

Abstract

Versatile methods for iridium-catalyzed, kinetic asymmetric substitution of racemic, branched allylic esters are reported. These reactions occur with a variety of aliphatic, aryl, and heteroaryl allylic benzoates to form the corresponding allylic substitution products in high yields (74–96%) with good to excellent enantioselectivity (84–98% ee) with a scope that encompasses a range of anionic carbon and heteroatom nucleophiles. These kinetic asymmetric processes occur with distinct stereochemical courses for racemic aliphatic and aromatic allylic benzoates, and the high reactivity of branched allylic benzoates enables enantioselective allylic substitutions that are slow or poorly selective with linear allylic electrophiles.
Allylic substitution catalyzed by metallacyclic iridium complexes derived from phosphoramidites1 has become a valuable method to prepare enantioenriched small molecules with broad scope by the addition of carbon2 and heteroatom3 nucleophiles to allylic esters. Although many linear allylic carbonates react to form products with high enantiomeric excess (eq 1), iridium-catalyzed allylic substitution of racemic, branched allylic esters typically occurs with much lower enantioselectivity (eq 2).4 Such allylic esters are attractive substrates for metal-catalyzed allylic substitution because they are readily prepared from a wide range of aldehydes and a vinyl Grignard reagent. Moreover, these allylic esters are less substituted at the alkene unit and typically react with rates that are faster than those of the corresponding linear isomers.4b–c
 |
(1) |
 |
(2) |
To date, enantioselective substitution of racemic, branched allylic electrophiles has been limited primarily to reactions of stabilized carbon nucleophiles catalyzed by complexes of Mo,5 Pd,6 or Rh7 and a few examples of allylic amination catalyzed by complexes of Rh8 and Pd.9 Examples of iridium-catalyzed allylic substitution of racemic, branched allylic electrophiles that occur with high enantioselectivity are rare. Helmchen reported allylic alkylation of racemic allylic acetates catalyzed by an iridium-phosphoramidite complex, but only one substrate reacted in greater than 80% ee.4a–b In addition, Carreira4c and Alexakis4d reported kinetic resolutions by etherification and alkylation of racemic, branched allylic carbonates or esters with catalysts generated in situ from [Ir(COE)2Cl]2 and a chiral diene ligand or [Ir(COD)Cl]2 and a chiral phosphoramidite ligand. However, these allylic etherifications occur with widely varying levels enantioselectivity, and the one example of allylic alkylation occurred in 84% ee. Palladium-catalyzed isomerization of branched allylic esters to linear allylic esters, followed by iridium-catalyzed enantioselective, allylic substitution of the linear isomer has been reported, 10 but this strategy requires removal of the palladium catalyst prior to the allylic substitution step and is limited to reactions of aromatic allylic esters that fully rearrange to the linear isomer.
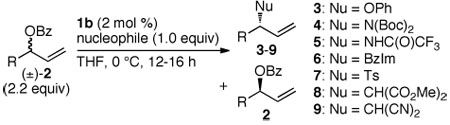
Here, we report enantioselective allylic substitution of racemic, branched allylic benzoates with a variety of anionic carbon and heteroatom nucleophiles in the presence of a single-component, metallacyclic iridium complex to form substitution products with high enantioselectivity. These kinetic asymmetric processes occur with distinct stereochemical courses for racemic aliphatic and aromatic allylic benzoates, and the higher reactivity of the branched allylic esters enables transformations of sterically demanding electrophiles.
Recently published mechanistic studies on allylic amination showed that enantiomeric, branched allylic amines coordinate to a metallacyclic iridium catalyst with much different binding constants. 11 These data implied that the metallacyclic iridium catalyst should select for reaction of one enantiomer of a racemic, branched allylic ester and form a substitution product with high enantiomeric excess, if the allylic substitution occurs with a high degree of retention of configuration. Prior reactions of enantioen-riched allylic esters with iridium catalysts formed the substitution product with a modest enantiomeric excess,4a–b but reactions of enantioenriched allylic esters catalyzed by metallacyclic iridium complexes could occur with a higher degree of retention of configuration. Such substitutions of racemic, branched allylic esters would contrast with typical kinetic resolutions that leave behind enantioenriched reactant but form products possessing modest enantiopurity.
To test if we could obtain substitution products having high enantiomeric excess from the selective reaction of one enantiomer of a racemic mixture of allylic esters, we conducted reactions of the racemic, branched allylic esters derived from 5-phenylpent-1-en-3-ol with sodium phenoxide (NaOPh) in the presence of our single-component, metallacyclic Ir-phosphoramidite complexes 1a11 and 1b3d (Figure 1). After testing several allylic carbonates and esters, we found that the reaction of NaOPh with 2.2 equivalents of allylic benzoate 2a in the presence of 2 mol % 1b formed allyl phenyl ether 3a in 83% yield and 95% ee at 0 °C (Table 1, entry 1).12
Figure 1.

Metallacyclic iridium-phosphoramidite complexes 1a and 1b.
Table 1.
Ir-Catalyzed Allylic Substitution of Racemic Aliphatic Allylic Benzoatesa
 | |||||
|---|---|---|---|---|---|
| entry | R (2) | nucleophile | product | Yield (%)b |
ee (%)c |
| 1 | BnCH2 (2a) | NaOPh | 3a | 83 | 95 |
| 2 | n-Pr (2b) | NaOPh | 3b | 86 | 92 |
| 3 | i-Pr (2c) | NaOPh | 3c | 76 | 98 |
| 4 | Cy (2d) | NaOPh | 3d | 86 | 96 |
| 5d | t-Bu (2e) | NaOPh | 3e | 88 | 96 |
| 6 | BnCH2 (2a) | LiN(Boc)2 | 4a | 96 | 93 |
| 7 | BnCH2 (2a) | KNHC(O)CF3 | 5a | 74 | 98 |
| 8 | BnCH2 (2a) | NaBzIm | 6a | 84 | 97 |
| 9 | BnCH2 (2a) | NaTs | 7a | 80 | 94 |
| 10 | BnCH2 (2a) | NaCH(CO2Me)2 | 8a | 82 | 94 |
| 11 | BnCH2 (2a) | NaCH(CN)2 | 9a | 77 | 88 |
See Supporting Information for experimental details.
Isolated yield of products 3–9. Branched-to-linear selectivities were >95:5.
The enantiomeric excess of 3–9 was determined by chiral HPLC methods.
Reaction was run in the presence of 4 mol % 1b at 50 °C for 24 h.
Reactions of a series of anionic nucleophiles with aliphatic allylic benzoates are summarized in Table 1. As shown in entries 2–4, NaOPh reacted with branched allylic benzoates 2b–2d (R = n-Pr, i-Pr, or c-hexyl) to form allyl aryl ethers 3b–3d in 76–86% yield and 92–98% ee. The reactions of the branched allylic esters occur faster than the analogous reactions of the linear allylic esters. This higher reactivity allowed for an unusual asymmetric allylic substitution process. The reaction of NaOPh with allylic benzoate 2e in which the allyl unit is substituted with a t-Bu group occurred at 50 °C to set a neopentyl stereocenter in the allyl aryl ether product 3e in 96% ee (entry 5). The reaction of NaOPh with the linear isomer of 2e did not occur under identical or more forcing conditions. Nucleophilic substitutions at neopentyl centers are challenging, and enantioselective substitutions at such a site are particularly rare.13
The enantioselective allylic substitution also occurred with anionic nitrogen, sulfur, and carbon nucleophiles in the presence of catalyst 1b, as summarized in entries 6–11. The reactions of 2a with the ammonia equivalents LiN(Boc)2 and KNHC(O)CF3, the heteroaromatic derivative sodium benzimidazolate (NaBzIm), the sulfur nucleophile sodium p-toluenesulfinate (NaTs), and the sodium salts of the carbon nucleophiles dimethyl malonate and malononitrile occurred with high enantioselectivity (88–98% ee). The resulting products 4a–9a were isolated in good yields (74–96%).
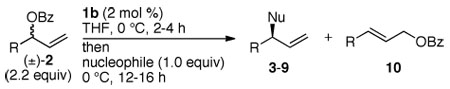
The reactivity of branched aromatic allylic benzoates was distinct from that of branched aliphatic allylic benzoates. Allylic substitution of branched aromatic allylic benzoates in the presence of catalyst 1b occurred in competition with isomerization to the linear isomer. The isomerization of one enantiomer of the allylic benzoate was faster than isomerization of the other, but the competitive processes resulted in reaction mixtures. These mixtures consisted of 1) the (R)-allylic substitution product with modest enantiopurity, 2) the achiral linear aromatic allylic benzoate, and 3) the (S)-enantiomer of the branched allylic benzoate possessing high enantiopurity.
These data are consistent with a system that reacts with the relative rates and equilibria in Scheme 1. The rate constant for nucleophilic attack (k5) on allyliridium complex A is competitive with the rate constant for collapse (k3) to form the linear allylic benzoate from A (k5 ≈ k3); the rate (controlled by K1k3) for the formation of the linear allylic benzoate from (R)-2 is greater than the rate for the formation (controlled by K2k4) of the linear allylic benzoate from (S)-2 (K1k3 ≫ K2k4);14 the rate constant for nucleophilic attack (k6) on allyliridium complex B is greater than the rate constant for collapse (k4) to form the linear allylic benzoate from B. We sought to exploit these relative rates to convert the more reactive (R)-enantiomer of the branched allylic benzoate 2 to the linear isomer, followed by allylic substitution of the remaining (S)-enantiomer of 2 with retention of configuration. This process would lead to the product of allylic substitution with high enantiomeric excess.
Scheme 1.

Strategy for Kinetic Asymmetric Allylic Substitution of Racemic, Branched Aromatic Allylic Benzoates.a
a [Ir] = [Ir(COD)(κ2−L2)(ethylene)] (1b), K1 = k1/k−1, and K2 = k2/k−2
To establish the foundation for this strategy, we evaluated the selectivity factors (s) for kinetic resolution of racemic aromatic allylic benzoates in the absence of a nucleophile and the stereochemical outcome from allylic substitutions of the less reactive enantiomer of the branched allylic benzoates. As anticipated from the preliminary experiment, reaction of the racemic allylic ester from 1-phenylprop-2-en-1-ol formed a mixture of the (S)-enantiomer of the starting material and the achiral linear allylic benzoate with a high s-value. The kinetic resolution of (±)-2f in the presence of 0.5 mol % of 1b occurred with a selectivity factor of 50. The linear allylic benzoate 10a formed in 49% yield, and the remaining branched allylic benzoate (S)-2f was isolated in 45% yield with 98% ee (eq 3).15 In addition, allylic substitution of the (S)-enantiomer of a representative branched allylic benzoate in the presence of 2b occurred with essentially complete retention of configuration. The reaction of (S)-2f (98% ee) with NaOPh and 2 mol % of 2b formed allyl aryl ether (S)-3f in 91% yield and 98% ee (eq 4).
 |
(3) |
 |
(4) |
Having established that kinetic resolution of racemic aromatic allylic benzoates by metallacycle 1b is highly selective and that the allylic substitution of the less reactive enantiomer occurs with complete retention of configuration, we developed a protocol to convert these racemic electrophiles to enantioenriched substitution products by sequential kinetic resolution and allylic substitution. The kinetic resolution of (±)-2f in the presence of 2 mol % of 1b at 0 °C in THF occurred in approximately 2 h. Addition of NaOPh to the resulting solution containing the less reactive enantiomer of the branched allylic benzoate (S)-2f and the linear cinnamyl benzoate 10a formed allyl aryl ether (S)-3f in 81% yield and 96% ee (Table 2, entry 1). The absolute stereochemistry of the product shows that the allyl aryl ether (S)-3f was formed by reaction of the phenoxide with the less reactive S-enantiomer of the allylic benzoate 2f, not by reaction with the linear allylic benzoate. The absolute stereochemistry of the product was the opposite of that resulting from etherification of linear aromatic allylic electrophiles.3g
Table 2.
Ir-Catalyzed Allylic Substitution of Racemic Aromatic Allylic Benozatesa
 | |||||
|---|---|---|---|---|---|
| entry | R (2) | nucleophile | prod- uct |
yield (%)b,c |
ee (%)d |
| 1 | Ph (2f) | NaOPh | 3f | 81 | 96 |
| 2 | 4-MeO-C6H4 (2g) | NaOPh | 3g | 90 | 95 |
| 3 | 4-F3C-C6H4 (2h) | NaOPh | 3h | 86 | 95 |
| 4 | 4-Br-C6H4 (2i) | NaOPh | 3i | 84 | 98 |
| 5 | 3-Br-C6H4 (2j) | NaOPh | 3j | 95 | 98 |
| 6 | 2-F-C6H4 (2k) | NaOPh | 3k | 87 | 94 |
| 7e | 2-Me-C6H4 (2l) | NaOPh | 3l | 83 | −84 |
| 8 | 3-pyridyl (2m) | NaOPh | 3m | 86 | 92 |
| 9 | Ph (2f) | LiN(Boc)2 | 4b | 92 | 93 |
| 10f | Ph (2f) | KNHC(O)CF3 | 5b | 76 | 92 |
| 11 | Ph (2f) | NaTs | 7b | 75 | 96 |
| 12e | 2-Me-C6H4 (2l) | NaTs | 7c | 93 | −94 |
| 13 | Ph (2f) | NaCH(CO2Me)2 | 8b | 87 | 98 |
| 14 | Ph (2f) | NaCH(CN)2 | 9b | 83 | 91 |
See Supporting Information for experimental details.
Isolated yield of products 3–9. Branched-to-linear selectivities were >95:5.
The yields of 10 (based on 2) ranged from 48–52% (entries 1–6, 8–11, and 13–14). The yield of 10 was 5–8% for reactions of 2l (entries 7 and 12). See Table S2 in the Supporting Information.
Enantiomeric excess of 3–9 determined by chiral HPLC methods.
Nucleophile was added prior to isomerization.
Allylic substitution was run at 0 °C to rt in the presence of 4 mol % 1b.
The scope of this combined enantioselective isomerization and allylic substitution is shown in Table 2. This process encompasses reactions of branched allylic benzoates containing a variety of aryl groups. The reactions of NaOPh with electron-rich, electron-neutral, and electron-deficient aromatic allylic benzoates containing para-, meta-, and ortho-substitution, as well as reactions of heteroaromatic allylic benzoates occurred in 83–90% yield and in 84–98% ee (entries 1–8).
Reactions of nitrogen, sulfur, and carbon nucleophiles with branched aromatic allylic benzoates also occurred in high yields and enantioselectivities. The reactions of LiN(Boc)2 and KNHC(O)CF3 with 2f formed protected allylamines 4b and 5b in 93% and 92% ee (entries 9 and 10). The reactions of NaTs with allylic benzoates 2f and 2l generated (S)-7b (R = Ph) and (R)-7c (R = 2-Me-C6H4) in good yields with excellent enantioselectivities (entries 11 and 12). Furthermore, the sodium salts of dimethyl malonate and malononitrile reacted with 2f to form allylic alkylation products 8b and 9b in 98% and 91% ee (entries 13 and 14).
In principle, enantioselective allylic substitution without isomerization is possible if the rate of nucleophilic attack on complex A (k5) greatly exceeds that for collapse to the linear allylic benzoate (k3) (Scheme 1). The identity of the nucleophile does not substantially alter the ratio of rate constants k5/k3. However, ortho substitution on the aromatic group (R) of 2 did substantially change the ratio of rate constants k5/k3. The branched aromatic allylic benzoate 2k containing a small ortho-fluoro group (entry 6) reacted like para- and meta-substituted branched allylic benzoates to form (S)-3k with high enantioselectivity. In contrast, branched aromatic allylic benzoates containing larger ortho substituents rearranged to the linear isomer much more slowly than did the ortho-fluoro-substituted, meta-substituted and para-substituted substrates. Thus, ortho-methyl derivative 2l underwent substitution in a manner that was similar to that of the aliphatic electrophiles; the reactions of NaOPh and NaTs with aromatic allylic benzoate 2l (R = o-Me-C6H4) formed allyl aryl ether (R)-3l in 83% yield and 84% ee and allylic sulfone (R)-7c in 93% yield and 94% ee by addition of the nucleophile at the same time as the catalyst (entry 7). The enantioselectivities of the reactions of the branched cinnamyl carbonates containing ortho substituents were substantially higher than those of the linear isomer, further underscoring the improved transformations that can be realized in some cases by use of the branched isomers.
In summary, we have developed versatile methods for iridium-catalyzed, kinetic asymmetric substitution of racemic allylic electrophiles. These reactions occur between a variety aliphatic, aromatic, and heteroaromatic allylic benzoates and a range of anionic heteroatom and carbon nucleophiles to form the corresponding allylic substitution products with good to excellent enantioselectivity. The high reactivity of branched allylic benzoates enables enantioselective allylic substitutions that are slow or poorly selective with linear allylic electrophiles. Moreover, the high-yield, one-pot synthesis of the branched allylic esters makes these reactions practical with 2.2 equiv of this reagent, particularly for processes in which the nucleophile is a valuable component. Efforts to extend the current methods to neutral nucleophiles and to reactions of additional allylic electrophiles are ongoing.
Supplementary Material
Acknowledgment
We thank the NIH for financial support of this work (GM55382 and GM58108 to J.F.H. and GM84584 to L.M.S.), Johnson-Matthey for gifts of IrCl3 and [Ir(COD)Cl]2, and Dr. Klaus Ditrich and BASF for gifts of chiral amines. M.U. thanks the JSPS for a fellowship.
Footnotes
Supporting Information Available: Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Kiener CA, Shu CT, Incarvito C, Hartwig JF. J. Am. Chem. Soc. 2003;125:14272. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]; (b) Leitner A, Shekar S, Pouy MJ, Hartwig JF. J. Am. Chem. Soc. 2005;127:15506. doi: 10.1021/ja054331t. [DOI] [PubMed] [Google Scholar]
- 2.For selected examples, see: Alexakis A, Polet D. Org. Lett. 2004;6:3529. doi: 10.1021/ol048607y. Lipowsky G, Miller N, Helmchen G. Angew. Chem. Int. Ed. 2004;43:4595. doi: 10.1002/anie.200460016. Graening T, Hartwig JF. J. Am. Chem. Soc. 2005;127:17192. doi: 10.1021/ja0566275. Weix DJ, Hartwig JF. J. Am. Chem. Soc. 2007;129:7720. doi: 10.1021/ja071455s. Liu W-B, He H, Dai L-X, You S-L. Org. Lett. 2008;10:1815. doi: 10.1021/ol800409d. Polet D, Rathgeb X, Falciola CA, Langlois J-B, El Hajjaji S, Alexakis A. Chem. Eur. J. 2009;15:1205. doi: 10.1002/chem.200801879.
- 3.For selected examples, see: Ohmura T, Hartwig JF. J. Am. Chem. Soc. 2002;124:15164. doi: 10.1021/ja028614m. Shu C, Leitner A, Hartwig JF. Angew. Chem. Int. Ed. 2004;43:4797. doi: 10.1002/anie.200460276. Weihofen R, Tverskoy O, Helmchen G. Angew. Chem. Int. Ed. 2006;45:5546. doi: 10.1002/anie.200601472. Stanley LM, Hartwig JF. J. Am. Chem. Soc. 2009;131:8971. doi: 10.1021/ja902243s. Pouy MJ, Stanley LM, Hartwig JF. J. Am. Chem. Soc. 2009;131:11312. doi: 10.1021/ja905059r. Stanley LM, Hartwig JF. Angew. Chem. Int. Ed. 2009;48:7841. doi: 10.1002/anie.200904338. Lopez F, Ohmura T, Hartwig JF. J. Am. Chem. Soc. 2003;125:3426. doi: 10.1021/ja029790y. Shu C, Hartwig JF. Angew. Chem. Int. Ed. 2004;43:4794. doi: 10.1002/anie.200460214. Lyothier I, Defieber C, Carreira EM. Angew. Chem. Int. Ed. 2006;45:6204. doi: 10.1002/anie.200602408. Ueda M, Hartwig JF. Org. Lett. 2009;12:92. doi: 10.1021/ol9023248.
- 4.(a) Bartels B, Helmchen G. Chem. Commun. 1999:741. [Google Scholar]; (b) Bartels B, García-Yebra C, Rominger F, Helmchen G. Eur. J. Inorg. Chem. 2002:2569. [Google Scholar]; (c) Fischer C, Defieber C, Suzuki T, Carreira EM. J. Am. Chem. Soc. 2004;126:1628. doi: 10.1021/ja0390707. [DOI] [PubMed] [Google Scholar]; (d) Polet D, Alexakis A, Tissot-Croset K, Corminboeuf C, Ditrich K. Chem. Eur. J. 2006;12:3596. doi: 10.1002/chem.200501180. [DOI] [PubMed] [Google Scholar]
- 5.(a) Trost BM, Hachiya I. J. Am. Chem. Soc. 1998;120:1104. [Google Scholar]; (b) Malkov AV, Spoor P, Vinader V, Kocovsky P. Tetrahedron Lett. 2001;42:509. [Google Scholar]; (c) Glorius F, Neuburger M, Pfaltz A. Helv. Chim. Acta. 2001;84:3178. [Google Scholar]; (d) Belda O, Moberg C. Acc. Chem. Res. 2004;37:159. doi: 10.1021/ar030239v. [DOI] [PubMed] [Google Scholar]
- 6.(a) Prétôt R, Pfaltz A. Angew. Chem. Int. Ed. 1998;37:323. doi: 10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; (b) Pàmies, Diéguez M, Claver C. J. Am. Chem. Soc. 2005;127:3646. doi: 10.1021/ja0425738. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi T, Okada A, Suzuka T, Kawatsura M. Org. Lett. 2003;5:1713. doi: 10.1021/ol0343562. [DOI] [PubMed] [Google Scholar]
- 8.Vrieze DC, Hoge GS, Hoerter PZ, Van Haitsma JT, Samas BM. Org. Lett. 2009;11:3140. doi: 10.1021/ol901031b. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hayashi T, Kishi K, Yamamoto A, Ito Y. Tetrahedron Lett. 1990;31:1743. [Google Scholar]; (b) You S-L, Zhu X-Z, Luo Y-M, Hou X-L, Dai L-X. J. Am. Chem. Soc. 2001;123:7471. doi: 10.1021/ja016121w. [DOI] [PubMed] [Google Scholar]
- 10.Shekhar S, Trantow B, Leitner A, Hartwig JF. J. Am. Chem. Soc. 2006;128:11770. doi: 10.1021/ja0644273. [DOI] [PubMed] [Google Scholar]
- 11.Marković D, Hartwig JF. J. Am. Chem. Soc. 2007;129:11680. doi: 10.1021/ja074584h. [DOI] [PubMed] [Google Scholar]
- 12.The remaining branched allylic benzoate 2a was isolated in 52% yield (based on 2.2 equiv of 2a) and 78% ee. The ee of the remaining 2a-e is necessarily modest in reactions conducted with 2.2 equiv of 2a-e that occur to form products 3–9 with high ee. However, the ee of remaining 2a was 94–97% with concomitant decreasing ee of 3a (75–93%) when performing the reactions of NaOPh with less than 2.2 equiv of 2a (1.8–2.0 equiv). See Table S1 in the Supporting Information for these data.
- 13.For recent examples of enantioselective ligand-accelerated allylic alkylation with Grignard reagents to set a neopentyl stereocenter, see: Jackowski O, Alexakis A. Angew. Chem. Int. Ed. 2010;49:3346. doi: 10.1002/anie.201000577.
- 14.The rates of formation of allyliridium complexes A and B from the linear allylic benzoate are considered to be negligible in this analysis because the linear allylic benzoate does not undergo conversion under these reaction conditions.
- 15.The selectivity factor (s) = ln{(1−c)(1−ee)}/ln{(1−c)(1+ee)} where c is the conversion of (±)-2f and ee is the enantiomeric excess of the remaining branched allylic benzoate 2f. Kagan HG, Fiaud JC. Topics in Stereochemistry. Vol. 18. New York: John Wiley & Sons; 1988. p. 249.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.