and the diagnostic codes used in this study have been previously assessed and found to be accurate [1, 20, 21, 28]. Additionally, and more importantly, the current study evaluates physician prescribing decisions when treating patients whom they believe to have IPF, regardless of whether that diagnosis is correct pathologically. It is also possible that the use of corticosteroids and cytotoxic agents could have been missed. Some patients may receive a single short course (typically 7 days or less) of treatment from a specialty provider which may not appear in the database. Under the National Health Service, subsequent prescriptions for continuation of therapy should be written by the general practitioner and should therefore be captured in this analysis. By evaluating the prescription information in the first 30 days after diagnosis rather than only at the time of diagnosis, the risk of misclassification is reduced.
As with all observational studies, it is possible that our results are confounded by additional factors that we have not measured. Additionally, these findings may not be generalizable outside of the UK. Finally, the effects of more recent studies demonstrating a potential benefit of corticosteroids/azathioprine/N-acetylcysteine and the most recent guidelines published in 2008 on prescribing behaviour cannot be assessed using these data.
Conclusions
We found that there has been a small decrease in the use of corticosteroids in the initial management of IPF, and that this decline appears to be related temporally to the publication of treatment guidelines. The use of cytotoxic agents among those treated with corticosteroids has increased significantly across the same time period consistent with current recommendations. However, corticosteroid monotherapy remains the most commonly prescribed initial treatment regimen.
Acknowledgments
During the conduct of this study, JM received support from the National Institutes of Health in the form of an institutional training grant (T32-GM075766-02) and a Ruth Kirchstein National Research Service Award (1F32-HL092741-01). This study was also supported in part by a Clinical and Translational Science Award from the National Institutes of Health (UL1-RR024134).
Competing interests
JM, ZC, and JC have no competing interests to declare. MK is the PI on a clinical trial sponsored by Gilead Sciences Inc, Seattle WA. SK has performed research and done consulting work funded by various pharmaceutical companies, none directly related to the topic of this manuscript.
REFERENCES
- 1.Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61:980–5. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richeldi L, Davies HR, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 2003;3:CD002880. doi: 10.1002/14651858.CD002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, Depaso WJ, Cain K, Hammar SP, Wetzel CE, Dreis DF, Hutchinson J, Pardee NE, Winterbauer RH. Azathioprine combined with prednisone in the treatment of idiopathic pulmonary fibrosis: a prospective double-blind, randomized, placebo-controlled clinical trial. Am Rev Respir Dis. 1991;144:291–6. doi: 10.1164/ajrccm/144.2.291. [DOI] [PubMed] [Google Scholar]
- 4.Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, MacNee W, Thomeer M, Wallaert B, Laurent F, Nicholson AG, Verbeken EK, Verschakelen J, Flower CD, Capron F, Petruzzelli S, De Vuyst P, van den Bosch JM, Rodriguez-Becerra E, Corvasce G, Lankhorst I, Sardina M, Montanari M. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2005;353:2229–42. doi: 10.1056/NEJMoa042976. [DOI] [PubMed] [Google Scholar]
- 5.Davies HR, Richeldi L, Walters EH. Immunomodulatory agents for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev. 2003;3:CD003134. doi: 10.1002/14651858.CD003134. [DOI] [PubMed] [Google Scholar]
- 6.Johnson MA, Kwan S, Snell NJ, Nunn AJ, Darbyshire JH, Turner-Warwick M. Randomised controlled trial comparing prednisolone alone with cyclophosphamide and low dose prednisolone in combination in cryptogenic fibrosing alveolitis. Thorax. 1989;44:280–8. doi: 10.1136/thx.44.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raghu G, Brown KK, Bradford WZ, Starko K, Noble PW, Schwartz DA, King TE., Jr A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350:125–33. doi: 10.1056/NEJMoa030511. [DOI] [PubMed] [Google Scholar]
- 8.Fiorucci E, Lucantoni G, Paone G, Zotti M, Li BE, Serpilli M, Regimenti P, Cammarella I, Puglisi G, Schmid G. Colchicine, cyclophosphamide and prednisone in the treatment of mild-moderate idiopathic pulmonary fibrosis: comparison of three currently available therapeutic regimens. Eur Rev Med Pharmacol Sci. 2008;12:105–11. [PubMed] [Google Scholar]
- 9.British Thoracic Society Standards of Care Committee. The diagnosis, assessment and treatment of diffuse parenchymal lung disease in adults. British Thoracic Society and Standards of Care Committee. Thorax. 1999;54(Suppl 1):S1–30. doi: 10.1136/thx.54.suppl_1.s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–64. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 11.Collard HR, Loyd JE, King TE, Jr, Lancaster LH. Current diagnosis and management of idiopathic pulmonary fibrosis: a survey of academic physicians. Respir Med. 2007;101:2011–16. doi: 10.1016/j.rmed.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubbard R, Venn A. The impact of coexisting connective tissue disease on survival in patients with fibrosing alveolitis. Rheumatology (Oxford) 2002;41:676–9. doi: 10.1093/rheumatology/41.6.676. [DOI] [PubMed] [Google Scholar]
- 13.Johnston ID, Prescott RJ, Chalmers JC, Rudd RM. British Thoracic Society study of cryptogenic fibrosing alveolitis: current presentation and initial management. Fibrosing Alveolitis Subcommittee of the Research Committee of the British Thoracic Society. Thorax. 1997;52:38–44. doi: 10.1136/thx.52.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munson JC, Kreider ME, Chen Z, Christie JD, Kimmel SE. Use of corticosteroids and corticosteroid-sparing agents in the initial management of idiopathic pulmonary fibrosis. Chest Meeting Abstracts. 2008;134:S20002. Abstract. [Google Scholar]
- 15.Munson JC, Kreider ME, Christie JD, Kimmel SE. The effect of treatment guidelines on the initial management of idiopathic pulmonary fibrosis. Pharmacoepidemiol Drug Saf. 2009;18:S144–5. doi: 10.1002/pds.1959. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbard R, Lewis S, Smith C, Godfrey C, Smeeth L, Farrington P, Britton J. Use of nicotine replacement therapy and the risk of acute myocardial infarction, stroke, and death. Tob Control. 2005;14:416–21. doi: 10.1136/tc.2005.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kell P, Hvidsten K, Morant S, Harnett J, Bridge S. Factors that predict changing the type of phosphodiesterase type 5 inhibitor medication among men in the UK. Br J Urol. 2007;99:860–3. doi: 10.1111/j.1464-410X.2006.06668.x. [DOI] [PubMed] [Google Scholar]
- 18.Mortimer KJ, Tata LJ, Smith CJ, West J, Harrison TW, Tattersfield AE, Hubbard RB. Oral and inhaled corticosteroids and adrenal insufficiency: a case-control study. Thorax. 2006;61:405–8. doi: 10.1136/thx.2005.052456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367:1075–9. doi: 10.1016/S0140-6736(06)68474-2. [DOI] [PubMed] [Google Scholar]
- 20.Gribbin J, Hubbard R, Smith C. Role of diabetes mellitus and gastro-oesophageal reflux in the aetiology of idiopathic pulmonary fibrosis. Respir Med. 2009;103:927–31. doi: 10.1016/j.rmed.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000;161:5–8. doi: 10.1164/ajrccm.161.1.9906062. [DOI] [PubMed] [Google Scholar]
- 22.Lewis JD, Bilker WB, Weinstein RB, Strom BL. The relationship between time since registration and measured incidence rates in the General Practice Research Database. Pharmacoepidemiol Drug Saf. 2005;14:443–51. doi: 10.1002/pds.1115. [DOI] [PubMed] [Google Scholar]
- 23.Maldonado G, Greenland S. Simulation study of confounder-selection strategies. Am J Epidemiol. 1993;138:923–36. doi: 10.1093/oxfordjournals.aje.a116813. [DOI] [PubMed] [Google Scholar]
- 24.Bradley B, Branley HM, Egan JJ, Greaves MS, Hansell DM, Harrison NK, Hirani N, Hubbard R, Lake F, Millar AB, Wallace WA, Wells AU, Whyte MK, Wilsher ML, British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee. Thoracic Society of Australia. New Zealand Thoracic Society. Irish Thoracic Society Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63(Suppl V):v1–58. doi: 10.1136/thx.2008.101691. [DOI] [PubMed] [Google Scholar]
- 25.Flaherty KR, Toews GB, Lynch JP, 3rd, Kazerooni EA, Gross BH, Strawderman RL, Hariharan K, Flint A, Martinez FJ. Steroids in idiopathic pulmonary fibrosis: a prospective assessment of adverse reactions, response to therapy, and survival. Am J Med. 2001;110:278–82. doi: 10.1016/s0002-9343(00)00711-7. [DOI] [PubMed] [Google Scholar]
- 26.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 27.Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–15. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 28.Hubbard RB, Smith C, Le Jeune I, Gribbin J, Fogarty AW. The association between idiopathic pulmonary fibrosis and vascular disease: a population-based study. Am J Respir Crit Care Med. 2008;178:1257–61. doi: 10.1164/rccm.200805-725OC. [DOI] [PubMed] [Google Scholar]