


Abstract
Oligonucleotide models of ribosomal RNA domains are powerful tools to study the binding and molecular recognition of antibiotics that interfere with bacterial translation. Techniques such as selective chemical modification, fluorescence labeling and mutations are cumbersome for the whole ribosome but readily applicable to model RNAs, which are readily crystallized and often give rise to higher resolution crystal structures suitable for detailed analysis of ligand–RNA interactions. Here, we have investigated the HX RNA construct which contains two adjacent ligand binding regions of helix h44 in 16S ribosomal RNA. High-resolution crystal structure analysis confirmed that the HX RNA is a faithful structural model of the ribosomal target. Solution studies showed that HX RNA carrying a fluorescent 2-aminopurine modification provides a model system that can be used to monitor ligand binding to both the ribosomal decoding site and, through an indirect effect, the hygromycin B interaction region.
INTRODUCTION
Aminoglycosides are natural products that bind to the small (30S) ribosomal subunit and interfere with bacterial translation. A highly substituted cyclohexane scaffold, 2-deoxystreptamine (2-DOS), which is conserved among structurally diverse aminoglycosides, confers specific RNA recognition (Figure 1) (1–3). Compounds from the two largest families of aminoglycosides, including neomycin and kanamycin, bind to the ribosomal decoding site (A-site) at an internal loop of helix h44 in 16S rRNA and reduce fidelity of mRNA translation (4,5). At least three other distinct binding sites for aminoglycosides of unusual structure exist in the 30S subunit (6). These include hygromycin B, streptomycin and spectinomycin. Hygromycin B, an aminoglycoside with a unique spiro-acetal structure (Figure 1), binds to a region of h44 immediately adjacent to the decoding site loop (4,7,8) and primarily inhibits translocation of mRNA and tRNAs on the ribosome but only marginally impacts decoding fidelity (8,9). Unlike the A-site target of aminoglycoside antibiotics, which harbors specific sequence differences between bacteria and eukaryotes, the hygromycin B binding site is conserved between lineages (Figure 1). While the utility of decoding site-binding aminoglycosides as antibiotics emerges from a combination of discrimination for the bacterial target RNA as well as their inability to permeate mammalian cells (10), hygromycin B lacks bacterial target specificity (11), preventing its use in anti-infective therapy but rendering it a widely used tool compound for selection in cell culture.
Figure 1.

Aminoglycosides and the ribosomal RNA helix h44 target. (a) Aminoglycosides of the neomycin and kanamycin families as well as hygromycin B share in common a conserved 2-deoxystreptamine (2-DOS) ring (highlighted in blue). (b) Overlay of crystal structures showing paromomycin (green) and hygromycin B (orange) bound to the h44 RNA (7,29). (c) Secondary structure of h44 in 16S rRNA which contains the decoding site loop (A1408/A1492/A1493). Residues that differ between the bacterial and eukaryotic sequence are in bold. Binding regions for aminoglycosides are marked in color: neomycin and kanamycin families, green; hygromycin B, orange. (d) Secondary structure of the HX RNA construct used in this work for crystallization and solution binding studies.
The adjacency of the hygromycin B binding region to the decoding site target of the neomycin and kanamycin antibiotics had inspired the design of aminoglycoside hybrid ligands that were conceived to bridge between the two sites and thereby interfere with ribosomal function (12). A first generation of hybrid ligands was synthesized that had activity as inhibitors of bacterial translation, however, none of the compounds showed potency superior to the natural aminoglycosides. Here, we investigated the bipartite HX oligonucleotide (Figure 1) as a target model for the adjacent aminoglycoside binding sites in helix h44 to support the discovery of synthetic antibacterial ligands. Similar RNA models, consisting of two oligonucleotides with distinct sequences, as well as ‘dimeric’ constructs which are formed from two identical strands and contain two decoding sites (13), have been widely used to study high-resolution structure, dynamics and drug binding of the ribosomal decoding site (14,15).
In this contribution, we describe fluorescence labeling in solution as well as X-ray crystallographic structure determination of HX RNA as a model of the aminoglycoside antibiotic binding region in helix h44. Our goals were to obtain a high-resolution crystal structure for the HX RNA, which could be compared to the h44 structure in the ribosome, and to demonstrate aminoglycoside binding to the model RNA. We show that, similar to the smaller decoding site, the HX RNA can be used as a faithful model of the extended aminoglycoside binding region of h44 that retains dynamic and ligand binding characteristics of the ribosomal target.
MATERIALS AND METHODS
RNA and aminoglycosides
Gel-purified and desalted synthetic oligonucleotides (HX-A: 5′-CCG CGC CCG UCA CAC CAC CCG; HX-B: 5′-GGG UGG UGA AGU CGU AAC GCG GC) as well as brominated derivatives [HX-ABr: 5′-CCG CGC CCG (5-Br-U)CA CAC CAC CCG; HX-BBr: 5′-GGG (5-Br-U)GG UGA AGU CGU AAC GCG GC] and 2AP-labeled RNA were purchased from Dharmacon (Lafayette, CO). Oligonucleotides HX-A and HX-B were reconstituted without further purification in 10 mM sodium cacodylate buffer, pH 6.5, mixed and annealed by heating to 75°C followed by snap cooling on ice to give reconstituted HX RNA. Aminoglycosides were purchased as sulfates from Sigma (St Louis, MO) and used without further purification.
Crystallization and X-ray diffraction data collection
For crystallization of HX RNA by hanging drop vapor diffusion, 1 μl of 0.2 mM HX RNA in annealing buffer was mixed with an equal amount of precipitating solution containing 50 mM sodium cacodylate pH 6.5, 1 mM magnesium chloride and 1.95 M ammonium sulfate. Hexagonal pyramidal-shaped crystals grew within two days at 16°C after equilibration against 700 μl of well solution containing 1.95 M ammonium sulfate. Crystals were soaked for ∼30 s in a cryoprotectant buffer containing the original reservoir solution as well as 20% PEG 200 and flash-cooled in liquid nitrogen. Diffraction data were collected in a nitrogen stream at 100 K on beamline 17-ID at the Advanced Photon Source (APS), Argonne National Laboratory (IL). Two hundred images were collected at a crystal-to-detector distance of 20 cm with an exposure time of 10 s per frame with 1° oscillation. Data were processed, integrated and scaled with the HKL2000 package (Table 1) (16).
Table 1.
Data collection and refinement statistics for HX RNA
| Data collection | |
|---|---|
| Wavelength (Å) | 0.919411 |
| Low-resolution limit (Å) | 2.3 |
| High-resolution limit (Å) | 28.7 |
| Redundancy | 12.5 (12.2) |
| Completeness (%) | 99.7 (100) |
| I/σ(I) | 36.22 (5.31) |
| Total reflections | 61 667 |
| Unique reflections | 4941 |
| Refinement | |
| Space group | P64 |
| Cell dimensions (Å) | |
| a | 57.46 |
| b | 57.46 |
| c | 58.43 |
| Rwork/Rfree | 0.191/0.231 |
| No. of atoms | |
| RNA atoms | 935 |
| Solvent atoms | 29 |
| Metal ions | 3 Mg2+ |
| Anions | 2 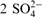
|
| Mean B factors (Å2) | |
| RNA | 57.84 |
| Solvent | 53.21 |
| Metal | 64.1 |
| Anion | 95.65 |
| RMSD | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.4 |
| Dihedral angles (°) | 17.11 |
Numbers in parentheses are for the highest-resolution shell.
Structure solution and refinement
Phases were calculated from single-wavelength anomalous diffraction data of the brominated RNA derivative using the CNS suite of programs (17,18). After positions of heavy atoms were identified, an electron density map was calculated that guided the manual construction of an initial three-dimensional structure model of the RNA in Coot (19). The orientation and position of this model were refined by rigid body refinement with each chain as a rigid group in the program Refmac (20) within the CCP4 package (21). The structure was further refined using several rounds of restrained minimization in Refmac and manual rebuilding in Coot based on the obtained 2Fo − Fc and Fo − Fc maps. During the model rebuilding, three magnesium cations and two sulfate anions were placed based on electron density. The progress of the model rebuilding and refinement was monitored using Rfree, which was calculated from a randomly chosen test set comprising 4.6% of the data. Final refinement was carried out with the PHENIX software (22) using combined TLS and individual isotropic atomic displacement parameters (Table 1). During the last cycles, the water picking protocol in PHENIX was added. The refined structure has been deposited with the Protein Data Bank (accession code 3LOA).
UV melting
UV melting experiments were performed on a thermostatted UV-2401PC spectrofluorometer (Shimadzu, Columbia, MD) according to established procedures (23). RNA at 750 nM concentration in 10 mM sodium cacodylate buffer, pH 6.5, was heated from 25–85°C at a rate of 0.25 degree/min. Depending on the experiment, 6 mM MgCl2 or 10 μM aminoglycoside were present. RNA samples were covered with a thin layer of silicone oil to prevent evaporation during heating. Absorption was recorded at 260 nm every 6 s.
Fluorescence spectroscopy
Fluorescence measurements were performed on a thermostatted RF-5301PC spectrofluorometer (Shimadzu, Columbia, MD) at 25°C. Emission spectra were recorded in 10 mM sodium cacodylate buffer, pH 6.5, at 1.5 μM RNA concentration and while irradiating at 310 nm (2AP). EC50 values were calculated with the Sigmaplot software (Systat Software, Point Richmont, CA) by fitting a dose response curve to the normalized fluorescence intensity plotted versus the log of aminoglycoside concentration. Normalized relative fluorescence was calculated by subtracting background signal measured in a titration of compound into buffer and normalization by the fluorescence intensity of the labeled free RNA (24).
RESULTS AND DISCUSSION
Crystal structure of HX RNA
The three-dimensional structure of the HX RNA was determined by X-ray crystallography at 2.3 Å resolution (Table 1). A series of bipartite oligonucleotide constructs, which comprised the ribosomal decoding site as well as the adjacent hygromycin binding region (Figure 1), were screened for crystallization. Ultimately, the HX RNA was chosen since it formed large crystals giving the best diffraction quality. This RNA contains additional terminal base pairs and 3′ unpaired nucleotide overhangs which facilitated packing in the crystal. The structure was solved by using phases calculated from single-wavelength anomalous diffraction of RNA containing two 5-bromo substituted uridines (U1406, U1487). In the crystal, the HX RNA adopts an overall straight double-stranded helix (Figure 2). Pseudo-continuous stacking of neighboring helices is stabilized by intermolecular base pair formation between complementary 3′ overhanging nucleotides. Three Mg2+ and two 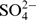 ions are present in the crystal structure. One cation and anion each are located at interfacial positions, participating in the crystal packing.
ions are present in the crystal structure. One cation and anion each are located at interfacial positions, participating in the crystal packing.
Figure 2.

Stereo views of the three-dimensional structure of HX RNA as determined by X-ray crystallography. (a) Overlay of the structure and the 2Fo − Fc electron density map contoured at 1.5σ. Only a core segment comprised of the decoding site loop and hygromycin B binding site is shown (residues G1401–C1409 and G1491–C1501). Bases that are not part of Watson–Crick pairs are highlighted in color (compare secondary structure in Figure 1d). (b) The upper stem region with non-canonical base interactions. (c) The decoding site internal loop with adenine residues highlighted in red. A water molecule mediating the interaction between A1493 and the A1492 phosphate group is shown as blue sphere.
The internal loop of the decoding site, which is comprised of the residues A1498, A1492 and A1493, adopts a conformation that is distinct from the arrangements seen in previously obtained structures of this region in context of the whole 30S ribosomal subunit (25,26) and similar model oligonucleotides that lacked the hygromycin B binding region (15,27). Packing of HX RNA in the crystal involves the A1492 base which is rotated away from the helix and interacts with a neighboring RNA molecule. The base of A1493 is oriented inside the minor groove, filling the void opposite to A1408, where it is stabilized by a single hydrogen bond between HN6 of A1493 and N1 of A1408 as well as stacking on G1494 and a water-mediated contact to the phosphate group of A1492. In structures of the decoding site in the ligand-free state of whole 30S subunits (25,26), where the conformation of residues in h44 is unaffected by crystal packing interactions, A1492 is oriented inside the RNA helix and, in one case (PDB ID 2AW2), forming a base pair with A1408 (26), while A1493 has been found in either of two states, rotated out or stacking inside on A1492. The positions of A1493 in the HX crystal structure and the ribosome are similar. In oligonucleotide models of the decoding site, the orientation of A1492 and A1493 is constrained by involvement of those residues in crystal packing contacts, which may bear little relevance to the conformational states in solution. An identical conformation of the two sensor adenines as found in the HX crystal structure has also been observed in the ‘empty’ decoding site of dimeric model constructs which were complexed with aminoglycoside ligands at only one of the two decoding sites (3,28). In addition to the three adenines of the decoding site loop, residues that are involved with aminoglycoside binding include the C1407–G1494 and U1406○U1495 pairs. The geometry of the non-canonical pair U1406○U1495, which contains a bromo modification (5-Br-U1406) in the HX RNA, is identical to that in the ribosome and decoding site model oligonucleotides.
In the hygromycin B binding region, the arrangement of bases corresponds to the secondary structure shown in Figure 1c, which reflects the configuration found in crystal structures of the ribosome and the 30S subunits (25,26). Non-canonical base pairs occur between co-planar residues C1402 and A1500 as well as C1403 and U1498. The base of A1499 docks at the minor groove edge of the C1403○U1498 pair while forming interactions with the purine of A1403 and the 2′-OH group of U1498. The flanking pairs G1401–C1501 and C1404–G1497 adopt regular Watson–Crick geometries. Superposition of HX RNA and the corresponding region of h44 in the crystal structures of the ribosomal 30S subunit, free and in complex with the aminoglycoside, reveals that the geometry of the hygromycin B binding site is nearly identical in the model RNA and the ribosomal subunit structures, attested by root mean square deviation (RMSD) values of 1.1–1.2 Å (Figure 3). At the major groove edge of the C1404–G1497 pair in the hygromycin B binding site of the HX RNA, a Mg2+ ion is found which is absent in crystal structures of the ligand-free 30S subunit. Considering the relatively low concentration of 1 mM Mg2+ in the crystallization conditions for HX RNA, it is conceivable that this metal binding site is physiologically relevant but not resolved in the crystal structures of the whole ribosome and the 30S subunit.
Figure 3.

Stereo view of a superposition of the hygromycin B binding site in HX RNA (white backbone) and the corresponding region in the crystal structures of the ribosomal 30S subunit, free (orange, PDB 1J5E; RMSD = 1.2 Å) (25) and in complex with the aminoglycoside ligand (green, PDB 1HNZ; RMSD = 1.1 Å) (7). Color coding of bases in the HX structure is the same as in Figure 2. The positions of an Mg2+ ion (yellow) as well as a bromine in 5-bromo uridine 1406 (blue) in the HX structure are indicated with spheres. RMSD values were calculated for superposition of residues C1403–C1409 and G1494–U1498; RMSD of 1J5E versus 1HNZ=0.4 Å.
The high similarity of the hygromycin B binding site geometry in the HX RNA compared to whole 30S subunits suggested that the small oligonucleotide might provide a useful model system for the study of ligand interactions at the ribosomal h44 helix. We next investigated the impact of aminoglycoside and metal ion binding on the thermal stability and conformational mechanics of the decoding site in the HX RNA.
Impact of ligand binding on the stability of the HX RNA hybrid
Thermal denaturation of HX RNA in buffer was monitored by measuring absorption at 260 nm while heating in the absence and presence of ligands (Figure 4a). Monophasic melting profiles were recorded for the free RNA and when hygromycin B or Mg2+ were present. A biphasic denaturation transition was observed after addition of paromomycin, a decoding site-binding aminoglycoside related to neomycin B (Figure 1a). Both aminoglycosides as well as Mg2+ led to increased melting temperatures. In all cases, the temperature shift was substantial, which indicated stabilization of the HX RNA through binding of the ligands. Hygromycin B at 10 μM increased the thermal stability of the RNA by 7°C while paromomycin at the same concentration had an even greater effect. The occurrence of two transitions in the denaturation profile suggested that a larger conformational change precedes the dissociation of the oligonucleotide hybrid complex for paromomycin but not for either hygromycin B or Mg2+.
Figure 4.

Solution studies of HX RNA. (a) UV melting profiles of HX RNA, free (blue; Tm=38.4°C) and in the presence of Mg2+ (6 mM, light green; Tm = 56.4°C), hygromycin B (10 µM, orange; Tm = 45.4°C), or paromomycin (10 μM, green; Tm,1 = 41.5°C, Tm,2 = 58.8°C). (b) Fluorescence of the 2AP label at position 1493 during formation of the HX RNA hybrid by titration of the 2AP-labeled single strand (HX-B) with increasing amounts of the other oligonucleotide (HX-A). (c) Titrations of 2AP1493 labeled HX RNA with Mg2+ (light green, EC50 = 17 ± 5 μM), hygromycin B (orange, EC50 = 3.1 ± 0.6 μM) or paromomycin (green, EC50 = 0.14 ± 0.01 μM). (d) Titrations of 2AP1493 labeled HX RNA with hygromycin B in the presence of Mg2+ (black: 0; blue: 10 μM; green: 100 μM; orange: 320 μM; red: 1000 μM).
Solution studies of fluorescently labeled HX RNA
The conformationally flexible adenine residues A1492 and A1493 play a key role in ribosomal decoding by acting as molecular sensors that monitor the formation of a correctly base-paired hybrid between the mRNA codon and the tRNA anticodon (5). The bases of the sensor adenines occupy positions inside or rotated out of the h44 helix. The conformational transition into the ‘flipped out’ state in which both A1492 and A1493 are rotated into the decoding site space is observed when mRNA and cognate tRNA are bound. The same transition is triggered by binding of aminoglycosides of the neomycin and kanamycin families to the decoding site loop (29). It has been suggested that aminoglycoside binding to the decoding site RNA lowers the energy barrier for the conformational transition of the sensor adenines and thereby interferes with discrimination between cognate and near-cognate anticodon interactions which ultimately increases the error rate during translation (5,30). Replacement of either A1492 or A1493 by fluorescent base analogs, such as 2-aminopurine (2AP) or pteridines, in oligonucleotide models of the decoding site has been used to study conformational states of the adenine sensors and to monitor conformational changes triggered by ligand binding (24,27,31), including aminoglycosides. Here, we have introduced 2AP as a fluorescent replacement of A1493 in HX RNA. Titration of 2AP1493 labeled oligonucleotide (HX-B) with increasing amounts of the complementary strand (HX-A) resulted in a steady increase of the 2AP fluorescence intensity up to a 1:1 strand ratio (Figure 4b), indicating full hybridization. As has been found before in model RNAs of the decoding site alone, in the hybridized HX RNA, the 2AP1493 preferentially adopted a conformation rotated out of the helix and directed into the solvent which led to increased fluorescence compared with that generated from the random coiled single strand (Figure 5, ‘free’ state) (24,27). With respect to the decoding site conformational mechanics, the HX RNA thus behaved undistinguishable to smaller model systems which were lacking the hygromycin B binding region.
Figure 5.

Model illustrating conformational changes in the decoding site of fluorescently labeled 2AP1493 HX RNA, free and upon binding of neomycin- and kanamycin-type aminoglycosides, hygromycin B, or Mg2+. See text for ‘Discussion’ section.
We next tested the response of 2AP1493 labeled HX RNA to addition of aminoglycosides and Mg2+. When paromomycin was titrated to HX RNA, a decrease of fluorescence occurred (Figure 4c), exactly as had been observed for smaller model RNAs of the decoding site (24,27,31). Upon binding of the aminoglycoside, A1492 is expelled from the internal loop and forced to stack on the rotated-out 2AP1493 base, which leads to fluorescence quenching (Figure 5, ‘Neo/Kan’ state). Similarly, titrations of HX RNA with tobramycin, a decoding site-binding aminoglycoside related to kanamycin (Figure 1a), resulted in fluorescence quenching (data not shown). The EC50 values of the dose response for fluorescence quenching by paromomycin (0.14 ± 0.01 μM) and tobramycin (1.5 ± 0.10 μM) were in good agreement with values previously reported for 2AP1493 labeled decoding site models (24,27,31). The HX construct thus retained the binding characteristics of decoding site model RNAs for aminoglycosides of the neomycin and kanamycin families. While simple decoding site RNAs do not monitor binding of hygromycin B, the HX RNA was responsive to this aminoglycoside, showing an increase of fluorescence (Figure 4c), which suggested that a higher exposure of the 2AP1493 label was induced by binding of this compound. We speculate that binding of hygromycin B to HX RNA led to reduced flexibility in the adjacent decoding site loop and immobilization of the sensor adenines in their more preferred conformations, oriented inside the RNA for A1492 and rotated-out for A1493, which resulted in an increase of fluorescence (Figure 5, ‘Hyg’ state). Previous studies of fluorescently labeled decoding site RNAs indicated that, in the absence of stabilizing ligands, A1493 samples conformational space in the interior and outside the RNA helix, albeit with a preference for the rotated-out orientation (24,27). Conformational flexibility of the decoding site internal loop might be more pronounced in the HX RNA due to the adjacent non-canonical base pairs. Binding of hygromycin B at these non-canonical pairs might stabilize the region above the decoding site loop and thereby reduce conformational flexibility of the sensor adenines. As a consequence, 2AP1493 is locked in the rotated-out position where its fluorescence is increased relative to free HX RNA. The orientation of A1492 and A1493 supported by our fluorescence data is consistent with the conformation of the sensor adenines observed in the crystal structure of the full Escherichia coli ribosome in complex with hygromycin B (8). A fluorescence increase was also observed when Mg2+ was added (Figure 4c), although the metal required a higher concentration to achieve the same effect as hygromycin B (EC50 values: Mg2+, 17 ± 5 μM; hygromycin B, 3.1 ± 0.6 μM). Binding of hygromycin B, but not paromomycin, to HX RNA was in direct competition with Mg2+ as demonstrated by titration curves recorded in the presence of increasing amounts of the metal (Figure 4d).
The HX RNA as a model for ligand binding studies at ribosomal helix h44
While crystal structures are now available for the bacterial ribosome, its subunits, and antibiotics complexes thereof, small oligonucleotide models of ribosomal RNA remain useful for investigations of RNA molecular recognition and ligand screening. Powerful techniques such as selective chemical modification, fluorescence labeling and mutations are cumbersome for the whole ribosome but readily applicable to model RNAs, which are readily crystallized and often give rise to higher resolution crystal structures suitable for detailed analysis of ligand–RNA interactions. Here, we have shown that the HX RNA is a useful model of two adjacent ligand binding regions in the ribosomal RNA helix h44. High-resolution crystal structure analysis confirmed that the HX RNA is a faithful structural model of the ribosomal target. Solution studies showed that HX RNA carrying a fluorescent 2AP1493 modification provides a model system that can be used to monitor ligand binding to both the ribosomal decoding site and, through an indirect effect, the hygromycin B interaction region (Figure 5). In the free HX RNA, a fluorescence signal of intermediate intensity originates from 2AP1493 sampling conformational space with a preference for orientations outside the RNA helix. Binding of aminoglycosides of the neomycin and kanamycin families to the decoding site internal loop induces a conformation in which fluorescence is quenched due to expulsion of A1492 and mutual stacking of bases 1492 and 1493. Ligand interaction in the region adjacent to the decoding site loop, which is the binding site for hygromycin B, stabilizes non-canonical base pairs and reduces flexibility of the sensor adenines such that 2AP1493 is locked in the rotated-out conformation where its gives rise to high fluorescence. This indirect effect on 2AP1493 fluorescence has been observed for the aminoglycoside hygromycin B as well as Mg2+. We are currently using the HX RNA in the screening for new ligands that interact with helix h44 at both the hygromycin B binding region and the decoding site. Such ligands will be useful as tool compounds to study ribosomal function and in the development of new antibacterials.
FUNDING
Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the IMCA-CAT beamline 17-ID at the Advanced Photon was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Faculty startup funds from the University of California, San Diego. Funding for open access charge: University of California, San Diego startup funds.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Chandler Ho for help with crystallization trials.
REFERENCES
- 1.Busscher GF, Rutjes FP, van Delft FL. 2-Deoxystreptamine: central scaffold of aminoglycoside antibiotics. Chem. Rev. 2005;105:775–791. doi: 10.1021/cr0404085. [DOI] [PubMed] [Google Scholar]
- 2.Hermann T. Drugs targeting the ribosome. Curr. Opin. Struct. Biol. 2005;15:355–366. doi: 10.1016/j.sbi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Francois B, Russell RJ, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moazed D, Noller HF. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 5.Ogle JM, Ramakrishnan V. Structural insights into translational fidelity. Annu. Rev. Biochem. 2005;74:129–177. doi: 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- 6.Yonath A. Antibiotics targeting ribosomes: resistance, selectivity, synergism and cellular regulation. Annu. Rev. Biochem. 2005;74:649–679. doi: 10.1146/annurev.biochem.74.082803.133130. [DOI] [PubMed] [Google Scholar]
- 7.Brodersen DE, Clemons W.M., Jr, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 8.Borovinskaya MA, Shoji S, Fredrick K, Cate JH. Structural basis for hygromycin B inhibition of protein biosynthesis. RNA. 2008;14:1590–1599. doi: 10.1261/rna.1076908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bakker EP. Aminoglycoside and aminocyclitol antibiotics: hygromycin B is an atypical bactericidal compound that exerts effects on cells of Escherichia coli characteristics for bacteriostatic aminocyclitols. J. Gen. Microbiol. 1992;138:563–569. doi: 10.1099/00221287-138-3-563. [DOI] [PubMed] [Google Scholar]
- 10.Hermann T. Aminoglycoside antibiotics: old drugs and new therapeutic approaches. Cell Mol. Life Sci. 2007;64:1841–1852. doi: 10.1007/s00018-007-7034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabanas MJ, Vazquez D, Modolell J. Dual interference of hygromycin B with ribosomal translocation and with aminoacyl-tRNA recognition. Eur. J. Biochem. 1978;87:21–27. doi: 10.1111/j.1432-1033.1978.tb12347.x. [DOI] [PubMed] [Google Scholar]
- 12.Vourloumis D, Winters GC, Simonsen KB, Takahashi M, Ayida BK, Shandrick S, Zhao Q, Han Q, Hermann T. Aminoglycoside-hybrid ligands targeting the ribosomal decoding site. Chembiochem. 2005;6:58–65. doi: 10.1002/cbic.200400197. [DOI] [PubMed] [Google Scholar]
- 13.Vicens Q, Westhof E. Crystal structure of paromomycin docked into the eubacterial ribosomal decoding A site. Structure. 2001;9:647–658. doi: 10.1016/s0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 14.Hermann T. A-site model RNAs. Biochimie. 2006;88:1021–1026. doi: 10.1016/j.biochi.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Westhof E. Molecular recognition between the ribosomal decoding site and natural or non-natural aminoglycosides. Nucleic Acids Symp. Ser. 2005;49:59–60. doi: 10.1093/nass/49.1.59. [DOI] [PubMed] [Google Scholar]
- 16.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 17.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 18.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 19.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.Collaborative Computational Project. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr., 50, 760–763. [DOI] [PubMed] [Google Scholar]
- 22.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 23.Puglisi JD, Tinoco I., Jr Absorbance melting curves of RNA. Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- 24.Parsons J, Hermann T. Conformational flexibility of ribosomal decoding-site RNA monitored by fluorescent pteridine base analogs. Tetrahedron. 2007;63:3548–3552. [Google Scholar]
- 25.Wimberly BT, Brodersen DE, Clemons W.M., Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 26.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 27.Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, Simonsen KB, Vourloumis D, Hermann T. Monitoring molecular recognition of the ribosomal decoding site. Angew Chem. Int. Ed. Engl. 2004;43:3177–3182. doi: 10.1002/anie.200454217. [DOI] [PubMed] [Google Scholar]
- 28.Murray JB, Meroueh SO, Russell RJ, Lentzen G, Haddad J, Mobashery S. Interactions of designer antibiotics and the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2006;13:129–138. doi: 10.1016/j.chembiol.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 29.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 30.Ogle JM, Brodersen DE, Clemons W.M., Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 31.Kaul M, Barbieri CM, Pilch DS. Fluorescence-based approach for detecting and characterizing antibiotic-induced conformational changes in ribosomal RNA: comparing aminoglycoside binding to prokaryotic and eukaryotic ribosomal RNA sequences. J. Am. Chem. Soc. 2004;126:3447–3453. doi: 10.1021/ja030568i. [DOI] [PubMed] [Google Scholar]