

Abstract
Individuals with autism spectrum disorders have impairments in social, communicative, and behavior development that are often accompanied by abnormalities in cognitive functioning, learning, attention, and sensory processing. In this report, we describe a 3-year-old male child with an autism spectrum disorder who carries a 2Mb deletion of chromosome 1q42. Array comparative genome hybridization revealed that this deletion involves at least three genes—DISC1, DISC2, and TSNAX—which have been found to be associated with neuropsychiatric disorders and are likely to play key roles in normal CNS development. Further studies revealed that the deletion was inherited from his unaffected mother. This suggests that other genetic and/or environmental factors, some of which may be sex specific, may modify the phenotypic effects of this deletion. While this case provides evidence for the potential role of DISC1, DISC2, and TSNAX in the development of autism spectrum disorders, it is equally clear that caution must be taken when providing families with prognostic information and genetic counseling regarding such deletions.
Keywords: DISC1, DISC2, TSNAX, autism spectrum disorder, chromosome 1q42
INTRODUCTION
Chromosome 1q42 contains at least three genes which have been implicated in neuropsychiatric disorders; disrupted in schizophrenia 1 and 2 (DISC1, DISC2), and translin-associated factor X (TSNAX). The first evidence of the role of DISC1 and DISC2 in human disease came from a report of a large Scottish family in which a balanced translocation disrupting these genes segregated with schizophrenia and related psychiatric disorders [St. Clair et al., 1990; Millar et al., 2000]. Miller et al. proposed that alteration of DISC1 and/or DISC2 activity caused by truncation and/or abnormal regulation of expression was causally linked to the psychiatric illness seen in translocation carriers. Since then, SNPs in DISC1 have been associated with schizophrenia, schizoaffective disorder, bipolar disorder, major depression and, most recently, autism and Asperger syndrome [Hodgkinson et al., 2004; Cannon et al., 2005; Thomson et al., 2005; Hashimoto et al., 2006; Kilpinen et al., 2008; Saetre et al., 2008]. DISC2, which overlaps exon 9 of DISC1 but is transcribed in the opposite direction, is thought to be a noncoding antisense transcript which negatively regulates DISC1 [Millar et al., 2000].
TSNAX (previously referred to as TRAX) is thought to play a role in dendritic trafficking of RNA molecules in neurons, a process thought to be critical in synaptic plasticity [Muramatsu et al., 1998; Li et al., 2008]. Haplotypes within and overlapping TSNAX have been found to be associated with schizophrenia, and marker D1S251, which lies between TSNAX and DISC1, has been shown to segregate with schizophrenia in Taiwanese families [Hennah et al., 2003; Hwu et al., 2003; Cannon et al., 2005].
Here we report a 3-year-old male with developmental delay and autistic behaviors who has an interstitial of deletion 1q42. The deletion spans approximately 2 Mb, involves DISC1, DISC2, and TSNAX and was inherited from his nonaffected mother. This case provides support for the possible involvement of these genes in the development of autism spectrum disorders and also suggests that other genetic and/or environmental factors act to modify the phenotype associated with this deletion.
CLINICAL REPORT
The patient is a 3-year-old male born to a 34-year-old Caucasian mother and a 56-year-old Pakistani father. He has four sisters—ages 3, 10, 12, and 16—all of whom are in good health. There is no family history of consanguinity, psychiatric disorders, or autism.
Prenatal ultrasound examinations revealed a varicose vein in his umbilical cord and concern for possible intrauterine growth retardation for which labor was induced at 36 weeks gestation. His birth weigh was 1,960 g (between the 10th and 25th centile). No dysmorphic features were noted and he was discharged to home on day 4 of life.
Although the majority of his gross motor developmental milestones were normal—including crawling at 6–8 months and walking at 13–14 months—he was unable to sit without support until almost a year of age. This prompted referral to an early childhood intervention program through which he received physical, occupational, and speech therapy starting at 11 months of age. His fine motor development was delayed with development of a pincer grasp occurring at 2.5 years of age. He continues to prefer to use a raking motion when picking up small objects and has difficulty with zippers, buttons, and scissors. His language development was similarly delayed with his first word being said at 2–2.5 years of age. He does not speak in full sentences but does use short two to three word phrases. There has been no evidence of regression.
He was evaluated by his local school district at 2 years, 8 months of age using the Childhood Autism Rating Scale (CARS), the Autism Behavior Checklist Record (ABC), and the Gilliam Autism Rating Scale and the Behavior Assessment System for Children, Second Edition (BASC-2) and was felt to demonstrate characteristics consistent with a diagnosis of Pervasive Developmental Disorder, Not Otherwise Specified (PDD-NOS).
At 3 years, 2 months of age he was evaluated in a neuropsychiatric clinic where his score on the Modified Checklist for Autism in Toddlers was suggestive of an autism spectrum disorder and he was diagnosed with PDD-NOS.
He was then referred to the Clinic for Autistic Spectrum Disorders at Texas Children’s Hospital where an evaluation at 3 years, 6 months of age confirmed the diagnosis of PDD-NOS. During this evaluation, he was noted to have impairments in communication and social interactions—including lack of spontaneous make-believe play, poor eye contact, failure to develop peer relationships appropriate to developmental level, and a lack of social and emotional reciprocity—but he did not consistently demonstrate restricted repetitive and stereotyped patterns of behavior and activities.
On the Bayley Scales of Infant and Toddler Development-Third Edition, he scored well below age expected levels with composite cognitive (75) and social-emotional (75) domain scores within the borderline deficient range and composite language (65) and motor (64) scores within the deficient range. His scores on the Adaptive Behavior Scale were also well below age expected levels. The Gilliam Autism Rating Scale was completed by his parents. Scores indicated that the probability of Autism was “Very Likely”. His mother completed the Child Behavior Checklist and noted clinical elevations on measures of attention problems and aggressive behaviors, and borderline clinical elevations for emotional reactivity. With respect to DSM-Oriented Scales, clinical elevations were noted for Anxiety Problems and Attention-Deficit/Hyperactivity Disorder Problems.
His most recent physical examination was at 3 years, 9 months. His weight was 20.6 kg (97th centile), his height was 109.7 cm (98th centile), and his FOC was 51 cm (75th centile). No dysmorphic features were noted. His neurologic examination revealed slightly increased tone in his lower extremities bilaterally with two beats of clonus in his right lower extremity. He also demonstrated decreased ability to fully dorsiflex his bilateral lower extremities beyond 90°.
Fragile X testing and sequencing of MECP2 were normal. An unenhanced head CT was normal at 3 years of age but an MRI evaluation revealed multifocal scattered areas of gliosis and/or abnormal myelination in the bilateral cerebral white matter of unclear etiology.T1 hyperintense signal abnormalities in the globus pallidi were also noted with minimal to negligible hyperintense signal on the T2 weighted images and no corresponding signal abnormalities were identified on the gradient echo exam. Since T1-signal hyperintensities of the basal ganglia can be associated with manganese deposition caused by hepatic disease, liver enzymes were checked and the patient was found to have mildly elevated AST, GGT, and unconjugated bilirubin levels at 70 U/L (normal 20–60 U/L), 23 U/L (normal 6–19 U/L), and 1.0 (normal <1), respectively, with a normal ALT and alkaline phosphatase and mildly decrease in albumin at 3.4 g/dl (normal 3.7–5.5 g/dl). On a separate occasion, the patient’s ammonia was slightly elevated at 57 μmol/L (normal range 22–48 μmol/L) during an acute illness. Further metabolic evaluation, including measurements of orotic acid, lactate, urine organic acids, plasma amino acids, urine amino acids, and acylcarnitines failed to identify an underlying metabolic disorder.
METHODS AND RESULTS
DNA was isolated from the patient’s whole blood and analyzed for copy number variants on a clinical basis in the Medical Genetics Laboratories at Baylor College of Medicine using Chromosomal Microarray Analysis (CMA) version 7.1. This array comparative genome hybridization (aCGH) analysis revealed a loss in copy number (deletion) in the 1q42.13q42.2 region of the long arm of chromosome 1, spanning a minimum of 1.986 Mb and a maximum of 2.030 Mb. The deletion was confirmed by FISH analysis. Parental FISH analysis identified the same deletion in the patient’s mother.
After appropriate research consent was obtained, a more detailed aCGH analysis was performed on the patient’s DNA using a high-resolution genome-wide aCGH using Human Genome CGH 244 Oligo Microarray Kit G4411B (Agilent, Wilmington, DE) according to the manufacturer’s protocol version 2.0. Arrays were scanned using an Agilent DNA Microarray Scanner. Data were extracted using Feature Extraction Software Version 9.1.3 (Agilent) and analyzed using CGH Analytics 3.4.40 Software (Agilent). Copy number changes were identified with the assistance of the Aberration Detection Method 2 algorithm (threshold 6.0) contained within the CGH Analytics 3.4.40 Software. This analysis revealed that the deletion involved at least 15 known genes including the 5′ portion of DISC1—exons 1 to 10—and all of DISC2 and TSNAX (Fig. 1). The minimum deleted region spanned from 228208265 to 230194378 and the maximum deleted region from 228178451 to 230212442 in genome build hg18. A search of the Database of Genomic Variants (http://projects.tcag.ca/variation/) revealed that several duplications but no deletions associated with DISC1, DISC2, and/or TSNAX have been identified in normal controls.
FIG. 1.
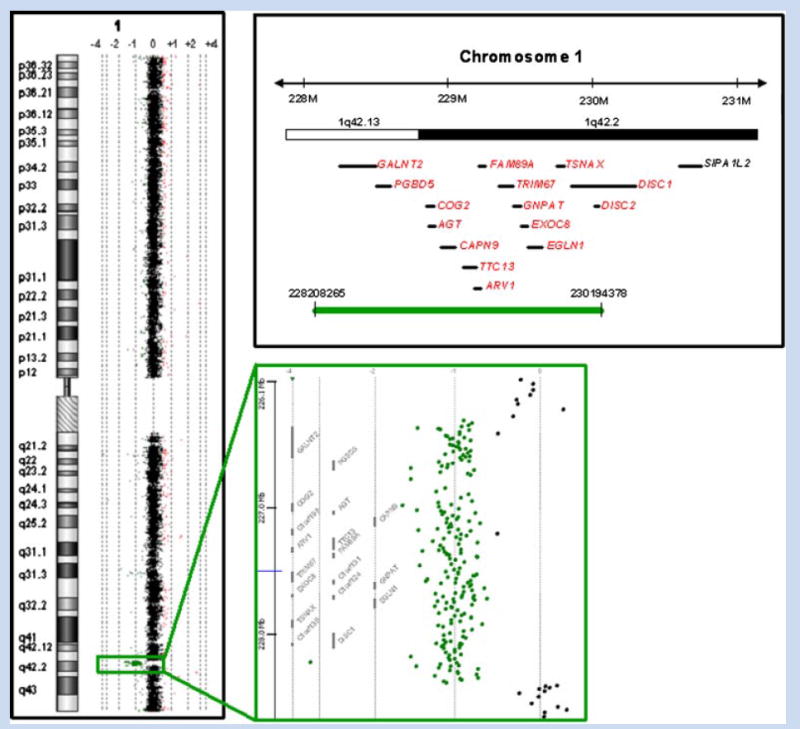
Array comparative genome hybridization data and a schematic representation of the maternally inherited ~2 Mb deletion present in our patient. The deletion encompasses all or portions of at least 15 known genes (shown in red) including the 5′ end of DISC1—exons 1 to 10—and all of DISC2 and TSNAX. The green bar represents the minimal deleted region. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.].
One possible cause for the discrepancy between the patient’s phenotype and that of his unaffected mother is that the copies of DISC1 and/or TSNAX that he inherited from his father contain deleterious changes. To investigate this possibility we sequenced the coding regions of DISC1 and TSNAX in our patient. Two nonsynonymous changes—L607F and E661K—were identified in DISC1. As expected these changes appeared to be homozygous due to the absence of a maternal allele. Neither of these changes was listed as a polymorphism in the SNP database. The functional significance of these changes was evaluated using the on-line tool PolyPhen which predicts the impact of an amino acid substitution on protein structure and function using physical and comparative considerations; both changes were predicted to be benign. No changes were identified in the coding sequencing of TSNAX. DISC2 was not screened for changes since it is thought to be a noncoding antisense transcript.
DISCUSSION
In this report we describe a patient with PDD-NOS—an autism spectrum disorder—who has a deletion of chromosome 1q42, which involves three genes, DISC1, DISC2, and TSNAX which have been implicated in the development of neuropsychiatric disorders.
Only recently has there been evidence for an association between autism and Asperger syndrome and DISC1 [Kilpinen et al., 2008]. DISC1 interacts with a number of proteins that play a role in brain development and function including PDE4B, PDE4D, LIS1, NDE1, and NDEL1 [Bradshaw et al., 2008]. PDE4B was found to be disrupted by a balanced translocation in a patient with schizophrenia and a cousin diagnosed with a psychotic disorder and, alongwith PDE4D, encodes a phosphodiesterase that degrades cAMP, a second messenger implicated in learning, memory, and mood [Millar et al., 2005]. DISC1, PDE4B, and PDE4D associate with LIS1, NDE1, and NDEL1, which forma complex that binds to dynein, and are known to play critical roles in neuronal migration and cortical development. Bradshaw et al. recently proposed that DISC1 may act as an assembly scaffold for all of these proteins and that the NDE1/NDEL1/LIS1/dynein complex is modulated by cAMP levels via cAMP-dependant protein kinase A (PKA) and PDE4 [Bradshaw et al., 2008].
While little is known about the role of DISC2 in neurodevelopment, it is thought to function as a noncoding antisense transcript which regulates the expression of DISC1 [Millar et al., 2005]. It is possible that haploinsufficiency of this gene may result in dysregulation of the remaining DISC1 gene in our patient.
TSNAX forms a heteromeric RNA-binding complex with the protein Translin which is thought to play a role in dendritic and axonal trafficking of RNA molecules and may also be involved in the regulation of transcription and cell proliferation [Li et al., 2008]. Translin-deficient knockout mice, which lack the TSNAX/Translin RNA-binding complex, have undetectable levels of TSNAX, exhibit sex-specific differences in tests of learning and memory, locomotor activity, anxiety-related behavior, and sensorimotor gating, and reduced levels of the norepinephrine and serotonin—which have prominent roles in regulating fear and anxiety-related behavior [Chennathukuzhi et al., 2003; Stein et al., 2006].
Although it is likely that haploinsufficiency of DISC1, DISC2, and/or TSNAX contributed to the neurologic phenotype of our patient, the same 2Mbdeletion was found in his unaffected mother. This discrepancy could be due to additional loss of function caused by deleterious mutations in our patient’s paternally inherited alleles. However, sequence analysis of the coding regions of our patient’s DISC1 and TSNAX genes revealed no such changes. Another possibility is that other genetic and/or environmental factors played a role in the development of this phenotype in our patient. Wenote that autism spectrum disorders are more prevalent in males than females with a ratio of about 6.8:1 in a recent study [Williams et al., 2008]. Although the etiology of this excess of males has yet to be fully identified, it is possible that these same sex-specific factors could explain why the patient’s mother is asymptomatic.
Several studies have suggested an association with advanced parental age and the development of autism spectrum disorder [Croen et al., 2007; Kolevzon et al., 2007; Durkin et al., 2008]. Given that the patient’s father was 53 at the time of his birth, it is possible that factors associated with advanced paternal age may have also contributed to his neurologic phenotype.
With the increasing use of aCGH in the diagnostic work-up of individuals with neuropsychiatric disorders and as a prenatal screening tool, it is likely that other individuals with deletions of 1q42 involving DISC1, DISC2, and/or TSNAX will be identified. While this case provides evidence for the potential role of these genes in the development of autism spectrum disorders, it is equally clear that caution must be taken when providing families with prognostic information and genetic counseling regarding such deletions that include this region.
References
- Bradshaw NJ, Ogawa F, Antolin-Fontes B, Chubb JE, Carlyle BC, Christie S, Claessens A, Porteous DJ, Millar JK. DISC1, PDE4B, and NDE1 at the centrosome and synapse. Biochem Biophys Res Commun. 2008;377:1091–1096. doi: 10.1016/j.bbrc.2008.10.120. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M, Gasperoni T, Tuulio-Henriksson A, Pirkola T, Toga AW, Kaprio J, Mazziotta J, Peltonen L. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- Chennathukuzhi V, Stein JM, Abel T, Donlon S, Yang S, Miller JP, Allman DM, Simmons RA, Hecht NB. Mice deficient for testis-brain RNA-binding protein exhibit a coordinate loss of TRAX, reduced fertility, altered gene expression in the brain, and behavioral changes. Mol Cell Biol. 2003;23:6419–6434. doi: 10.1128/MCB.23.18.6419-6434.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croen LA, Najjar DV, Fireman B, Grether JK. Maternal and paternal age and risk of autism spectrum disorders. Arch Pediatr Adolesc Med. 2007;161:334–340. doi: 10.1001/archpedi.161.4.334. [DOI] [PubMed] [Google Scholar]
- Durkin MS, Maenner MJ, Newschaffer CJ, Lee LC, Cunniff CM, Daniels JL, Kirby RS, Leavitt L, Miller L, Zahorodny W, Schieve LA. Advanced parental age and the risk of autism spectrum disorder. Am J Epidemiol. 2008;168:1268–1276. doi: 10.1093/aje/kwn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Numakawa T, Ohnishi T, Kumamaru E, Yagasaki Y, Ishimoto T, Mori T, Nemoto K, Adachi N, Izumi A, Chiba S, Noguchi H, Suzuki T, Iwata N, Ozaki N, Taguchi T, Kamiya A, Kosuga A, Tatsumi M, Kamijima K, Weinberger DR, Sawa A, Kunugi H. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum Mol Genet. 2006;15:3024–3033. doi: 10.1093/hmg/ddl244. [DOI] [PubMed] [Google Scholar]
- Hennah W, Varilo T, Kestilä M, Paunio T, Arajärvi R, Haukka J, Parker A, Martin R, Levitzky S, Partonen T, Meyer J, Lönnqvist J, Peltonen L, Ekelund J. Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex-dependent effects. Hum Mol Genet. 2003;12:3151–3159. doi: 10.1093/hmg/ddg341. [DOI] [PubMed] [Google Scholar]
- Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, Malhotra AK. Disrupted in schizophrenia 1 (DISC1): Association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet. 2004;75:862–872. doi: 10.1086/425586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwu HG, Liu CM, Fann CS, Ou-Yang WC, Lee SF. Linkage of schizophrenia with chromosome 1q loci in Taiwanese families. Mol Psychiatry. 2003;8:445–452. doi: 10.1038/sj.mp.4001235. [DOI] [PubMed] [Google Scholar]
- Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, Nieminen-von Wendt T, von Wendt L, Paunio T, Peltonen L. Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry. 2008;13:187–196. doi: 10.1038/sj.mp.4002031. [DOI] [PubMed] [Google Scholar]
- Kolevzon A, Gross R, Reichenberg A. Prenatal and perinatal risk factors for autism: A review and integration of findings. Arch Pediatr Adolesc Med. 2007;161:326–333. doi: 10.1001/archpedi.161.4.326. [DOI] [PubMed] [Google Scholar]
- Li Z, Wu Y, Baraban JM. The Translin/Trax RNA binding complex: Clues to function in the nervous system. Biochim Biophys Acta. 2008;1779:479–485. doi: 10.1016/j.bbagrm.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, Devon RS, Clair DM, Muir WJ, Blackwood DH, Porteous DJ. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, Malloy MP, Chubb JE, Huston E, Baillie GS, Thomson PA, Hill EV, Brandon NJ, Rain JC, Camargo LM, Whiting PJ, Houslay MD, Blackwood DH, Muir WJ, Porteous DJ. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Ohmae A, Anzai K. BC1 RNA protein particles in mouse brain contain two y-, h-element-binding proteins, translin and a 37 kDa protein. Biochem Biophys Res Commun. 1998;247:7–11. doi: 10.1006/bbrc.1998.8657. [DOI] [PubMed] [Google Scholar]
- Saetre P, Agartz I, De Franciscis A, Lundmark P, Djurovic S, Kähler A, Andreassen OA, Jakobsen KD, Rasmussen HB, Werge T, Hall H, Terenius L, Jönsson EG. Association between a disrupted-in-schizophrenia 1 (DISC1) single nucleotide polymorphism and schizophrenia in a combined Scandinavian case-control sample. Schizophr Res. 2008;106:237–241. doi: 10.1016/j.schres.2008.08.024. [DOI] [PubMed] [Google Scholar]
- St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- Stein JM, Bergman W, Fang Y, Davison L, Brensinger C, Robinson MB, Hecht NB, Abel T. Behavioral and neurochemical alterations in mice lacking the RNA-binding protein translin. J Neurosci. 2006;26:2184–2196. doi: 10.1523/JNEUROSCI.4437-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson PA, Wray NR, Millar JK, Evans KL, Hellard SL, Condie A, Muir WJ, Blackwood DH, Porteous DJ. Association between the TRAX/DISC locus and both bipolar disorder and schizophrenia in the Scottish population. Mol Psychiatry. 2005;10:657–668. doi: 10.1038/sj.mp.4001669. [DOI] [PubMed] [Google Scholar]
- Williams E, Thomas K, Sidebotham H, Emond A. Prevalence and characteristics of autistic spectrum disorders in the ALSPAC cohort. Dev Med Child Neurol. 2008;50:672–677. doi: 10.1111/j.1469-8749.2008.03042.x. [DOI] [PubMed] [Google Scholar]