

Abstract
The HOG mitogen-activated protein kinase pathway mediates the osmotic stress response in Saccharomyces cerevisiae, activating genes like GPD1 (glycerol phosphate dehydrogenase), required for survival under hyperosmotic conditions. Activity of this pathway is regulated by Sln1p, a homolog of the “two-component” histidine kinase family of signal transduction molecules prominent in bacteria. Sln1p also regulates the activity of a Hog1p-independent pathway whose transcriptional output can be monitored using an Mcm1p-dependent lacZ reporter gene. The relationship between the two Sln1p branches is unclear, however, the requirement for unphosphorylated pathway intermediates in Hog1p pathway activation and for phosphorylated intermediates in the activation of the Mcm1p reporter suggests that the two Sln1p branches are reciprocally regulated. To further investigate the signals and molecules involved in modulating Sln1p activity, we have screened for new mutations that elevate the activity of the Mcm1p-dependent lacZ reporter gene. We find that loss of function mutations in FPS1, a gene encoding the major glycerol transporter in yeast activates the reporter in a SLN1-dependent fashion. We propose that elevated intracellular glycerol levels in the fps1 mutant shift Sln1p to the phosphorylated state and trigger the Sln1-dependent activity of the Mcm1 reporter. These observations are consistent with a model in which Sln1p autophosphorylation is triggered by a hypo-osmotic stimulus and indicate that the Sln1p osmosensor is tied generally to osmotic balance, and may not specifically sense an external osmolyte.
Yeast cells maintain elaborate mechanisms for adaptation and growth under a variety of adverse environmental conditions. For example, an increase in the osmolarity of the medium elicits an osmotic stress response that includes transient cell cycle arrest, restructuring of the actin cytoskeleton, and an elevation in the intracellular glycerol concentration (1–4). Glycerol is produced intracellularly to partially offset the rise in external osmolarity thus preventing water loss that would lead to dehydration and eventual death (5–9). Although the exact mechanism by which cells sense a change in osmotic pressure is not known, components of the intracellular signaling pathway that responds to osmotic stress response have begun to be elucidated (10–12). In Saccharomyces cerevisiae, the response to osmotic stress is mediated, in part, by the Hog1p MAP1 kinase pathway (10). Two membrane-associated osmosensors, Sln1p and Sho1p, regulate the activity of the Hog1p pathway, although the mechanism by which they do so differs (11). Sho1p associates with and activates the Ste11p MEK kinase which phosphorylates the Pbs2p MEK, which in turn activates the MAP kinase Hog1p (10, 12). Sln1p, on the other hand, is a yeast homolog of “two-component” regulators prominent in bacterial signal transduction (13). The two-component designation refers to the two modules, a sensor/transmitter (histidine kinase) and a response regulator (receiver plus output) module that comprise the basic signaling unit. Two-component signal transduction involves autophosphorylation of the transmitter module on a histidine residue followed by phosphotransfer to an aspartate residue of the receiver module (14). Sln1p controls the activity of the Hog1p MAP kinase pathway via a phosphorelay system involving Ypd1p (a phosphorelay intermediate) and Ssk1p (a response regulator with an output domain). The dephosphorylated form of the Ssk1p response regulator activates the MEK kinases, Ssk2p and Ssk22p which then activate the Pbs2p MEK and Hog1p MAP kinase (11, 15, 16). Hog1p is thought to phosphorylate transcription factors responsible for turning on a family of osmoresponse genes that includes glycerol phosphate dehydrogenase (GPD1) which is involved in the synthesis of glycerol (8).
Recently, we have shown that Sln1p also regulates the activity of a Hog1p-independent pathway whose transcriptional output can be monitored using a lacZ reporter gene we call P-lacZ, which consists of a CYC1-lacZ fusion in which the UAS of CYC1 has been replaced by a palindromic-binding site (P site) for the Mcm1p transcription factor (17, 18). Potential and known target genes of Mcm1p encode proteins involved in cell type determination, cell wall and membrane integrity, cellular metabolism, and cell cycle progression (19). Null mutations in SLN1 decrease P-lacZ reporter gene expression whereas activator mutations in the gene (sln1*) increase P-lacZ reporter gene expression. As is the case for the HOG pathway, Sln1p control of the P-lacZ reporter appears to require the kinase and phosphotransfer functions of Sln1p (17). The sln1* mutations that result in increased P-lacZ reporter gene activity map to positions likely to affect phosphorylation. More compelling evidence for the role of phosphorylation and phosphotransfer in sln1* activation of the P-lacZ reporter comes from the osmo-sensitive phenotype of these mutants which is a reflection of reduced HOG pathway activity (17). The demonstration of a second pathway controlled by Sln1p led us to propose that the Sln1p osmosensor sends two signals in response to changes in osmolarity. Activation of Hog1p accounts for the increase in glycerol, whereas changes in Mcm1p activity might account for changes in parameters of cell growth (e.g. cell cycle progression, cell wall composition, and expansion) that are postulated to occur in response to osmotic stress.
In their natural environment, yeast are exposed not only to increased osmolarity but also to decreased osmolarity. Cellular adjustment to decreased osmolarity involves a protein kinase C-like protein (PKC) encoded by the PKC1 gene (20) which regulates a MAP kinase cascade consisting of the BCK1 MEK kinase (21, 22), MKK1 and MKK2 MEKs (23), and the MAP kinase, MPK1 (24, 25). In contrast to HOG pathway mutants which fail to grow under high osmolarity conditions, mutants lacking PKC pathway function exhibit cell lysis at low osmolarity (26). The cell lysis phenotype and corresponding reduction in β-glucan content in the cell walls of pkc1 mutants (27) indicates that the PKC pathway is required, in part, for cell wall integrity. Consistent with the idea that the PKC kinase cascade is an osmosensory system required for growth under low osmolarity, Davenport et al. (28) have shown that Mpk1p is rapidly tyrosine phosphorylated in response to hypo-osmotic conditions. Interestingly, the Hog1 and Mpk1 pathways appear to cross-talk; tyrosine phosphorylation of Mpk1 was inhibited in cells exposed to an elevated osmotic environment.
Although there is now a considerable body of information about the pathways that lead to changes in gene expression and adaptation to osmotic stress environments, the mechanism by which osmotic stress is sensed remains obscure. To further investigate the signals and molecules involved in modulating Sln1 activity, we have undertaken a genetic screen to find new mutations that mimic the effect of sln1* mutations in elevating the activity of the P-lacZ reporter gene. We report here the isolation of loss of function mutations in FPS1, a gene encoding a major glycerol transporter of the cell. Mutations in FPS1 are known to reduce glycerol efflux resulting in increased intracellular glycerol levels, thus disturbing normal osmotic balance (29). The identification of Fps1p as a regulator of a Sln1p pathway activity suggests that the Sln1 protein may generally detect and respond to deviations from the normal osmotic gradient and may not specifically bind or sense external osmolytes.
EXPERIMENTAL PROCEDURES
Yeast Strains and Media
All yeast strains used in this work (Table I) are from our strain collection or were constructed for this study. The fps1 deletion was introduced by one-step transformation (30) using the 4.9-kb BamHI-HindIII fragment from pWT1040. The replacement was confirmed by Southern hybridization analysis of BamHI-HindIII digested genomic DNA using the 2.5-kb BamHI-HindIII fragment from the FPS1 gene as a probe. Conversion of the LEU2 gene disruption marker to kanR in various strains was accomplished as described previously (31). Kan replacements were confirmed on the basis of the newly gained Leu− and G418 resistance phenotypes. Disruption of the GPD1 locus was accomplished by transformation of the appropriate yeast strains with a 3.8-kb gpd1Δ::LEU2 disruption fragment isolated from plasmid pJF1070.
Table I.
Yeast strains used in this study
| Straina | Relevant genotype | Ref. or Notes |
|---|---|---|
| JF1331 | MATa his4–917 lys2–128δ leu2–1 trp1Δ1 ura3–52; pGY48 | |
| JF1562 | MATa his3Δ200 leu2Δ1 ura3–52 trp1Δ63 CanR CyhR | CanR, CyhR derivative of FY251 (52); JF1564 is JF1562 carrying pGY48 |
| JF1564 | ||
| JF1565 | MATα his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR | JF1566 and JF1567 are derivatives of JF1565 |
| JF1566 | JF1566 has pGY48 integrated at URA3 and JF1567 carries the free plasmid | |
| JF1567 | ||
| JF1593 | MATa kar1–1 his4 leu2 trp1Δ1 or 62 ura3–52 | |
| JF1595 | MATα kar1–1 his4 ade2–101 leu2 trp1Δ1 or 62 ura3–52 | |
| JF1628 | MATa arg4Δ::URA3 his3Δ200 leu2–1 trp1Δ63 ura3–52 CyhR; pGY48 | arg4Δ::URA3, CyhR derivative of FY251 (52) |
| JF1681 | MATα nrp1002 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | Progeny of first backcross with JF1628 |
| JF1709 | MATα nrp0831 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | Progeny of first backcross with JF1628 |
| JF1713 | MATα sln1–22 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | sln1–22 derivative of JF1567; two-step replacement |
| JF1717 | MATα nrp0427 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | Progeny of first backcross with JF1628 |
| JF1722 | MATα TRP1+ his3Δ200 leu2Δ1 ura3–52 lys2Δ201 CanR CyhR; pGY48 | TRP1+ derivative of JF1567; one-step replacement |
| JF1727 | MATα nrp0932 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | Progeny of first backcross with JF1628 |
| JF1729 | MATα nrp0831 his3Δ200 leu2Δ1 ura3–52 TRP1+ lys2Δ201 CanR CyhR; pGY48 | TRP1+ derivative of JF1709; one-step replacement |
| JF1732 | MATα fps1Δ::LEU2 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | fps1Δ::LEU2 derivative of JF1567; one-step replacement |
| JF1733 | MATα fps1Δ::LEU2 sln1–22 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | fps1Δ::LEU2 derivative of JF1713; one-step replacement |
| JF1738 | MATa skn7Δ::TRP1 his4–917 lys2–128δ leu2–1 trp1Δ1 ura3–52; pGY48 | skn7Δ::TRP1 derivative of JF1331; one-step replacement |
| JF1765 | MATα ssk1Δ::kan sln1Δ::TRP1 his4–917 lys2–128δ leu2 trp1Δ1 ura3–52; pGY48 | |
| JF1770 | MATa fps1Δ::LEU2 skn7Δ::TRP1 his4–917 lys2–128δ leu2–1 trp1Δ1 ura3–52; pGY48 | fps1Δ::LEU2 derivative of JF1738; one-step replacement |
| JF1777 | MATa ssk1Δ::kan his4–917 lys2–128δ leu2–3,112 trp1Δ1 ura3–52; pGY48 | |
| JF1779 | MATα fps1Δ::kan his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY48 | fps1Δ::kan derivative of JF1732; one-step replacement |
| JF1780 | MATα fps1Δ::LEU2 ssk1Δ::kan sln1Δ::TRP1 his4–917 lys2–128δ leu2 trp1Δ1 ura3–52; pGY48 | fps1Δ::LEU2 derivative of JF1765; one-step replacement |
| JF1781 | MATa ypd1Δ::TRP1 ssk1Δ::kan his4–917 lys2–128δ leu2–3,112 trp1Δ1 ura3–52; pGY48 | ypd1Δ::TRP1 derivative of JF1777; one-step replacement |
| JF1783 | MATa fps1Δ::LEU2 ssk1Δ::kan his4–917 lys2–128δ leu2–3,112 trp1Δ1 ura3–52; pGY48 | fps1Δ::LEU2 derivative of JF1777; one-step replacement |
| JF1784 | MATa fps1Δ::LEU2 his4–917 lys2–128δ leu2–1 trp1Δ1 ura3–52; pGY48 | fps1Δ::LEU2 derivative of JF1331 |
| JF1825 | MATα his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY88 | fps1Δ::LEU2 derivative of JF1566; one-step replacement |
| JF1826 | MATa gpd1Δ::LEU2 his3Δ200 leu2Δ1 ura3–52 trp1Δ63 CanR CyhR | gpd1Δ::LEU2 derivative of JF1562; one-step replacement |
| JF1827 | MATa fps1Δ:::LEU2 gpd1Δ::LEU2 his3Δ200 leu2Δ1 trp1Δ63 ura3–52 CanR CyhR; pGY88::URA3 | Progeny of a cross between JF1825 and JF1826 |
| JF1832 | MATα fps1Δ::kan his3Δ200 leu2Δ1 ura3–52 trp1Δ63 lys2Δ201 CanR CyhR; pGY88::URA3 | JF1779 with integrated P-lacZ reporter |
All strains used in this study are derived from laboratory strain S288C.
Conversion of the SLN1+ allele to sln1–22 was accomplished by two-step replacement (32). Plasmid pWT980 which carries the sln1–22 allele was linearized within the SLN1 sequences using NruI. Ura+ transformants in which the plasmid was integrated at the SLN1 locus were then subject to selection on 5-fluoroorotic acid to isolate recombinants containing only the sln1–22 allele. The presence of the sln1–22 allele was confirmed by sequence analysis (primer: 5′-AGAATGTTGAACTTGGAGGGC-3′) of a 0.5-kb polymerase chain reaction product generated from yeast genomic DNA template (primers: SLN1 3200 (5′-GCCACATCAAGTTGAGAATTCCCCACAGTCAAAGACG-3′) and SLN1 4119 (5′-CGCGCAAGCTTTTGATTTCTC-3′)).
Integration of the P-lacZ reporter was accomplished by linearizing the P-lacZ integrating vector, pGY88 within URA3 using ApaI, and transforming the strain to Ura+. Successful integration at the URA3 locus and the number of plasmids integrated in each strain was examined by Southern analysis of PstI digested DNA. A 1-kb lacZ polymerase chain reaction product (primers: lacZ 280F (5′-CGGTTACGATGCGCCCATCTACACC-3′) and lacZ 1291R (5′-GGATCATCGGTCAGACGATTCATTGG -3′)) was used for probe synthesis. The presence of multiple copies of the reporter was revealed by hybridization of the probe to a novel 7-kb PstI fragment.
The media were prepared as described by Sherman et al. (33) and included synthetic complete medium (SC) lacking one or more specific amino acids (e.g. SC-uracil) and rich medium (YPD). Plates for the detection of β-galactosidase contained 50 mg of 5-bromo-4-chlor-3-indolyl β-D-galactopyranoside (X-gal)/ml and were prepared as described by Larson et al. (34).
Plasmids
The reporter plasmids pGY48 (35) and pBG12 (TEF2 lacZ) (36) were previously described. The integrating derivative of pGY48, pGY88, was constructed by deleting a 2.2-kb EcoRI fragment containing the 2-μm origin. The CYC1-lacZ reporter is pLG669Z (37), and the ΔUAS-lacZ reporter is pLG670Z, a derivative of pLG669Z which lacks the XhoI fragment encompassing the CYC1 UAS. Additional P site-dependent reporters included pJF947 (1xP), pCM771 (4xP), and pCM772 (4xmutP). The P site and mut P site oligonucleotides used in construction of these reporters were as follows: 5′-TCGAGTTTCCTAATTAGGAAAC-3′ (P) and 5′-TCGAGTTTCCTAATTAATAAAC-3′ plus 5′-TCGAGTTTATTAATTAGGAAAC-3′ (mutP). LEU2 CEN plasmids used in the genetic screen for new nrp mutants included SLN1 (pGY111) (18), GAL11 (pJF765) (35), and KEX2 (pSBKX, R. Fuller).
pGY107 is a 2-μm plasmid containing an MCM1 gene with a Myc epitope tag. The Myc epitope was inserted into a new EcoRV site that was introduced by site-directed mutagenesis between amino acids 3 and 4 of the cloned MCM1 gene. The Myc epitope was encoded by complementary 30-base pair oligonucleotides which were annealed to generate a blunt end fragment for cloning into the EcoRV site. The presence and orientation of Myc sequences was confirmed by DNA sequence analysis. The modified MCM1 gene was then subcloned as a 3.5-kb XbaI-XhoI fragment into pRS316 (URA3, CEN) (38). The plasmid complemented the α-halo defect of an mcm1-1 mutant.
pWT1037 consists of a 2.6-kb BamHI-HindIII fragment containing FPS1 cloned into the BamHI and HindIII sites of the LEU2 CEN vector, pRS315 (38). pWT1039 consists of a 2.8-kb HindIII-EcoRI fragment containing SDH2 and YLL042C cloned into the HindIII and EcoRI sites of pRS315. pWT1040 is a derivative of pWT1037 from which an internal 0.9-kb XhoI-PstI fragment was deleted and replaced with a 3.3-kb XhoI-PstI LEU2 fragment excised from plasmid YEp13.
pJF1070 is a LEU2 marked disruption of the GPD1 gene. A GPD1 fragment encompassing the entire open reading frame was polymerase chain reaction amplified using a primer pair corresponding to open reading frame YDL022W (Research Genetics) and genomic DNA as template. A 1.9-kb AvaI-BamHI restriction fragment was cloned into pUC19 (Promega Biotech) to generate the intermediate plasmid, pJF1068. A deletion of 268 base pairs was introduced into the GPD1 open reading frame on pJF1068 by SalI digestion and ligation. A 2.2-kb SalI-XhoI LEU2 fragment was subsequently ligated into the SalI site to create pJF1070.
pWT250 is a derivative of pRS424 (38), a 2-μm TRP1 marked plasmid into which was cloned a 5-kb EcoRI fragment containing the GPD1 gene isolated from YEpGPD1 (8). Plasmid pWT980 used in converting SLN1+ strains to sln1–22 consists of a 2.1-kb EagI-XbaI fragment containing the sln1–22 alteration cloned into pRS406 URA3 integrating vector.
Genetic Screen
A kar1 (JF1593 or JF1595) strain carrying the plasmid to be introduced was mated to candidate mutants (which also carry two recessive drug resistance alleles). Cytoductants were selected on drug plates (selecting for the persistence of the nrp nucleus and loss of the kar1 nucleus) lacking leucine (selecting for the presence of the plasmid) and tested for their X-gal phenotype. The starting strain, JF1567 was subject to ethyl methanesulfonate mutagenesis (18) sufficient to cause 65% killing. Approximately 54,600 surviving colonies were screened on X-gal plates to find 550 mutants with increased blue color. Mutants were crossed by a wild type strain of the opposite mating type and diploids tested for their X-gal phenotype. Fifty diploids out of 550 exhibited a mutant (blue phenotype) and were thus put aside on the basis of the dominance of the mutation. SLN1 and GAL11 plasmids were introduced into each of the 500 remaining mutants by a cytoduction strategy (see “Results”). Nine were complemented by the SLN1 plasmid, and 22 were complemented by the GAL11 plasmid. From previous studies we were aware that mutations in the KEX2 gene also (nonspecifically) increase the color of reporter bearing strains on X-gal plates. Mutations in the KEX2 gene were in fact the major class in the preliminary screen that resulted in three sln1* alleles. Consequently, we also introduced a KEX2 plasmid into each mutant. Three hundred mutants were complemented by the KEX2 plasmid or by more than one plasmid. One hundred forty-two mutants could not be complemented by any of the three plasmids. Each of the remaining mutants was tested for its P site specificity. The P-lacZ reporter was first eliminated from each strain by selection on 5-fluoroorotic acid plates and then a TEF2-lacZ reporter introduced. Each strain was examined on X-gal media and cultured for quantitation of β-galactosidase levels in liquid assays. Twelve mutants exhibited a 2-fold or greater increase in P-lacZ activity but less than a 2-fold increase in TEF2-lacZ reporter activity.
Sporulation
Cells were cultured in YPD or SC media overnight. Approximately 8 × 107 cells were harvested by centrifugation and washed twice with water. Cells were resuspended in sporulation medium (1% potassium acetate and 10 μg/ml amino acids required by the strain). Sporulation cultures were incubated with aeration at room temperature for 5–8 days.
Isolation and Quantitation of RNA
Cells were grown to a concentration of 1 × 107 cells/ml at 30 °C in the designated media. Sorbitol (Fluka) was added to log phase cultures to a final concentration of 1.0 M as follows. A 5.0 M sorbitol stock was first added to conditioned YPD to a final concentration of 2.0 M. YPD cultures grown to 1 × 107 cells/ml were then diluted 1:1 with the 2.0 M conditioned YPD/sorbitol to a final concentration of 1.0 M sorbitol. Preparation of RNA, electrophoresis, blotting, and hybridization were performed as described previously (18). 32P-Labeled probes were prepared using random primers (39, 40). Quantitation was performed using a PhosphorImager (Molecular Dynamics).
Immunodetection of Mcm1 Protein
Cells were harvested in exponential growth. Extracts were prepared using a previously described trichloroacetic acid extraction (41). Samples were subjected to SDS-polyacrylamide gel electrophoresis (10%; 4.5% stacking gel), transferred to nitrocellulose, and the blot was probed with anti-Myc monoclonal antibody 9E10 (1:1000 dilution). Immune complexes were visualized by ECL chemiluminescence (Amersham Corp.)
Intracellular Glycerol Measurements
Intracellular glycerol was assayed enzymatically with a commercial glycerol determination kit (Boehringer-Mannheim Biochemicals). Extracts were prepared essentially as described (42). Cells were grown to log phase in selective media. Three 10-ml aliquots were separately filtered onto 0.45-μm cellulose nitrate filters (Whatman), washed with 5 ml of cold YPD, and resuspended in 2 ml 0.5 M Tris-HCl, pH 7.5. One-ml samples were heated to 95 °C for 10 min and cell debris pelleted by low-speed centrifugation. Pooled aliquots were used in determining glycerol levels according to manufacturer’s specifications. Protein extracts were prepared from the remaining cells. Cells were vortexed four times (30 s each) in the presence of glass beads and cell debris pelleted by high speed centrifugation. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad). Glycerol concentrations were normalized to total protein. Fold increase in glycerol concentration was determined by normalizing to the glycerol concentration in wild type cells. Data represent the average of six trials using a minimum of three transformants.
RESULTS
Isolation of New nrp Mutants
A strain carrying the MCM1-dependent reporter, P-lacZ exhibits a pale blue phenotype on plates containing the chromogenic substrate, X-gal (50 mg/liter). Previously, we have described recessive alleles of SLN1 (nrp2-1, nrp2-2, and nrp2-3; renamed sln1–21, 22, and 23) and GAL11 that exhibit increased P-lacZ reporter activity (18, 35). To identify additional NRP (negative regulators of P-lacZ) genes, we repeated the screen using a strategy designed to rapidly detect (and discard) additional SLN1 and GAL11 alleles. SLN1 and GAL11 plasmids (LEU2 selectable marker) were introduced separately into each individual mutant using a cytoduction strategy based on the dominant nuclear fusion defect of the kar1-1 mutant (43) (see “Experimental Procedures”). Using this procedure, it was possible to simultaneously screen hundreds of candidates on a small number of Petri plates for mutants whose phenotype failed to be complemented by the SLN1 or GAL11 genes. A similar tactic was used to determine which mutants had a specific effect on the P-lacZ reporter. In this case mutants were first cured of the P-lacZ plasmid and subsequently mated to a kar1 strain containing the TEF2-lacZ reporter, a reporter whose transcription is independent of Mcm1p.
Of the 12 mutants exhibiting a 2-fold or greater elevation in P-lacZ activity, four showed clear 2:2 segregation of the X-gal phenotype suggesting that the mutation was due to a change at a single locus. Each of these four was examined more thoroughly for the specificity of the reporter gene activation phenotype by transformation with Mcm1-independent reporter plasmids, TEF2-lacZ and CYC1-lacZ, as well as a reporter lacking a UAS insert (Table II). Two mutants, nrp0831 and nrp0932, continued to exhibit a specific increase in Mcm1-dependent reporter gene expression, whereas nrp0427 and nrp1002 were eliminated from further consideration on the basis that they were not P-lacZ specific. Subsequent analysis revealed that the increased P-lacZ activity in the nrp0932 mutation was due to an effect of the mutation on the copy number of the reporter plasmid (data not shown) and we therefore eliminated it from further study. The specificity of the reporter gene activation phenotype in nrp0831 was confirmed by comparing the effect of the nrp0831 mutation on a reporter carrying 4 copies of the P site or 4 copies of a mutated version of the P site. The activating effect of the nrp0831 mutation was eliminated in the mutated P site reporter (nrp0831/wt = 1.1).
Table II.
Reporter specificity of the nrp mutants
| Strain | Genotype |
β-Galactosidase activitya |
|||
|---|---|---|---|---|---|
| No UAS- pLG670Z |
CYC1-lacZ pLG669Z |
TEF2-lacZb | P-lacZb | ||
| JF1565 | Wild type | 12.8 (4.7) | 51.4 (15.9) | 116.0 (28.2) | 146.0 |
| JF1717 | nrp0427 | 16.9 (4.4) | 103.0 (23.7) | 210.0 (52.6) | 433.0 |
| JF1709 | nrp0831 | 18.1 (8.5) | 67.2 (25.7) | 132.0 (49.7) | 327.0 |
| JF1727 | nrp0932 | 12.1 (1.9) | 33.0 (7.8) | 106.0 (21.1) | 367.0 |
| JF1681 | nrp1002 | 45.3 (10.9) | 132.0 (26.4) | 14.2 (3.5) | 330.0 |
Activities are reported as Miller units and are the averages of six measurements involving at least three transformants in all cases except for the values listed for the P-lacZ reporter (top), which represent a single measurement.
Plasmids were as follows: TEF2-lacZ, pBG12; P-lacZ, pGY48.
Cloning of NRP0831 by Complementation
The X-gal phenotype of the nrp0831 mutation is recessive (Fig. 1B, bottom) so it was possible to clone the gene by complementation. A CEN based genomic library was introduced by transformation into the nrp0831 strain, JF1709, a product of a primary backcross. Sixteen thousand transformants were screened on X-gal plates for restoration of the pale blue (wild type) phenotype. Plasmids were rescued from each of the seven transformants that were white on X-gal plates and each could be shown to confer upon the starting strain a pale blue phenotype after retransformation. Partial sequence analysis of each of four potential NRP0831 candidates revealed that the four plasmids covered a common region of chromosome XII encompassing the open reading frames of two known genes, FPS1 and SDH2, as well as one additional complete open reading frame (YLL042C) and part of the VPS13 gene (Fig. 1A). Subclones containing a 2.6-kb BamHI to HindIII fragment that includes the entire FPS1 coding region and 300-base pair upstream sequence complemented the nrp0831 X-gal phenotype, whereas a 2.8-kb EcoRI to HindIII fragment including SDH2 and YLL042C failed to complement the mutant phenotype (Fig. 1B, top).
Fig. 1. Identification of nrp0831 complementing sequences on chromosome XII.

Diagram of the transcriptional orientation and boundaries of the open reading frames included on the genomic library plasmid complementing the nrp0831 mutant. Complementation by the original library clone and two subclones (shown below the map) is shown in the upper part of the table. FPS1 plasmid, pWT1037; SDH2 plus YLL042 plasmid, pWT1039; vector, pRS315. Recessiveness of the nrp0831 mutation is shown in the lower part of the table. β-Galactosidase values are given in Miller units and are the averages of n assays.
The nrp0831 Mutation Is an Allele of FPS1
To verify the identification of the FPS1 gene as a regulator of P-lacZ reporter gene activity, P-lacZ activity was measured in strains containing a deletion of the FPS1 gene. The fps1Δ mutation caused an increase in P-lacZ activity similar in magnitude to the one in the nrp0831 mutant (Fig. 1B, middle). These results suggest that mutant nrp0831 contains a loss of function mutation in the FPS1 gene. The nrp0831/fps1Δ diploid (Fig. 1B, bottom) was sporulated and 52 tetrads dissected. 158 out of 158 of the viable spores were blue on X-gal media, showing 100% cosegregation of FPS1 with the nrp0831 locus, consistent with the hypothesis that the nrp0831 mutation is an allele of FPS1 (data not shown).
The increased P-lacZ reporter activity in fps1Δ mutants was also reflected at the RNA level. RNA was prepared from wild type and mutant strains carrying a single integrated copy of the the P-lacZ reporter plasmid, and lacZ levels quantitated following Northern (RNA) hybridization analysis. After normalization to URA3 transcript levels at 5, 10, and 20 μg of RNA concentrations lacZ RNA levels were shown to be approximately 2-fold higher (fps1Δ/wt 5 μg, 457.2/213.9 = 2.1; 10 μg, 488.7/218.3 = 2.2; 20 μg, 650.8/310.1 = 2.1) in the fps1Δ mutant than in the wild type strain (Fig. 2).
Fig. 2. Effect of mutation in the FPS1 gene on transcript levels from the P-lacZ reporter.
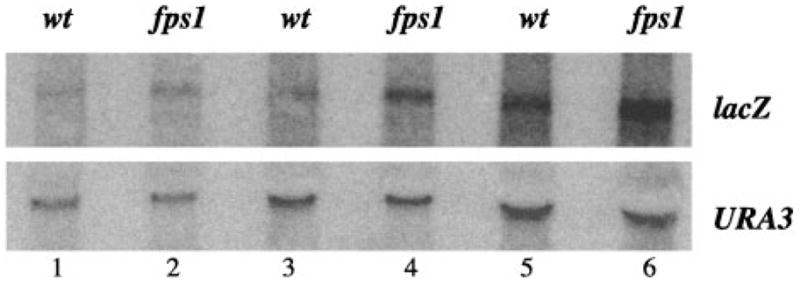
An equal amount of RNA (5 μg, lanes 1 and 2; 10 μg, lanes 3 and 4; 20 μg, lanes 5 and 6) from wild type (JF1566, lanes 1, 3, and 5) and fps1Δ (JF1825, lanes 2, 4, and 6) strains was subjected to Northern (RNA) hybridization using lacZ and URA3 probes sequentially. Transcript levels were quantitated using PhosphorImage analysis (Molecular Dynamics). lacZ transcript levels were normalized to URA3 levels, to control for loading differences between lanes.
To examine whether the increase in P-lacZ activity in the fps1Δ mutant was due to a change in the activity of Mcm1p rather than to alterations in Mcm1 protein levels (18), strains bearing a Myc epitope-tagged MCM1 gene were evaluated using anti-Myc immunoblot analysis. As observed previously for the sln1* mutants (29) (Fig. 3), Mcm1 protein levels were not significantly affected in the nrp0831 background, nor in strains deleted for FPS1 (Fig. 3).
Fig. 3. Mcm1 protein levels are unaffected by mutations in FPS1.
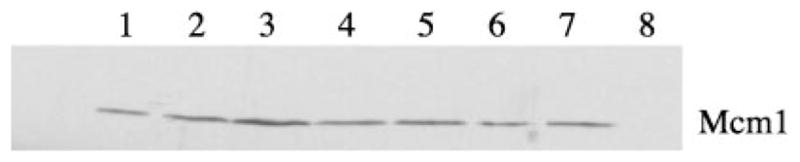
Protein extracts prepared from wild type (JF1567, lanes 1–3), nrp0831 (JF1709, lane 4), fps1Δ (JF1732, lane 5), sln1–22 (JF1567, lane 6), and sln1–22 fps1Δ (JF1733, lane 7) strains carrying pGY107, the 2-μm MCM1-Myc plasmid (lanes 1–7) and the wild type strain lacking pGY107 (JF1709, lane 8) were subjected to SDS-polyacrylamide gel electrophoresis in duplicate. One gel was treated with Coomassie Brilliant Blue (not shown) to examine loading and an identical gel was treated with anti-Myc antibody for Western analysis as described in detail under “Experimental Procedures.” A 2-s exposure following chemiluminescence detection is shown. Lanes 1, 2, and 3 were loaded with 0.5, 1, and 2 × protein, respectively; while lanes 4–8 each contained 1 × protein.
The fps1Δ-dependent Increase in P-lacZ Reporter Activity Requires Sln1 and Skn7
The elevation in P-lacZ activity in an fps1 mutant is similar to the effect of sln1* activating mutations (17, 18). To investigate the relationship between SLN1 and FPS1, we measured P-lacZ activity in the double mutant. The results of this analysis indicate that the effect of the sln1* mutation is enhanced by deletion of FPS1 (Table III). While the additivity of the sln1* and fps1 phenotypes is consistent with the existence of two independent pathways leading to activation of the P-lacZ reporter, an alternate explanation may be that the sln1* mutation and the fps1 mutation each only partially shift the pool of Sln1 protein to the phosphorylated form, and that the combined effects of the two mutations increases the signaling through a single pathway leading to P-lacZ activation. To distinguish between these possibilities we examined whether the fps1 effect was dependent on Sln1p and on Sln1p pathway intermediates.
Table III.
Effect of the FPS1 mutation on sln1* activation of the Mcm1-dependent reporter
| Straina | Genotype | β-Galactosidase activityb |
|---|---|---|
| JF1567 | Wild type | 261.0 (14.3) |
| JF1732 | fps1Δ | 768.0 (49.0) |
| JF1713 | sln1* | 495.0 (15.2) |
| JF1733 | fps1Δ sln1* | 1118.0 (39.3) |
The sln1* designation in the strains used in this experiment represents the sln1–22 mutation.
Activities are reported as Miller units and are the averages of six measurements involving at least three transformants in all cases.
Tests of the requirement for SLN1 were done in an ssk1Δ background to suppress the otherwise lethal sln1Δ mutation (15). Although, in this experiment, the ssk1 mutation depressed P-lacZ levels by 50%, the effect of the fps1 mutation is still apparent in the SLN1+ ssk1Δ strain (Table IV, rows 3 and 4). In the sln1Δ mutant background, in contrast, elevation in P-lacZ levels due to fps1Δ was eliminated (Table IV, rows 5 and 6). The data in Table IV indicates that the Sln1 protein is required for the fps1 phenotype.
Table IV.
Role of SLN1 in the fps1Δ phenotype
| Strain | Relevant genotype | fps1Δ/FPS1+ | β-Galactosidasea |
|---|---|---|---|
| JF1331 | FPS1 SLN1 SSK1 | 172.0 (14.8) | |
| JF1784 | fps1Δ SLN1 SSK1 | 2.0 | 354.0 (54.1) |
| JF1777 | FPS1 SLN1 ssk1Δ | 77.3 (8.4) | |
| JF1783 | fps1Δ SLN1 ssk1Δ | 3.5 | 272.0 (26.5) |
| JF1765 | FPS1 sln1Δ ssk1Δ | 168.0 (18.6) | |
| JF1780 | fps1Δ sln1Δ ssk1Δ | 0.8 | 135.0 (21.7) |
Activities are reported as Miller units and are the averages of six measurements involving at least three transformants in all cases.
Sln1p regulates the activity of two separate pathways in a reciprocal fashion. The Hog1p-dependent osmotic response pathway is kept inactive by phosphorylated Sln1 pathway intermediates (15), while a second (Hog1-independent) pathway monitored using the P-lacZ reporter is activated in response to phosphorylation of Sln1p (17). Likewise accumulation of unphosphorylated Sln1p increases HOG pathway activity (15) and decreases activity of the second branch.2 We have recently shown that Sln1p activation of the P-lacZ reporter is mediated by the Skn7 receiver protein.3 To determine the involvement of Skn7p in fps1Δ-mediated activation of the P-lacZ reporter, a skn7 fps1 double mutant was constructed. The data in Table V show that Skn7p is required for the fps1Δ phenotype. Deletion of the SKN7 gene suppressed the fps1Δ-mediated enhancement of P-lacZ reporter activity.
Table V.
Effect of skn7 mutation on the fps1Δ phenotype
| Strain | Relevant genotype | fps1Δ/FPS1+ | β-Galactosidase activitya |
|---|---|---|---|
| JF1331 | FPS1 SKN7 | 120.0 (17.0) | |
| JF1784 | fps1Δ SKN7 | 2.1 | 248.0 (50.0) |
| JF1738 | FPS1 skn7Δ | 93.7 (6.8) | |
| JF1770 | fps1Δ skn7Δ | 1.1 | 99.0 (18.0) |
Activities are reported as Miller units and are the averages of six measurements involving at least three transformants in all cases.
Effect of FPS1 Mutation Is Due to Osmotic Imbalance Caused by Accumulation of Intracellular Glycerol
Since FPS1 encodes a glycerol facilitator and the Hog1p pathway regulates the production of glycerol in response to osmotic stress, it seemed likely that the fps1 effect on P-lacZ activity would also require the Hog1p-dependent production of glycerol. GPD1 encodes the major NADH-dependent glycerol phosphate dehydrogenase activity in yeast and has been shown to be required for the increase in intracellular glycerol in fps1 mutants (42). We constructed an fps1Δ gpd1Δ double mutant to test whether the 2-fold increase in intracellular glycerol levels seen in fps1-Δ mutants is required for the fps1 phenotype. We found, as expected, that the elevated P-lacZ phenotype in fps1 mutants was reversed by the gpd1 mutation (Table VI).
Table VI.
Effect of GPD1 dosage on glycerol levels and the fps1Δ phenotype
| Straina | Genotype | Plasmid | Relative intracellular glycerolb | Relative β-galactosidase activityc |
|---|---|---|---|---|
| JF1566 | Wild type | None or vector | 1.0 | 100 |
| JF1825 | fps1Δ | None or vector | 2.0 | 170 |
| JF1833 | gpd1Δ | None | Not tested | 92 |
| JF1827 | fps1Δ gpd1Δ | None | Not tested | 112 |
| JF1566 | Wild type | GPD1 | 2.6 | 92 |
| JF1825 | fps1Δ | GPD1 | 4.5 | 254 |
All strains carry the integrated P-lacZ reporter plasmid, pGY88.
Relative intracellular glycerol levels were determined by normalizing to the glycerol level of the wild type strain, JF1566. The actual average glycerol concentration for JF1566 was 0.27 g of glycerol/g of protein. Glycerol concentrations were determined as described under “Experimental Procedures.” Values are the means of six determinations using at least three different transformants or colonies. Standard deviations are less than 20% of the mean.
Relative β-galactosidase activities were normalized to the wild type strain, JF1566. Values are the mean of six determinations using at least three different transformants or colonies. Standard deviations are less than 20% of the mean.
FPS1 is required for normal efflux of glycerol (29). Under hyper-osmotic stress conditions, the channel closes to allow accumulation of glycerol in the cell. Under normal growth conditions the channel is open. In fps1 deletion mutants, glycerol efflux is prevented and the normal intracellular glycerol concentration increases by a factor of two (29). We used two approaches to determine whether the fps1 mutation activates the P-lacZ reporter due to a direct effect of increased glycerol levels, or, alternatively, whether elevated internal osmolarity might provide an osmotic imbalance signal that affects downstream target genes, perhaps including the P-lacZ reporter.
To determine if P-lacZ reporter activity is a reflection of intracellular glycerol concentrations, we compared β-galactosidase levels in wild type and fps1-Δ strains plus and minus the high copy GPD1 plasmid, pWT250. In an FPS1 strain, the GPD1 plasmid is expected to elevate both intracellular and extracellular glycerol concentrations (29, 42). However, in the fps1-Δ mutant, efflux is blocked and only intracellular glycerol increases (29, 42). As expected, intracellular glycerol concentrations were elevated in both the wild type (2.6x) and fps1-Δ (4.5x) strains carrying the GPD1 plasmid (Table VI). Despite the 2.6-fold increase in intracellular glycerol concentration in the wild type strain, P-lacZ activity was increased only in the fps1-Δ strain carrying the GPD1 plasmid and not at all in the wild type strain (Table VI). These data suggest that the P-lacZ reporter activity is not tied to intracellular glycerol levels, but may instead reflect the differential between intracellular and extracellular osmotic strength.
If the fps1 phenotype were due to osmotic imbalance due to accumulation of intracellular glycerol, increasing the osmotic strength of the medium would be predicted to counteract the signal and suppress the phenotype. Since the stability of the β-galactosidase protein (t1/2 > 20 h) (44) would have precluded detecting a decline in β-galactosidase activity after transient exposure of cells to sorbitol, we measured lacZ mRNA (t1/2 ~ 30 min) (45, 46) levels in this experiment. An fps1Δ strain carrying an integrated P-lacZ reporter was cultured in YPD media and harvested at indicated times following the addition of 1.0 M sorbitol. lacZ message levels were measured by Northern (RNA) hybridization analysis (Fig. 4). At short times after addition of sorbitol there was a striking and reproducible decrease in lacZ expression in the fps1Δ mutant. The reduction in reporter gene expression by sorbitol addition in the fps1 mutant indicates that the fps1Δ effect is likely the consequence of osmotic imbalance, rather than of the accumulation of glycerol per se.
Fig. 4. Effect of sorbitol on P-lacZ transcript levels in the fps1 mutant.
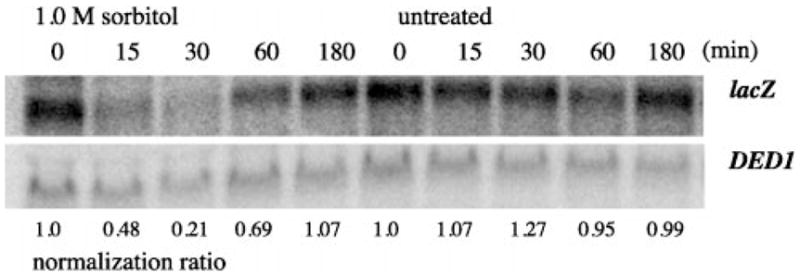
RNA was harvested from early log phase cells treated with 1.0 M sorbitol or untreated for the times (min) indicated and subjected to Northern hybridization analysis. Levels of lacZ hybridization were quantitated by PhosphorImage analysis (Molecular Dynamics) of the blot. Hybridization of the same blot to the DED1 probe was used for normalization. Values shown below the image were calculated as the ratio of lacZ/DED1 hybridization at a given time divided by the ratio of lacZ/DED1 hybridization at time 0 for a given growth condition.
DISCUSSION
Our genetic screen for modulators of Sln1 activation of P-lacZ reporter gene has led to the identification of the FPS1 gene. FPS1 encodes a MIP family channel protein which functions in glycerol transport (29). The permeability of the yeast plasma membrane for glycerol has two components, an FPS1-independent component attributable to passive diffusion, and an FPS1-dependent component representing facilitated diffusion (29). The Fps1p channel is closed under hyperosmotic stress and opens rapidly when hyperosmotic conditions are reversed (29). The Fps1 protein may also play a distinct role in controlling glycerol production, perhaps by associating with enzymes involved in glycerol metabolism, since overexpression of FPS1 unexpectedly leads to increased glycerol levels (29). Fps1p appears to play a central role in modulating intracellular glycerol concentrations under conditions of osmotic stress.
Sln1p has been shown to be a yeast osmosensor, however, the mechanism by which Sln1p senses a change in osmolarity is not known. Cell fractionation studies indicate that Sln1p is a membrane-associated protein.4 Assuming a plasma membrane localization, the role of the putative extracellular or periplasmic domain could be to bind or sense an osmolyte. However, the nature of the cellular response to osmolarity in Escherichia coli and in yeast suggest that there is wide array of salts, sugars, and other small molecules that constitute the osmotic environment and that the response to osmolarity is independent of the identity of any of these molecules. Thus, it is unlikely that the stimulus will take the form of any single osmolyte that binds to an extracellular domain. More general models of osmosensing have been suggested. For example, the existence of mechano-sensitive ion channels in the yeast plasma membrane that are activated by stretching of the membrane has led to the inference that yeasts are able to sense and respond to physical forces like osmotic pressure (47). Another possible form of stimulation of the osmotic sensors is suggested by the changes in cell shape and structure that occur in response to hyper-osmotic stress. For example, partial dehydration may alter the interaction between the plasma membrane and the cell wall. It has been previously proposed that a cell-wall component may serve as a ligand for a plasma membrane receptor (10). Changes in turgor pressure might cause an alteration in the interaction between ligand and receptor to create a signal.
Whether the stimulus is sensed extracellularly or as a perturbation in normal wall-membrane interactions, subsequent steps in signal transmission are likely to involve structural changes in the Sln1 protein that stimulate or repress the autokinase activity of the cytoplasmic histidine kinase domain. The discovery that a Sln1 regulated pathway can be activated by a signal generated in an fps1 mutant may provide some insight into the osmosensing mechanism. Given the known characteristics of the fps1 mutant, two distinct but related stimuli can be postulated. Analysis of membrane composition in fps1 mutants revealed alterations in phospholipid and glycolipid fractions in the fps1 mutants (48), suggesting that membrane composition itself may mimic the effect of osmotic stress on membrane composition, fluidity, or relationship to the wall. Alternatively, the increase in intracellular glycerol levels in an fps1 mutant might provide the signal. Our data showing a reduction in levels of P-lacZ activity in the gpd1 fps1 double mutant indicates that the increased level of intracellular glycerol in the fps1 mutant does, in fact, contribute to the P-lacZ phenotype in fps1 mutants.
How might an elevation in intracellular glycerol concentration lead to an increase in P-lacZ activity? One possibility is that increased levels of glycerol might stimulate P-lacZ expression by directly affecting the activity of a transcription factor involved in P-lacZ expression. However, this is unlikely because we have shown that Sln1p and the downstream receiver, Skn7p are required for the fps1 phenotype. In testing the requirement for Sln1p in the fps1 phenotype, we made use of a sln1Δ ssk1Δ double mutant. The ssk1 deletion was necessary to suppress the lethality of the sln1Δ mutation. However, the use of the ssk1 mutation complicated the interpretation of the results of this test since, in this experiment, the ssk1 mutation alone reduced P-lacZ activity by 50%. Comparison of fps1Δ ssk1Δ and the FPS1 ssk1Δ strains shows that the fps1 mutation caused a 3.5-fold increase in reporter activity despite the absence of Ssk1p, but did not cause an elevation in reporter activity in the absence of Sln1p. Our conclusion that Sln1p is required for the fps1 phenotype is further substantiated by analysis of the skn7 mutant strain (Table V). Like sln1, the skn7 mutation eliminated the P-lacZ activating effect due to fps1 deletion and is consistent with the requirement for Sln1 and Skn7 in the fps1 phenotype.
The decrease in reporter activity attributable to the ssk1 mutation may be due to reduced glycerol production in that genetic background. Although glycerol levels have never been directly measured in a strain retaining the Sho1p osmosensor but lacking the Sln1p contribution to Hog1p activation, both the kinetics and the responsiveness of Hog1p tyrosine phosphorylation to various osmotic stress conditions are altered in strains in which Sln1p pathway-specific molecules are defective (11).
The dependence of the fps1 phenotype on SLN1 and SKN7 indicates that fps1 activation of the P-lacZ reporter is mediated by the Sln1-Skn7 pathway. The additivity of the sln1* and fps1 mutations is likely to reflect the sum of the effects of the individual mutations on levels of phosphorylated Sln1p in the cell. Our previous studies suggest that the sln1* mutation shifts the normal Sln1—0/Sln1-P equilibrium to favor Sln1-P. Sln1-P levels may be further boosted due to an additional effect on the Sln1 equilibrium by accumulation of intracellular glycerol in fps1 mutants as depicted in the model in Fig. 5.
Fig. 5. Model illustrating the effect of FPS1 deletion on the activities of Sln1 regulated pathways.

In the absence of Fps1p, intracellular glycerol levels rise relative to extracellular levels. This differential in osmotic strength may be sensed by the cell as a hypo-osmotic environment. This activates Sln1p kinase activity and the Sln1p phosphorelay pathway and leads to the Ypd1p-dependent phosphorylation of the two receiver molecules, Ssk1p and Skn7p. Phosphorylation inactivates Ssk1, whereas phosphorylation of Skn7 leads to its activation. The phosphorylated form of Skn7 acts either directly or indirectly to activate transcription of the P-lacZ reporter.
How intracellular glycerol changes the activity of the Sln1 protein is not clear. One possibility is that glycerol, which is produced by the product of a Hog1p-dependent gene, could play a direct role in modulating the activity of Sln1p. Glycerol, itself, might stimulate Sln1p kinase activity, leading to an increase in the relative levels of Sln1-P in the cell and a corresponding decrease in Hog1 pathway activity. This would in effect lead to a negative feedback loop allowing the cell to precisely modulate the levels of intracellular glycerol that accumulate in response to osmotic stress. Alternatively, increased levels of intracellular glycerol might increase the normal osmotic gradient beyond a certain threshold causing the cell to perceive a hypo-osmotic environment. Although a slight osmotic gradient is required both to drive the entry of water important for cell growth (6, 49) and to create the turgor pressure needed to force wall expansion (50, 51), the magnitude of the “normal” gradient may be constrained. Accumulation of excess intracellular glycerol in the fps1 mutant may exceed this constraint, thus triggering a hypo-osmotic response.
Two experiments addressed this possibility. First, we used a high copy plasmid expressing the GPD1 gene to increase glycerol levels in wild type and fps1 mutants. The presence of the GPD1 plasmid caused intracellular glycerol levels to increase 2.6-fold in the wild type strain and 4.5-fold in the fps1 strain (Table VI). If P-lacZ activity were simply responding to glycerol levels, we would expect increased reporter activity in both strains. However, the activity of the P-lacZ reporter was increased only in the fps1 mutant. Since the fps1 mutant is defective in glycerol efflux, the extra intracellular glycerol contributed by GPD1 overexpression might exacerbate the osmotic imbalance already created by the fps1 mutation, whereas the increased intracellular glycerol concentration in the wild type strain would be rapidly dissipated. Hence, the elevation in P-lacZ activation in the fps1 mutant but not in the wild type strain carrying the GPD1 plasmid is consistent with a model in which the P-lacZ reporter responds to osmotic imbalance.
Results of experiments in which the presumptive gradient caused by the fps1 mutant was collapsed by addition of sorbitol to the medium also favor the interpretation that it is not the increased glycerol levels per se, but rather the osmotic imbalance that is important for P-lacZ activation in the fps1 mutant. The 2-fold elevation in P-lacZ levels in the fps1 mutant (Fig. 1) is eliminated within 15 min of sorbitol addition (Fig. 4). P-lacZ transcript levels continue to decrease, reaching a minimum at 30 min at 25% of fps1 levels (50% of wild type levels). lacZ levels could be seen to increase following sorbitol treatment beginning at 60 min. By 180 min, levels were comparable to those measured at the beginning of the experiment prior to addition of sorbitol, presumably because the normal osmotic balance has been restored.
Taken together, our results are consistent with a model (Fig. 5) in which the fps1 signal is interpreted by Sln1 (presumably triggering Sln1 phosphorylation) and transmitted through Skn7 to undefined downstream genes, ultimately stimulating expression of the P-lacZ reporter. The signal generated by the fps1 mutation appears to be osmotic imbalance due to accumulation of intracellular glycerol. Elevated intracellular glycerol levels may mimic a hypo-osmotic stimulus. Saito and colleagues (15, 16) have shown that hyper-osmotic conditions shift the equilibrium between phosphorylated and unphosphorylated Sln1 to favor the unphosphorylated form. The sln1* activating mutations we previously described appear to shift the equilibrium to favor the phosphorylated form of Sln1, counteracting the normal hyper-osmotic response (17). In the present study we show that loss of the Fps1p glycerol facilitator protein mimics the effect of activating mutations in SLN1, suggesting that the Sln1p signal need not be extracellular. The model outlined in Fig. 5 proposes that hyper-osmotic conditions trigger Sln1p dephosphorylation and Hog1 pathway activation, whereas hypo-osmotic conditions trigger Sln1 phosphorylation and Mcm1p pathway activation. Further studies will be required to determine how the Sln1p phosphorylation status is modulated by hyper- and hypo-osmotic conditions.
Acknowledgments
We acknowledge the assistance of Guoying Yu in constructing and characterizing the MCM1-Myc plasmid; Greg Gingerich and Cherie Malone for strain construction and technical assistance; Stefan Hohmann for the GPD1 plasmid, YEpGPD1, and Robert Fuller for the KEX2 plasmid; Ira Herskowitz and members of the Fassler and Deschenes laboratories for helpful discussions; and Robert Malone and Lois Weisman for critical review of the manuscript.
Footnotes
This work was supported by American Cancer Society Grant VM-148.
The abbreviations used are: MAP kinase, mitogen-activated protein kinase; MEK, MAP kinase kinase; MEK kinase, MAP kinase kinase kinase; P site, Mcm1-binding site; P-lacZ, Mcm1-dependent reporter; UAS, upstream activation sequence; kb, kilobase pairs; X-gal, 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside; MIP, major intrinsic protein of lens fiber gap junctions in mammals; PKC, protein kinase C.
S. Dean and J. Fassler, unpublished observations.
Li, S., Ault, A., Malone, C. L., Raitt, D., Dean, S., Johnston, L. H., Deschenes, R. J., and Fassler, J. S. (1998) EMBO J. 17, 6952–6962.
H. Lin, C. Malone, and R. Deschenes, unpublished observations.
References
- 1.Mager WH, Varela JCS. Mol Microbiol. 1993;10:253–258. doi: 10.1111/j.1365-2958.1993.tb01951.x. [DOI] [PubMed] [Google Scholar]
- 2.Brewster JL, Gustin M. Yeast. 1994;10:425–439. doi: 10.1002/yea.320100402. [DOI] [PubMed] [Google Scholar]
- 3.Chowdhury S, Smith KW, Gustin MC. J Cell Biol. 1992;118:561–571. doi: 10.1083/jcb.118.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blomberg A, Adler L. J Bacteriol. 1989;171:1087–1092. doi: 10.1128/jb.171.2.1087-1092.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blomberg A. Yeast. 1997;13:529–539. doi: 10.1002/(SICI)1097-0061(199705)13:6<529::AID-YEA103>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 6.Martinez de Maranon M, Marechal PA, Gervais P. Biochem Biophys Res Commun. 1996;227:519–523. doi: 10.1006/bbrc.1996.1539. [DOI] [PubMed] [Google Scholar]
- 7.Attfield PV. Nature Biotech. 1997;15:1351–1357. doi: 10.1038/nbt1297-1351. [DOI] [PubMed] [Google Scholar]
- 8.Albertyn J, Hohmann S, Thevelein JM, Prior BA. Mol Cell Biol. 1994;14:4135–4144. doi: 10.1128/mcb.14.6.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nevoigt E, Stahl U. FEMS Microbiol Rev. 1997;21:231–241. doi: 10.1111/j.1574-6976.1997.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 10.Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. Science. 1993;259:1760–1762. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- 11.Maeda T, Takekawa M, Saito H. Science. 1995;269:554–558. doi: 10.1126/science.7624781. [DOI] [PubMed] [Google Scholar]
- 12.Posas F, Saito H. Science. 1997;276:1702–1705. doi: 10.1126/science.276.5319.1702. [DOI] [PubMed] [Google Scholar]
- 13.Ota IM, Varshavsky A. Science. 1993;262:566–568. doi: 10.1126/science.8211183. [DOI] [PubMed] [Google Scholar]
- 14.Stock JB, Stock AM, Mottonen JM. Nature. 1990;344:395–400. doi: 10.1038/344395a0. [DOI] [PubMed] [Google Scholar]
- 15.Maeda T, Wurgler-Murphy S, Saito H. Nature. 1994;369:242–245. doi: 10.1038/369242a0. [DOI] [PubMed] [Google Scholar]
- 16.Posas F, Wurgler-Murphy SM, Maeda T, Witten EA, Thai TC, Saito H. Cell. 1996;86:865–875. doi: 10.1016/s0092-8674(00)80162-2. [DOI] [PubMed] [Google Scholar]
- 17.Fassler JS, Gray WM, Malone CL, Tao W, Lin H, Deschenes RJ. J Biol Chem. 1997;272:13365–13371. doi: 10.1074/jbc.272.20.13365. [DOI] [PubMed] [Google Scholar]
- 18.Yu G, Deschenes RJ, Fassler JS. J Biol Chem. 1995;270:8739–8743. doi: 10.1074/jbc.270.15.8739. [DOI] [PubMed] [Google Scholar]
- 19.Kuo MH, Grayhack E. Mol Cell Biol. 1994;14:348–359. doi: 10.1128/mcb.14.1.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin DE, Fields FO, Kunisawa R, Bishop JM, Thorner J. Cell. 1990;62:213–224. doi: 10.1016/0092-8674(90)90360-q. [DOI] [PubMed] [Google Scholar]
- 21.Costigan C, Gehrung S, Snyder M. Mol Cell Biol. 1992;12:1162–1178. doi: 10.1128/mcb.12.3.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KS, Levin DE. Mol Cell Biol. 1992;12:4396–4405. doi: 10.1128/mcb.12.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irie K, Takase M, Lee KS, Levin DE, Araki H, Matsumoto K, Oshima Y. Mol Cell Biol. 1993;13:3076–3083. doi: 10.1128/mcb.13.5.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KS, Irie K, Gotoh Y, Watanabe Y, Araki H, Nishida E, Matsumoto K, Levin DE. Mol Cell Biol. 1993;13:3067–3075. doi: 10.1128/mcb.13.5.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres L, Martin H, Garcia-Saez MI, Arroyo J, Molina M, Sanchez M, Nombela C. Mol Microbiol. 1991;5:2845–2854. doi: 10.1111/j.1365-2958.1991.tb01993.x. [DOI] [PubMed] [Google Scholar]
- 26.Levin DE, Bartlett-Heubusch E. J Cell Biol. 1992;116:1221–1229. doi: 10.1083/jcb.116.5.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu J, Yoda K, Yamasaki M. Mol Gen Genet. 1994;242:641–648. doi: 10.1007/BF00283417. [DOI] [PubMed] [Google Scholar]
- 28.Davenport KR, Sohaskey M, Kamada Y, Levin DE, Gustin MC. J Biol Chem. 1995;270:30157–30161. doi: 10.1074/jbc.270.50.30157. [DOI] [PubMed] [Google Scholar]
- 29.Luyten K, Albertyn J, Skibbe WF, Prior BA, Ramos J, Thevelein JM, Hohmann S. EMBO J. 1995;14:1360–1371. doi: 10.1002/j.1460-2075.1995.tb07122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothstein RJ. Methods Enzymol. 1983;101:202–209. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 31.Guldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothstein R. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 33.Sherman F, Fink GR, Hicks JB. Methods in Yeast Genetics. Cold Spring Harbor Laboratories; Cold Spring Harbor, NY: 1986. pp. 163–167. [Google Scholar]
- 34.Larson GP, Itakura K, Ito H, Rossi JJ. Gene (Amst) 1983;22:31–39. doi: 10.1016/0378-1119(83)90061-6. [DOI] [PubMed] [Google Scholar]
- 35.Yu G, Fassler JS. Mol Cell Biol. 1993;13:63–71. doi: 10.1128/mcb.13.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gray WM, Fassler JS. Mol Cell Biol. 1996;16:347–358. doi: 10.1128/mcb.16.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guarente L, Ptashne M. Proc Natl Acad Sci U S A. 1981;78:2199–2203. doi: 10.1073/pnas.78.4.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feinberg AP, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 40.Feinberg AP, Vogelstein B. Anal Biochem. 1984;137:266–267. doi: 10.1016/0003-2697(84)90381-6. [DOI] [PubMed] [Google Scholar]
- 41.Yaffe MP, Schatz G. Proc Natl Acad Sci U S A. 1984;81:4819–4823. doi: 10.1073/pnas.81.15.4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Philips J, Herskowitz I. J Cell Biol. 1997;138:961–974. doi: 10.1083/jcb.138.5.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conde J, Fink GR. Proc Natl Acad Sci U S A. 1976;73:3651–3655. doi: 10.1073/pnas.73.10.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bachmair A, Finley D, Varshavsky A. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 45.Purvis IJ, Bettany AJE, Loughlin L, Brown AJP. Nucleic Acids Res. 1987;15:7951–7962. doi: 10.1093/nar/15.19.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purvis IJ, Loughlin L, Bettany AJ, Brown AJP. Nucleic Acids Res. 1987;15:7963–7974. doi: 10.1093/nar/15.19.7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gustin MC, Zhou XL, Martinac B, Kung C. Science. 1988;242:762–766. doi: 10.1126/science.2460920. [DOI] [PubMed] [Google Scholar]
- 48.Sutherland FCW, Lages F, Lucas C, Luyten K, Albertyn J, Hohmann S, Prior BA, Kilian SG. J Bacteriol. 1997;179:7790–7795. doi: 10.1128/jb.179.24.7790-7795.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dale JE, Sutcliffe JF. In: Plant Physiology: A Treatise. Steward FC, editor. IX. Academic Press; NY: 1986. pp. 1–48. [Google Scholar]
- 50.Ortega JKE, Zehr EG, Keanini RG. Biophys J. 1989;56:465–475. doi: 10.1016/S0006-3495(89)82694-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cosgrove D. Annu Rev Plant Physiol. 1986;37:377–405. doi: 10.1146/annurev.pp.37.060186.002113. [DOI] [PubMed] [Google Scholar]
- 52.Winston F, Dollard C, Ricupero-Hovasse SL. Yeast. 1995;11:53–55. doi: 10.1002/yea.320110107. [DOI] [PubMed] [Google Scholar]