

Abstract
Comparing the structures of proteins is crucial to gaining insight into protein evolution and function. Here, we align the sequences of multiple protein structures by a dynamic programming optimization of a scoring function that is a sum of an affine gap penalty and terms dependent on various sequence and structure features (SALIGN). The features include amino acid residue type, residue position, residue accessible surface area, residue secondary structure state and the conformation of a short segment centered on the residue. The multiple alignment is built by following the ‘guide’ tree constructed from the matrix of all pairwise protein alignment scores. Importantly, the method does not depend on the exact values of various parameters, such as feature weights and gap penalties, because the optimal alignment across a range of parameter values is found. Using multiple structure alignments in the HOMSTRAD database, SALIGN was benchmarked against MUSTANG for multiple alignments as well as against TM-align and CE for pairwise alignments. On the average, SALIGN produces a 15% improvement in structural overlap over HOMSTRAD and 14% over MUSTANG, and yields more equivalent structural positions than TM-align and CE in 90% and 95% of cases, respectively. The utility of accurate multiple structure alignment is illustrated by its application to comparative protein structure modeling.
Keywords: multiple structure alignment, dynamic programming, guide tree, RMSD, structure overlap
Introduction
Alignment of the 3D structures of proteins is of significant importance in structural biology, because it helps categorize known structures to establish evolutionary and/or functional relationships (Mizuguchi et al., 1998; Brenner et al., 2000; Marti-Renom et al., 2001; Sujatha et al., 2001; Bhaduri et al., 2004; Wilson et al., 2009). As for any comparison, protein structure alignment can also be seen as an optimization of a scoring function, which in this case depends on the structures of the compared proteins. A frequently used scoring function is the number of residues superposed within a certain cutoff distance, although more complex functions including multiple sequence and structure features have also been described (Sali and Blundell, 1990; Taylor, 1999). A number of different optimization techniques have been used (Holm and Sander, 1995; Shindyalov and Bourne, 1998; Taylor, 1999; Ortiz et al., 2002) to optimize the scoring functions for structure comparison, most prominently dynamic programming (Taylor, 1999).
Most known protein structures are related to a number of other known structures (Murzin et al., 1995; Orengo et al., 1997), highlighting the need for methods that can simultaneously compare multiple structures. Although most methods can align only pairs of structures, methods such as MNYFIT (Sutcliffe et al., 1987), COMPARER (Sali and Blundell, 1990), MULTIPROT (Shatsky et al., 2004), CE-MC (Guda et al., 2004), MUSTANG (Konagurthu et al., 2006), MASS (Dror et al., 2003), MAMMOTH-mult (Lupyan et al., 2005) and MATT (Menke et al., 2008) can also align multiple structures. Multiple alignments are usually superior to pairwise alignments because they more accurately describe the variations within and between groups of related protein structures. Multiple alignments can help assess structural similarity and identify regions of conformational flexibility. The conformations of the flexible regions, usually loops, may provide insight into functional aspects of proteins, e.g. the substrate specificity of different serine proteases is governed by the conformation of the binding loops (Hedstrom, 2002). Multiple structure alignments may also result in more accurate comparative protein structure models than pairwise alignments (Fernandez-Fuentes et al., 2007; Chakravarty et al., 2008; Larsson et al., 2008). The utility of multiple templates for comparative modeling hinges on the accuracy of their multiple structure alignment, which is constructed before aligning it with the target sequence (Chakravarty et al., 2008).
Here, we describe SALIGN, an automated dynamic programming method for creating multiple structure alignments. A major motivation was to create alignments of multiple template structures for comparative modeling by satisfaction of spatial restraints, as implemented in MODELLER (Sali and Blundell, 1993). To obtain spatial restraints for modeling, both inter-residue distances and residue dihedral angles are transferred from the template structures to the target sequence aligned with the templates. Therefore, the more conserved are these features in the multiple template alignment, the more accurate is the corresponding comparative model. As a consequence, SALIGN was developed to construct multiple structure alignments that take into consideration both sequence and structure features.
We first describe the computation of pairwise and multiple alignments of structures using SALIGN (Materials and methods). Next, we compare its accuracy with those of several other state-of-the-art methods and illustrate its use in comparative modeling with an example of a model built using multiple templates (Results). Finally, we discuss potential limitations and improvements of the method (Discussion).
Materials and methods
SALIGN is implemented in the program MODELLER version 9v7 (http://salilab.org/modeller/) (Sali and Blundell, 1993). Although the SALIGN algorithms to align sequences as well as sequences with structures have been described elsewhere (Marti-Renom et al., 2004; Madhusudhan et al., 2006), here we describe the alignment of multiple protein structures. SALIGN is inspired by COMPARER (Sali and Blundell, 1990); however, unlike COMPARER, SALIGN utilizes an iterative procedure that renders it insensitive to parameter values. It is benchmarked on a large multiple structure alignment database, and is optimized for alignment accuracy. SALIGN creates pairwise alignments by dynamic programming using a scoring matrix that is a linear combination of sequence and structure feature distances. Multiple alignments are then constructed by assembling individual pairwise alignments. To maximize alignment accuracy, this procedure is carried out in two stages: first, an initial alignment is created, followed by an iterative refinement in the second stage (Fig. 1). The following sections describe the procedure in more detail.
Fig. 1.

SALIGN schema for multiple structure alignment. See Materials and methods.
Measures of alignment accuracy
For a superposition of two structures, the structure overlap (SO) is defined as the percentage of aligned residues that are within a given cutoff distance; the normalization factor is the length of the shorter of the two sequences. By default, SO at a cutoff distance of 3.5 Å is used (SO3.5).
For multiple structure alignments, the SO is the average SO for all pairwise alignments implied by the multiple alignment. We then quantify the accuracy of a multiple structural alignment by the average structural overlap at cutoffs of 1, 2, 3 and 4 Å (the quality score), which is similar to the GDT_TS score (Zemla et al., 2001).
Pairwise alignment of protein structures
SALIGN uses standard linear dynamic programming to align pairs of structures or ‘sub-alignments’ (i.e. a fixed previously obtained alignment of multiple structures). The scoring matrix for dynamic programming is a linear combination of six distance matrices, and the gap penalty is a linear function of gap length, depending on the gap opening and extension penalties (below). Each distance matrix contains distances (dissimilarities) in a sequence or structure feature between all inter-molecular pairs of residue positions in the two compared structures or sub-alignments (below). The final scoring matrix D is
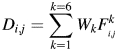 |
(1) |
where Wk is the weight associated with feature distance Fki,j when aligning residue positions i and j.
Sequence and structure features
The features include amino acid residue type, residue position, residue accessible surface area, residue secondary structure state and the conformation of a short segment centered on the residue. In principle, any feature of protein sequence and/or structure can be utilized to create the dynamic programming matrix for subsequent generation of the optimal pairwise alignment, as long as the feature is attributable to individual amino acid residues and a measure of distance can be defined between different states of the feature. Next, we briefly describe the six feature distances used in this study.
Residue type
Feature distance 1 is the dissimilarity version of the BLOSUM62 matrix (Henikoff and Henikoff, 1992).
Residue spatial position
Feature distance 2 is the Euclidean distance between pairs of aligned residues. The distance is computed between one selected atom (typically the Cα atom) from each residue. Because the distance between the atoms depends on the relative positions and orientations of the compared structures, this feature is used in refining an existing alignment (Sali and Blundell, 1990). The dynamic programming scoring matrix created by this feature distance is used to generate a new structure alignment from which the feature distances are recomputed. The refinement of an initial alignment progresses iteratively until convergence (number of structurally equivalent positions does not change between iterations and the change in the relative orientation of the fitted structures is small). The gap penalties used in this step are different from those used in generating the initial and final alignments (below).
Residue solvent accessibility
Feature distance 3 is the difference in side-chain solvent accessibility. Residues are categorized into three side-chain accessibility classes based on their percentage side-chain solvent accessibility (s): buried (s < 15%), semi-exposed (s is between 15% and 30%) and exposed (s > 30%). The value of the distance is 0 between identical classes and for residue pairs for which the absolute difference in their s values is <5%, 1 for neighboring classes and 2 for a buried-exposed match.
Residue secondary structure state
Feature distance 4 is the residue secondary structure state, distinguishing between α-helix, β-strand and other. The feature distance is 0 for equal states, 1 for ‘helix’ or ‘strand’ matched to the ‘other’ state and 2 for ‘helix’ matched to ‘strand’.
Residue local conformation
Feature distance 5 measures local conformational difference by the distance root-mean-square deviation (dRMSD) between the selected atoms (by default, Cα atoms) from segments of five residues centered on the two matched residues.
User specified feature
Feature distance 6 is a user specified distance for which an external matrix that describes the position–position dissimilarity can be input. In the current study, this feature distance is not used.
Averaging feature distances
When constructing a multiple alignment, each side of the alignment can have more than one structure (i.e. a sub-alignment). When two such blocks of structures are compared with one another, individual feature distances are computed either by calculating the distance between averaged features or by averaging the feature distances over all possible individual comparisons between structures in each of the two blocks. Specifically, the residue spatial position feature is calculated by first averaging the coordinates of the selected atoms in each block, followed by computing the Euclidean distance between the averaged structures. The residue–residue substitution score, the solvent accessibility distance, the secondary structure state difference and the dRMSD are all averaged over pairwise comparisons between structures in one block and those in the other block.
Gap penalties
SALIGN uses two sets of affine gap penalties:
| (2) |
| (3) |
Gap_1D [Eq. (2)] penalizes the creation (u1D) and extension (v1D) of gaps of length l in the amino acid sequence, and is used in the creation of initial alignments (below). Gap_3D [Eq. (3)] is the equivalent gap penalty for the refinement stage (below), penalizing the creation and extension of gaps when using the residue spatial position feature. For instance, when v3D = 1.5, pairs of positions are identified as equivalent when they have their selected atoms at most two times this value (3 Å) apart in the current superposition.
Multiple alignment following a guide tree
SALIGN uses the ‘guide-tree’ algorithm for collating pairwise alignments into a multiple alignment (Feng and Doolittle, 1987). Given a set of N proteins, N(N− 1)/2 all-against-all pairwise alignments are first computed. From the resulting matrix of alignment scores, a tree (dendrogram) is constructed (Fitch and Margoliash, 1967). A multiple alignment is then computed by progressively aligning pairs of ‘sub-alignments’, following the tree, starting with the closest pair of structures.
Alignment iterations
To achieve the best possible final alignment, the entire process of constructing pairwise alignments (and, if necessary, combining them into a multiple alignment) is iterated (Fig. 1). A set of prospective initial alignments is first built using a dynamic programming matrix consisting of only the residue substitution and local conformation features (features 1 and 5), sampling over a range of gap penalties and dynamic programming matrix offsets (Table I). Of the sampled alignments, the one with the best SO3.5 is chosen as the initial alignment. If no alignment yields an SO3.5 of at least 70%, the pool of initial alignments is widened by another search using only the residue local conformation feature (feature 5) and sampling over a wider range of parameters. The initial alignment is then refined using a scoring matrix constructed using the spatial position feature (feature 2) alone to obtain the final alignment; this refinement hinges crucially on the initial alignment.
Table I.
Gap penalty and dynamic programming matrix offset values explored at each alignment stage
| Feature weights | Alignment stage | Alignment type | Parameter values explored |
|---|---|---|---|
| (1, 0, 0, 0, 1, 0) | Initial | Local | 1D gap opening −150, −100, −50, 0 |
| 1D gap extension −50, 0 | |||
| Matrix offset varied from −3 to 0 in steps of 0.3 | |||
| (0, 0, 0, 0, 1, 0) | Initial | Local | 1D gap opening varied from 0 to 2.2 in steps of 0.3 |
| 1D gap extension varied from 0.1 to 2.3 in steps of 0.3 | |||
| Matrix offset varied from −3 to 0 in steps of 0.3 | |||
| (0, 1, 0, 0, 0, 0) | Final | Global | 3D gap opening 0, 1, 2, 3 |
| 3D gap extension 2, 3, 4, 5 |
The alignment procedure is iterative, exploring multiple values for both the creation of initial alignments and their final refinement.
Local and global alignments
SALIGN can use both the Needleman–Wunsch (Needleman and Wunsch, 1970) global alignment algorithm and the Smith–Waterman local alignment algorithm (Smith and Waterman, 1981). In this study, the initial alignments are local, whereas their refinement relies on global alignment.
Data sets
The accuracy of the multiple alignments was tested using the 402 HOMSTRAD alignments (Mizuguchi et al., 1998) (February 2007 release) that consisted of three or more proteins. For the pairwise alignment benchmark, the 9539 pairwise alignments implied by these HOMSTRAD family alignments were culled to a smaller set of 1204 pairwise alignments, by including only alignments with an RMSD between 2.0 and 3.0 Å and an SO3.5 between 20% and 70%. Multiple structure alignments of the HOMSTRAD families produced by SALIGN are available at http://salilab.org/salign/.
Results
We first establish the accuracy of SALIGN to align pairs of structures. The SALIGN algorithm was applied to pairs of whole PDB chains for each of the 1204 pairwise alignments in the benchmark set, using as the initial alignment an ungapped ‘alignment’ of the two chains. For comparison with other state-of-the-art protein structure alignment programs, the same PDB chain pairs were given to TM-align (Zhang and Skolnick, 2005) and CE (Shindyalov and Bourne, 1998), run with default parameters. The accuracies of the resulting alignments were compared by calculating the number of equivalent structural positions at 3.5 Å (Fig. 2). Of the 1204 alignments, SALIGN is equal to or better than TM-align and CE in 1086 (90%) and 1147 cases (95%), respectively.
Fig. 2.

Comparison of the accuracies of 1204 pairwise alignments obtained from SALIGN with those from TM-align and CE. Alignment accuracy is plotted for all three methods as the number of structurally equivalent Cα positions at a cutoff of 3.5 Å.
Next, we test SALIGN on multiple alignments. For each of the 402 HOMSTRAD families with three or more members, the multiple alignment was calculated with SALIGN. The HOMSTRAD family alignment, with all gaps removed, was used as the initial alignment. The resulting alignments were compared against the original HOMSTRAD alignments and those calculated using the MUSTANG program. SALIGN is significantly better than both alignment methods, improving the average structural overlap score by 15% compared with HOMSTRAD and 14% compared with MUSTANG, when averaged over all 402 families (Fig. 3).
Fig. 3.

Comparison of the accuracies of multiple alignments obtained from SALIGN with those from MUSTANG and HOMSTRAD. For 402 HOMSTRAD families with three or more members, the SALIGN quality score is compared with those of HOMSTRAD and MUSTANG. SALIGN is significantly more accurate than both alignment methods, improving the quality score by 15% on average compared with HOMSTRAD and 14% compared with MUSTANG.
By design, the accuracy of SALIGN multiple structure alignments depends on the accuracy of pairwise alignments. Pairwise alignments implied by multiple alignments are on average less accurate than those constructed directly, in agreement with a previous study (Raghava et al., 2003). To elaborate, we calculated the quality score for each pairwise alignment implied by the 402 SALIGN multiple alignments (without realignment). These scores were then compared against alignments obtained from using SALIGN on the pairs alone (Fig. 4). In 5992 cases (from 347 of the 402 families), the pairwise alignments implied by the SALIGN multiple alignments are worse than the corresponding SALIGN pairwise alignments. Interestingly, there are also 1353 cases (from 309 of the 402 families) where a pairwise alignment implied by the SALIGN multiple alignment improves upon the quality score of the direct pairwise alignment by SALIGN.
Fig. 4.

Comparison of SALIGN pairwise alignments with pairwise alignments implied by multiple alignments. The quality score of each pairwise alignment from all of the SALIGN multiple alignments is compared against that of each SALIGN direct pairwise alignment.
Finally, the utility of multiple structure alignments for comparative modeling was investigated. For a given HOMSTRAD family, a multiple alignment was constructed using SALIGN as detailed above. The last sequence in the alignment was arbitrarily designated as the target sequence for comparative modeling, whereas the other structures were the templates. Models of the target were then built with the standard ‘automodel’ protocol in MODELLER using (i) all templates simultaneously and (ii) each of the templates individually. A model was also constructed using multiple templates with the HOMSTRAD alignment. The accuracy of each model was assessed by SO3.5 with respect to the native structure. For example, when applied to the ‘hormone_rec’ HOMSTRAD family, the model of 1a28A using all four of the other structures (1lbdA, 2lbdA, 1prgA and 3ertA) resulted in 223 equivalent positions with a Cα RMSD error of 1.64 Å. In contrast, the model built using the HOMSTRAD multiple alignment resulted in only 203 equivalent positions with a Cα RMSD error of 1.50 Å. When building models using the templates individually, 1lbdA, 2lbdA, 1prgA and 3ertA yielded 158 (Cα RMSD 1.67 Å), 182 (1.64 Å), 184 (1.77 Å) and 193 (1.44 Å) equivalent positions, respectively. In this example, no separate procedure was used to align the target with template(s). The target–template alignments were those implied by the SALIGN and HOMSTRAD multiple structure alignments, to focus on the impact of using multiple versus single templates without considering sequence–structure alignment errors.
Discussion
We have described and tested a dynamic programming method to construct multiple structure alignments (SALIGN). The scoring function is a sum of an affine gap penalty and distance terms dependent on various sequence and structure features. These features include amino acid residue type, residue position, residue accessible surface area, residue secondary structure state and the conformation of a short segment centered on the residue. SALIGN can in principle include other residue features in constructing the dynamic programming scoring matrix (Sali and Blundell, 1990).
SALIGN was compared against TM-align and CE for pairwise alignment accuracy, and against HOMSTRAD and MUSTANG for multiple alignment accuracy. These state-of-the-art methods have been carefully benchmarked by their authors against many other methods not assessed against SALIGN in the current study (Holm and Sander, 1995; Kihara and Skolnick, 2003; Guda et al., 2004; Ochagavia and Wodak, 2004; Shatsky et al., 2004; Ye and Godzik, 2005).
We showed that SALIGN performs comparably or better than TM-align and CE on pairs of structures (Fig. 2). SALIGN was then tested for its accuracy of multiple structure alignments over a data set of 402 HOMSTRAD structure families with three or more structures. SALIGN outperformed HOMSTRAD and MUSTANG (in terms of quality score) in 376 and 375 families, respectively. Of these, SALIGN had quality scores that were more than 5% points better in 159 and 161 cases, respectively. By the same measure, SALIGN underperformed HOMSTRAD and MUSTANG in only 10 and 12 families, respectively. Of these, SALIGN underperformed HOMSTRAD and MUSTANG by more than 5 points in two and three cases, respectively; in all other underperforming cases, the quality score difference was <2 points. In the three relative failures of SALIGN (the DEAD, PH and tRNA-synt_2b families), the reason for poor accuracy can be attributed to one particular structure being misaligned in the multiple alignment in each of the families. This problem could in principle be fixed by a more exhaustive search over the parameter space for the globally optimal alignment.
The conservation of the features across protein structure families varies. SALIGN hence samples different linear combinations of feature distances to construct alignments (Table I) because a certain linear combination that yields optimal results for one family may not do so when applied to a different family. For instance, the best initial multiple alignment for the HOMSTRAD family ‘igcon’ was obtained using a linear combination of the residue type and local conformation features, whereas for the HOMSTRAD family ‘ghf18’, the best initial alignment was obtained when only the residue local conformation measure was considered. If only a single optimized set of parameters were used in generating the alignments, the quality score for approximately one half of the alignments would be worse than that in HOMSTRAD (data not shown). Clearly, using the quality score to discern between alignments produced by different parameter sets significantly improves the multiple alignment accuracy. One single initial alignment may sample as many as 720 different parameter values (Table I). The corresponding increase in multiple alignment accuracy comes at a price of computational time, e.g. an alignment of five 280-residue structures may take up to 12 min of CPU time.
We illustrated the efficacy of using SALIGN as a tool to align multiple templates in a comparative modeling exercise. The sequence of 1a28A was chosen as the target sequence with 1ldbA, 2ldbA, 1prgA and 3ertA serving as templates. The model built using multiple templates gave the best sequence coverage (223 residues at 1.64 Å Cα RMSD). No single template could cover as many residues and produce models that were as close to the native structure in terms of RMSD error. The model built using the HOMSTRAD multiple alignment had a marginally better RMSD value of 1.50 Å, but covered 20 residues fewer.
Individual pairwise alignments implied by the multiple alignment can be different from direct pairwise alignments. For SALIGN, 5992 and 1353 implied pairwise alignments (out of 8909) are slightly less accurate and slightly more accurate, respectively, than the direct pairwise alignments (Fig. 4). The accuracy of the implied pairwise alignments can be compromised as a result of averaging structure features and distances during combination of sub-alignments to form a multiple alignment. Different strategies to combine sub-alignments may need to be explored to further improve the accuracy of the SALIGN multiple alignments. When an implied pairwise alignment is more accurate than the direct pairwise alignment, it is mostly because the parameters used in generating the direct pairwise alignment (Table I) were suboptimal. A wider search in the parameter space should help offset this discrepancy, at the cost of a longer computational time.
SALIGN is already used for the construction of the DBAli database (Marti-Renom et al., 2001, 2007) to produce multiple structure alignments of its 11 605 structure families (http://salilab.org/DBAli/ and http://sgu.bioinfo.cipf.es/). Although we have focused here on the accuracy of multiple structure alignments, the method can in principle also be used to detect structural similarity in the first place. Our immediate focus, however, has been the use of SALIGN to generate multiple structure alignments for comparative modeling. SALIGN also completes the alignment suite in MODELLER, which now consists of different methods to perform multiple sequence–sequence, sequence–structure and structure–structure alignments. The corresponding web server will be available soon at http://salilab.org/salign/.
Funding
This work was supported by the National Institutes of Health (R01 GM54762-11, U54 GM62529).
Acknowledgements
We are grateful to Hannes Braberg, Fred Davis and other members of the Sali lab who helped test SALIGN and gave insightful criticisms. We also thank Ursula Pieper and Keren Lasker for helpful discussions and reading the manuscript.
Footnotes
Edited by Valerie Daggett
References
- Bhaduri A., Pugalenthi G., Sowdhamini R. BMC Bioinformatics. 2004;5:35. doi: 10.1186/1471-2105-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S.E., Koehl P., Levitt M. Nucleic Acids Res. 2000;28:254–256. doi: 10.1093/nar/28.1.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty S., Godbole S., Zhang B., Berger S., Sanchez R. BMC Struct. Biol. 2008;8:31. doi: 10.1186/1472-6807-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror O., Benyamini H., Nussinov R., Wolfson H. Bioinformatics. 2003;19(Suppl. 1):i95–i104. doi: 10.1093/bioinformatics/btg1012. [DOI] [PubMed] [Google Scholar]
- Feng D.F., Doolittle R.F. J. Mol. Evol. 1987;25:351–360. doi: 10.1007/BF02603120. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fuentes N., Rai B.K., Madrid-Aliste C.J., Fajardo J.E., Fiser A. Bioinformatics. 2007;23:2558–2565. doi: 10.1093/bioinformatics/btm377. [DOI] [PubMed] [Google Scholar]
- Fitch W.M., Margoliash E. Science. 1967;155:279–284. doi: 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- Guda C., Lu S., Scheeff E.D., Bourne P.E., Shindyalov I.N. Nucleic Acids Res. 2004;32:W100–W103. doi: 10.1093/nar/gkh464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L. Chem. Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- Henikoff S., Henikoff J.G. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Sander C. Trends Biochem. Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Kihara D., Skolnick J. J. Mol. Biol. 2003;334:793–802. doi: 10.1016/j.jmb.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Konagurthu A.S., Whisstock J.C., Stuckey P.J., Lesk A.M. Proteins. 2006;64:559–574. doi: 10.1002/prot.20921. [DOI] [PubMed] [Google Scholar]
- Larsson P., Wallner B., Lindahl E., Elofsson A. Protein Sci. 2008;17:990–1002. doi: 10.1110/ps.073344908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupyan D., Leo-Macias A., Ortiz A.R. Bioinformatics. 2005;21:3255–3263. doi: 10.1093/bioinformatics/bti527. [DOI] [PubMed] [Google Scholar]
- Madhusudhan M.S., Marti-Renom M.A., Sanchez R., Sali A. Protein Eng. Des. Sel. 2006;19:129–133. doi: 10.1093/protein/gzj005. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M.A., Ilyin V.A., Sali A. Bioinformatics. 2001;17:746–747. doi: 10.1093/bioinformatics/17.8.746. [DOI] [PubMed] [Google Scholar]
- Marti-Renom M.A., Madhusudhan M.S., Sali A. Protein Sci. 2004;13:1071–1087. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom M.A., Pieper U., Madhusudhan M.S., Rossi A., Eswar N., Davis F.P., Al-Shahrour F., Dopazo J., Sali A. Nucleic Acids Res. 2007;35:W393–W397. doi: 10.1093/nar/gkm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke M., Berger B., Cowen L. PLoS Comput. Biol. 2008;4:e10. doi: 10.1371/journal.pcbi.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi K., Deane C.M., Blundell T.L., Overington J.P. Protein Sci. 1998;7:2469–2471. doi: 10.1002/pro.5560071126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A.G., Brenner S.E., Hubbard T., Chothia C. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Needleman S.B., Wunsch C.D. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Ochagavia M.E., Wodak S. Proteins. 2004;55:436–454. doi: 10.1002/prot.10587. [DOI] [PubMed] [Google Scholar]
- Orengo C.A., Michie A.D., Jones S., Jones D.T., Swindells M.B., Thornton J.M. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Ortiz A.R., Strauss C.E., Olmea O. Protein Sci. 2002;11:2606–2621. doi: 10.1110/ps.0215902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghava G.P., Searle S.M., Audley P.C., Barber J.D., Barton G.J. BMC Bioinformatics. 2003;4:47. doi: 10.1186/1471-2105-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. J. Mol. Biol. 1990;212:403–428. doi: 10.1016/0022-2836(90)90134-8. [DOI] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Shatsky M., Nussinov R., Wolfson H.J. Proteins. 2004;56:143–156. doi: 10.1002/prot.10628. [DOI] [PubMed] [Google Scholar]
- Shindyalov I.N., Bourne P.E. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Sujatha S., Balaji S., Srinivasan N. Bioinformatics. 2001;17:375–376. doi: 10.1093/bioinformatics/17.4.375. [DOI] [PubMed] [Google Scholar]
- Sutcliffe M.J., Haneef I., Carney D., Blundell T.L. Protein Eng. 1987;1:377–384. doi: 10.1093/protein/1.5.377. [DOI] [PubMed] [Google Scholar]
- Taylor W.R. Protein Sci. 1999;8:654–665. doi: 10.1110/ps.8.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D., Pethica R., Zhou Y., Talbot C., Vogel C., Madera M., Chothia C., Gough J. Nucleic Acids Res. 2009;37:D380–D386. doi: 10.1093/nar/gkn762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Godzik A. Bioinformatics. 2005;21:2362–2369. doi: 10.1093/bioinformatics/bti353. [DOI] [PubMed] [Google Scholar]
- Zemla A., Venclovas C., Moult J., Fidelis K. Proteins. 2001;45(Suppl. 5):13–21. doi: 10.1002/prot.10052. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Skolnick J. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]