


Abstract
Oxidized DNA bases, particularly 7,8-dihydro-8-oxoguanine (8-oxoG), are endogenously generated in cells, being a cause of carcinogenic mutations and possibly interfering with gene expression. We found that expression of an oxidatively damaged plasmid DNA is impaired after delivery into human host cells not only due to decreased retention in the transfected cells, but also due to selective silencing of the damaged reporter gene. To test whether the gene silencing was associated with a specific change of the chromatin structure, we determined the levels of histone modifications related to transcriptional activation (acetylated histones H3 and H4) or repression (methylated K9 and K27 of the histone H3, and histone H1) in the promoter region and in the downstream transcribed DNA. Acetylation of histone H4 was found to be specifically decreased by 25% in the proximal promoter region of the damaged gene, while minor quantitative changes in other tested chromatin components could not be proven as significant. Treatment with an inhibitor of histone deacetylases, trichostatin A, partially restored expression of the damaged DNA, suggesting a causal connection between the changes of histone acetylation and persistent gene repression. Based on these findings, we propose that silencing of the oxidatively damaged DNA may occur in a chromatin-mediated mechanism.
INTRODUCTION
DNA damage has a causal role in carcinogenesis, ageing and several neurological syndromes. The cytotoxic consequences of DNA damage have been linked with degenerative changes in organs during ageing (1). Most of the toxic effects of DNA lesions probably arise from their interference with replication and transcription (2). At the same time, DNA lesions that are relatively well tolerated during replication and transcription generally are not cytotoxic, but often lead to mutations. This is the case for many of the DNA lesions that arise from oxidation of DNA bases by endogenously generated reactive oxygen species (ROS). Among these lesions, 7,8-dihydro-8-oxoguanine (8-oxoG) is one of the most frequent and the best studied. Removal of 8-oxoG from the chromosomal DNA occurs via a very efficient base excision repair (BER) pathway initiated by the DNA glycosylase OGG1 (3–5) that ensures repair within several hours in mammalian cells (6). If unrepaired, 8-oxoG can be efficiently, but inaccurately bypassed by replicating DNA polymerases, leading to C→A substitutions (7,8) in the newly synthesized DNA strand. 8-OxoG does not strongly inhibit transcription by RNA polymerase II as demonstrated in various reconstituted transcription systems in vitro (9–11), and apparently has no direct effect on the transcription of plasmids introduced into cells by transfection (12). However, expression of oxidatively damaged plasmid DNA containing 8-oxoG as a predominant base lesion is compromised in cells from patients with Cockayne syndrome, which carry mutations in CSA or CSB genes (13). Transcription of plasmid DNA containing 8-oxoG bases is also affected in mouse embryonic fibroblasts with mutated CSB (14), most probably due to interference of base excision repair with ongoing transcription (15). The effect of oxidative DNA base lesions on transcription is thus relevant for the human pathological condition, namely Cockayne syndrome. Moreover, oxidative base damage apparently can affect gene expression even in the absence of genetic defects (15). Taken together, the observed effects suggest that even without constituting an elongation block for RNA polymerases, the oxidative DNA base lesions have a general role for regulation of transcription.
The transcriptional status of genes in the nucleus is governed by the chromatin structure at the regulatory promoter regions and the transcription start site (16). It has been shown that various kinds of DNA damage induce local changes in the chromatin structure (17–19). In chromosome regions that are critical for gene regulation, such chromatin changes will probably cause an altered gene expression. In fact, DNA double-strand breaks in the gene promoter have already been shown to induce multiple chromatin changes initiating permanent gene silencing in a small fraction of the repaired genes (20). However, the presence of distinctive chromatin structures at DNA damage sites of different nature and their potential relevance for regulation of gene expression remain to be established. In the present study, we aimed at understanding the role of histone modifications for the transcription of oxidatively damaged DNA.
MATERIALS AND METHODS
Plasmids and DNA damage conditions
pDsRed-Monomer-N1 (pDsRed) plasmid was from Clontech (Saint-Germain-en-Laye, France). pEGFP-mODC-ZA (pZA) that encodes for enhanced green fluorescent protein has been described previously (15). The plasmids used do not replicate in HeLa cells and express the specified reporter proteins under the control of the human cytomegalovirus (CMV) immediate early promoter. Covalently closed pZA was damaged with light from a halogen lamp (Philips PF811) in the presence of 0.8 µM methylene blue (Sigma-Aldrich, Taufkirchen, Germany), as described previously (15). Methylene blue was removed by repeated extraction with a double volume of 1-butanol followed by DNA precipitation with ethanol. Average numbers of single-strand breaks (SSBs) in plasmid DNA were calculated by the Poisson formula from the relative frequencies of covalently closed and nicked molecules determined after separation of the two DNA forms by gel electrophoresis. Oxidized guanines were quantified as SSB induced by treatment with an excess of the specific DNA glycosylase Fpg, as previously described (15). Linear dependence of generated damage on the light dose has been confirmed over the range of light exposure times (data not shown), and the plasmid containing three Fpg-sensitive base modifications (unless otherwise specified) has been used in transfection experiments. The control undamaged plasmid has been dark-incubated with methylene blue.
Transfection and protein expression analyses
Exponentially growing HeLa cells were taken for transfections. Unless otherwise specified, the cells were transfected in full medium in six-well plates (Nunc, Wiesbaden, Germany) with a mixture containing 400 ng of undamaged pDsRed plasmid DNA, 400 ng of pZA plasmid DNA (non-damaged or containing three oxidized guanine bases generated by methylene blue plus light), and Effectene® transfection reagent (Qiagen, Hilden, Germany). If intended for chromatin analyses, transfections were performed in 94-mm Petri dishes, and the amount of each plasmid scaled up to 2 µg. The transfection reagent was removed after 4 h. Eight hours after transfection start the cells were detached. One part was fixed for flow cytometric analyses as previously described (21), and the remainder was split into different wells that were harvested and fixed at the indicated times after transfection. The expression levels of DsRed-Monomer and green fluorescent protein (GFP) were analysed by flow cytometry using BD FACSCalibur™ and CellQuest Pro software (Beckton Dickinson, GmbH, Heidelberg, Germany). The median GFP fluorescence was determined in transfected cells, defined by high expression of DsRed-Monomer protein as described previously (15) applying a FL-2 threshold level of 30. The fractions of cells with inactivated GFP gene were determined as percentages of DsRed-positive cells that failed to express GFP.
Determination of plasmid survival in the cells and mRNA expression levels
Cells were split in three equal parts 8 h after transfections and incubated in six-well plates for additional 16 h. For RNA isolation, one part was lysed with TRIZOL® Reagent (Invitrogen, Karlsruhe, Germany), according to the manufacturer’s instructions. The second part was incubated for 3 h at 50°C in 0.5% sodium dodecyl sulphate supplemented with 100 mg/l Proteinase K and used for isolation of total DNA by standard phenol–chlorophorm extraction procedures followed by ethanol precipitation. The third part was used for protein expression analyses, as described in the previous section. The plasmid copy numbers in DNA samples were determined by real-time quantitative PCR. After normalization on the recovered amounts of the reference pDsRed plasmid in each sample, the surviving fractions of the damaged pZA plasmid were then calculated relative to the recovery of undamaged pZA plasmid. RNA samples were treated with DNase I (Fermentas, St. Leon-Rot, Germany) for 30 min at 37°C. RNA integrity was verified by denaturing agarose gel electrophoresis. Reverse transcription (RT) reactions were performed with a Revert Aid™ First Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany) using random hexamer primers. cDNA samples were diluted 1:10, 1:150, and 1:1500 and quantified by real-time quantitative polymerase chain reaction (PCR) with gene-specific primers chosen within the single exons. Standard curves were built by serial dilutions of DNA isolated in parallel from the same transfection samples. The absence of contamination with chromosomal or plasmid DNA was confirmed by the absence of PCR products prior to RT.
Isolation of nuclei for the chromatin analyses and plasmid-ChIP
Four million cells were plated per 94-mm Perti dish 16 h prior to transfections. The cells were transfected as described above. At the day of transfection, 20 million cells were plated per 175 cm2 flask counting one flask per each plate of the transfected cells. These cells were used as a carrier during the isolation of nuclei. 24 h after transfections, each plate of the transfected cells was processed simultaneously with one flask of the carrier cells. The growing cells were cross-linked by adding 1% formaldehyde directly to the conditioning medium for 17 min at room temperature with gentle shaking. Cross-linking reactions were stopped with 0.125 M glycine, and the cells then kept on ice for the whole procedure. Monolayers were washed once with ice-cold phosphate-buffered saline (PBS) supplemented with 0.5 mM phenylmethanesulphonylfluoride, and harvested by scraping in a smaller volume of PBS supplemented with protease inhibitors. At this point, transfected cells were mixed with the carrier cells in a total volume of 25–30 ml and collected by centrifugation. Nuclei were further isolated from the cell pellets exactly following the ‘chromatin immunoprecipitation (ChIP) assay’ protocol by A. Kouskouty and I. Kyrmizi (PROT11, The EPIGENOME Network of Excellence, www.epigenome-noe.net), and finally resuspended in 1 ml of NUC buffer (15 mM HEPES pH 7.5, 60 mM KCl, 15 mM NaCl, 0.34 mM Sucrose, 0.15 mM β-mercaptoethanol). Aliquots were 40-fold diluted with 1 M NaOH, and OD260 was measured after incubation at room temperature for 5 min. The samples were divided in portions of 25 OD260 units and processed immediately for further applications or stored at –70°C for up to 1 month.
Preparation of soluble chromatin and equilibrium centrifugation
25 OD260 units of nuclei preparations were supplemented with 10 mM CaCl2 and digested with micrococcal nuclease (Fermentas, St. Leon-Rot, Germany). Digestions were performed with 40 U micrococcal nuclease (MNase) for 8 min at 37°C. The reactions were stopped by 10 mM ethylenediaminetetraacetic acid (EDTA) and 1% sodium dodecyl sulphate (SDS). Digested nuclei (20 OD260 units) were adjusted to 2 ml with TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA) containing 1% SDS and additionally sheared using a Bachofer GM 70 HD ultrasonic processor (Bachofer, GmbH, Reutlingen, Germany) equipped with a microtip. The samples were kept on ice/water slurry and sonicated 6 min at 20% power, then 6 minutes at 50% power at 40% duty cycle. Equal DNA fragmentation between the chromatin samples was checked in small aliquots after cross-link reversal by incubation at 65°C overnight followed by electrophoresis in 1.5% agarose gels.
If the chromatin samples were planned to be analysed by CsCl density gradient centrifugation, SDS was substituted for N-lauroylsarcosine during the chromatin solubilization procedure. CsCl solutions of 58%, 45% and 34% (w/w) were prepared in TE buffer and gradients were pre-formed by overlaying 1.65 ml of each solution and 1.65 ml of solubilized chromatin. Ultracentrifugation was performed in an Optima™ ultracentrifuge (Beckmann Instruments GmbH, München, Germany) with an AN-60TI rotor at 55 kRPM for 24 h at 20°C.
ChIP
Antibodies used for ChIP are listed in Supplementary Table S1. The sonicated chromatin samples were incubated at room temperature to dissolve the precipitated SDS, then vortexed and centrifuged for 10 min at maximum speed in a benchtop microcentrifuge to remove insoluble material. The buffer was then adjusted to radioimmunoprecipitation assay (RIPA) buffer [50 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS]. Preclearing with ProteinA/Protein G-Sepharose beads, chromatin immunoprecipitation, and cross-link reversal procedures were essentially as previously described (22) with few improvements. Namely, 0.9 OD 260 units of soluble chromatin were taken for incubation with each antibody and 0.27 OD260 units kept separately as a ‘30% input’ after the preclearing step. Following cross-link reversal and proteinase K treatment, DNA was purified from the 100 ul supernatants and from the ‘input’ samples by a MinElute reaction cleanup kit (Qiagen, Hilden, Germany). Finally, DNA was eluted in 40 ul Tris–HCl, pH 8.0 for direct use as a template in PCR reactions.
Real-time quantitative PCR
We used a Light Cycler 1.5 and FastStart DNA MasterPLUS SYBR Green I kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer’s instructions. The oligonucleotides employed as PCR primers and the specific PCR parameters are listed in Supplementary Tables S2 and S3. All oligonucleotides were from Eurofins MWG Operon (Ebersberg, Germany). All primers were controlled by melting curve analyses and agarose gel electrophoresis to produce a single specific PCR product. For each measurement, at least four standards prepared as dilutions of the relevant ChIP input DNA were amplified to generate a standard curve (linear regression coefficients were not <0.98). The measurements were done at least in duplicates. Recovered amounts (R) of all analysed DNA fragments were thus measured as fractions of input DNA and expressed for convenience as per mille (‰) of input. To correct for overall ChIP efficiencies between the samples processed independently within a single ChIP experiment, the recovery of each DNA fragment originating from the pZA plasmid was calculated relative to that of the promoter region of the reference pDsRed plasmid in the same ChIP sample: Rrel = R(pZA)/R(pDsRed). Finally, mean relative recoveries for n ChIP experiments 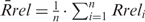 were calculated separately for the non-damaged (oG = 0) and damaged (oG = 3) pZA and for each analysed DNA fragment. Two-tailed Student’s t-test was applied to calculate P-values for the observed differences.
were calculated separately for the non-damaged (oG = 0) and damaged (oG = 3) pZA and for each analysed DNA fragment. Two-tailed Student’s t-test was applied to calculate P-values for the observed differences.
RESULTS
Gene inactivation by oxidative guanine lesions
Oxidative guanine lesions were generated in covalently closed pZA plasmid encoding for enhanced GFP by exposure to visible light in presence of the photosensitizer methylene blue. In double-stranded DNA, this treatment yields 8-oxoG as a predominant base modification, while oxidative pyrimidine products, sites of base loss and SSBs occur at many fold lower frequencies (23,24,15). Expression of GFP from the damaged or undamaged pZA plasmid was analysed at various times after delivery into the HeLa cells. To distinguish between the non-transfected cells and those with impaired GFP expression, a second (undamaged) reporter plasmid encoding for DsRed-Monomer protein was used as a tracer. Fluorescence of DsRed can be conveniently detected by flow cytometry and does not interfere with the GFP measurement. The cells expressing only DsRed-Monomer thus localize in the upper left quadrant of the fluorescence scatter plot (Figure 1A), while most of the cells expressing both DsRed-Monomer and GFP are found in the upper right quadrant, and the non transfected cells remain in the lower left quadrant.
Figure 1.

Gene inactivation by oxidative guanine lesions. (A) Dot density plots obtained by flow-cytometric analyses of the expression of DsRed-Monomer and green fluorescent protein (GFP) in HeLa cells at indicated times after co-transfection with equal amounts of pZA and pDsRed plasmids. The plasmid encoding for GFP was either not damaged (oG = 0) or contained on average three oxidized guanines (oG = 3). The transfected cells that do not express GFP localize in the upper left quadrants (marked with asterisks). (B) Quantitative analyses of the data presented in A representing percentages of the transfected cells located in the upper left quadrants (left) and median GFP fluorescence in the cells with a high DsRed-Monomer expression level (right). (C) Detection of oxidized guanine bases (oG) in the damaged plasmids by incubation with Fpg DNA glycosylase. The numbers of oxidized guanines were determined from the ratios of covalently closed circular (ccc) to open circular (oc) forms as described in ‘Materials and Methods’ section.
A representative time-course experiment demonstrating the effect of DNA damage on GFP expression is shown in Figure 1A. Each sample of the transfected cells was split into five parts to be analysed at different times after transfection. When undamaged pZA was used for transfection, virtually all cells (99.8% or more) expressing the tracer plasmid were also expressing GFP and were found in the upper right quadrant at all times. In contrast, cells transfected with the damaged pZA showed a different pattern of protein expression. Eight hours after transfection, GFP was detected in 99.2% of the transfected cells. However, the cells were gradually loosing the GFP expression during the time course as it is seen by their redistribution from the upper right to the upper left quadrant in the fluorescence scatter-plots (Figure 1A). The fraction of GFP-negative cells was then steadily increasing during the whole time of surveillance of 48 h. The most rapid decrease in GFP expression was observed between 8 and 32 h post-transfection (Figure 1B). This tendency was confirmed by calculating the average GFP fluorescence among the cells expressing high levels of DsRed-Monomer protein (see ‘Materials and Methods’ section). The DNA damage caused a 1.8-fold decrease in median GFP fluorescence 8 h after transfection. This ratio grew to 5-, 9- and more than 17-fold (16, 24 and 48 h after transfection, respectively) (Figure 1B, right chart). The results did not vary significantly between different plasmid preparations and were not influenced by the interexperimental differences in overall transfection efficiency as confirmed by at least five independent experiments.
GFP expression decreased in clear dependence on the damaging dose applied to the pZA plasmid. A very similar dose-dependent response was also observed in the expression of DsRed-Monomer when pDsRed plasmid was damaged (Supplementary Figure S1). A time course of DsRed-Monomer expression in the cells transfected with oxidatively damaged pDsRed plasmid also showed a gradual and time-dependent decrease in expression of the damaged gene (Supplementary Figure S2). The results thus indicate that oxidative base damage in DNA triggers a cellular mechanism, which affects the gene expression specifically in the damaged DNA. Strikingly, the gene expression of the damaged plasmid did not recover after two or three cell divisions. Persistent inactivation of the oxidatively damaged reporter gene therefore appears as a late consequence rather than direct effect of the DNA damage.
Effect of oxidative base damage on the amounts of plasmid DNA retained in transfected cells and the levels of mRNA expression
The decreased expression of the oxidatively damaged plasmid DNA can hardly be attributed to the impaired delivery to the host cells during transfection. Firstly, quantity and topology of the damaged DNA were not much different from the undamaged plasmid (Figure 1C). Second, expression of the damaged DNA was detected in more than 99% of the transfected cells at early time after transfection. Consequently, the damaged plasmid underwent at least one or several rounds of transcription after its delivery to the nucleus and was later modified in a manner that caused gene expression to decrease. We therefore hypothesized that the recognition of the oxidative base damage by DNA repair enzymes (e.g. OGG1 or NEIL1) or by other DNA maintenance proteins would promote either nucleolytic degradation or transcriptional silencing of the damaged DNA. To define the effect that a reduced amount of plasmid would have on the measured reporter gene expression, we performed transfections with decreasing amounts of pZA plasmid encoding for GFP, keeping the amount of the tracer pDsRed plasmid constant. We found good linear correlation between the pZA amount used for transfection and GFP fluorescence output (Figure 2A). Thus, eventual degradation of the damaged plasmid in the cell in principle could be a cause of the decreased reporter protein levels. To test this possibility, we measured the amounts of plasmid DNA residing in the cells and, in parallel, GFP fluorescence 24 h after transfections (Figure 2B). GFP expression levels in the cells transfected with non-damaged pZA plasmid (Figure 2B, upper panel) were proportional to the amounts of plasmid DNA recovered from the cells. Surviving fractions of the plasmid DNA residing in the cells were considerably decreased if DNA modifications were present (Figure 2B, lower panel). Clearly, the yields of GFP fluorescence in the cells transfected with damaged pZA plasmid were decreased to a much bigger extent than the amounts of the residing plasmid DNA (Figure 2B, lower panel). The data thus show that oxidative DNA damage not only promotes degradation of 40–60% of the plasmid DNA in the transfected cells, but also causes an even stronger decrease of expression of the remaining plasmid DNA.
Figure 2.

Plasmid survival and reporter gene expression in HeLa cells. The cells were transfected with 400 ng pDsRed plasmid (non-damaged reference) together with the indicated amounts of pZA plasmids (non-damaged or damaged, as specified). (A) Relative GFP fluorescence as a function of the amounts of non-damaged pZA plasmid used for transfection. Means of three independent experiments (±SD). (B) Comparison of DNA amounts recovered from the cells (open columns) and GFP protein expression (black columns) for the range of transfection inputs of the non-damaged (oG = 0) and damaged (oG = 3) pZA plasmid. All values are expressed relative to the values obtained after transfection with 400 ng of undamaged pZA plasmid. (C) Messenger RNA expression 24 h after transfections with non-damaged (oG = 0) and damaged (oG = 3) pZA plasmids. cDNA copy numbers (mean and data range) were determined by real-time PCR relative to the copy numbers of DNA recovered from the transfected cells. Mean GAPDH cDNA copy number is used for normalisation between the samples (duplicates).
We further tested to which extent the levels of mRNA produced from the plasmid gene were influenced by oxidative DNA damage. The levels of gene-specific transcripts were quantified by reverse transcription and real-time quantitative PCR relative to the DNA extracted from the transfected cells. Nearly the same levels of GFP and DsRed-Monomer transcripts were detected in the cells co-transfected with equal amounts of non-damaged pZA and pDsRed plasmids (Figure 2C). At the same time, co-transfection with the same amounts of non-damaged pDsRed and oxidatively damaged pZA resulted in a 6- to 7-fold excess of DsRed-Monomer over GFP transcripts, both measured relative to amounts of surviving plasmid DNA (Figure 2C, right panel). We thus conclude that degradation of the plasmid DNA can only partly account for the decreased transgene expression caused by the oxidative DNA base damage, while at least half of the effect has to be attributed to a mechanism modulating RNA synthesis. A recent communication reported an association of transiently transfected DNA with histones already within several hours after delivery to the host cells (25). This timing coincides with the decrease of expression of the damaged DNA observed in the present study. We therefore further assessed the potential role of chromatin structure in the regulation of transcription of the damaged DNA.
Association of transiently transfected plasmids with chromatin
We first tested whether the transiently transfected plasmid DNA becomes assembled into chromatin structure. The nuclei isolated from cells 24 h after transfection were treated with micrococcal nuclease (MNase) to solubilize chromatin. Purified pDsRed plasmid was added as an internal control representing naked DNA during the MNase digestion of the nuclei. Digested nuclei lysate was fractionated by equilibrium centrifugation in a CsCl gradient, and the fractions were analysed by agarose gel electrophoresis (Figure 3, top). Since the plasmid DNA makes a very small contribution to the total amount of DNA, only the distribution of chromosomal DNA is seen in the agarose gel. Most of DNA was found in fraction 4 followed by the fractions 5 and 6, thus indicating a peak of the chromatin fraction in the density gradient. To determine the location of the transfected plasmid (pZA) and of the naked plasmid DNA (pDsRed) in the same gradient, the fractions were diluted and analysed by semi-quantitative PCR with specific primers. The gel photos show the product analyses of the PCR reactions that were performed for the indicated number of cycles. The cycle numbers were determined empirically for each primer pair to avoid saturation of the PCR. Alignment of the two gels clearly shows that the DNA originating from the transfected pZA plasmid was shifted to higher gradient fractions, in comparison to the naked pDsRed DNA that has not been transfected (Figure 3, lower two gels). The observed distribution of the transfected plasmid is an indicator for its stable association with nucleoproteins within the chromatin, which results in a decreased buoyant density of the macromolecular complexes. Quantification of the PCR product bands indicates that naked DNA was almost exclusively found in the bottom of the gradient, in accordance with the relatively high density of the naked DNA (∼1.7 g/ml). A minor amount of naked DNA was smeared across the gradient, and approximately 10% was found as a small peak in the protein-rich fractions 9 and 10, being probably trapped by interaction with non-chromatin proteins (Figure 3). Remarkably, the distribution of the transfected DNA was completely different. Transfected DNA was clearly enriched in the fractions 4–6, with the maximal signal observed in fraction 4. Approximately 30% of the transfected DNA was found in the fractions 1 and 2, probably indicating the absence of proteins or a lower density of chromatin in this subpopulation of the plasmid molecules. Localization of the transiently transfected plasmid DNA within the chromatin fractions was observed in three independent experiments, while the naked pZA plasmid—similarly to pDsRed—was predominantly found in the lower gradient fractions (data not shown). Altogether, the data suggest that plasmid DNA is largely organized in chromatin 24 h after transfection. Thus, transcription of the transiently transfected transgene occurs in a chromatin context and potentially can be influenced by the chromatin structure.
Figure 3.

Association of the transfected plasmid with chromatin. Nuclei isolated from HeLa cells 24 h after transfection with undamaged pZA were mixed with pure pDsRed plasmid and treated with MNase as described in ‘Materials and Methods’ section. (Top) Agarose gel electrophoresis of the fractions obtained by isopycnic centrifugation of the MNase-treated nuclei and of the products of PCR performed with diluted fractions of the same CsCl gradient (numbers indicate the gradient fractions starting from the bottom). M, molecular size marker; nt, no template; p, plasmid DNA. (Bottom) Distribution of the two plasmids (transfected and naked) between the gradient fractions.
Histone components of the transiently transfected GFP gene
We then quantified the histone modifications that could potentially influence the activated or repressed state of the transiently transfected plasmid gene in the absence of damage. Acetylation of the histones H3 and H4 was analysed, since both acetylated histones are generally found in the promoters of actively transcribed genes in human cells and correlate well with an activated gene status (26,16). Among the chromatin marks frequently associated with gene repression, dimethylation of the histone H3 lysine residue 9 and trimethylation of lysine 27 were chosen, since these modifications are known to promote gene silencing via heterochromatin formation (26,27). In addition, histone H1 was assessed as a marker of general chromatin compaction.
The data show that acetylated histone H4 (H4Ac) is associated with the transiently transfected pZA plasmid, as the recoveries of both the promoter and 3′UTR DNA fragments were strongly enriched after immunoprecipitation with anti-AcH4 antibody, in comparison to the non-immune IgG (Figure 4). This tendency was confirmed in five independent experiments (Supplementary Figure S3). However, the degree of association of the plasmid DNA with H4Ac was not very high in comparison to the chromosomal gene loci. Thus, similar levels of H4Ac were detected in the chromatin of MYOD1 and AFM genes (Figure 4) that have very low expression levels in HeLa cells (as shown in Figure 2C). At the same time, the promoter region of the actively expressed housekeeping GAPDH gene typically contained 3–4-fold higher levels of AcH4 (Figure 4). Acetylated histone H3 (AcH3) was barely detectable in the chromatin associated with the plasmid DNA. The recoveries of the plasmid fragments were only marginally higher than those obtained with non-immune IgG (between 0.3 and 9 per mille of the total input DNA, depending on the ChIP experiment), and the differences between the specific and non-specific antibodies were not significant. If bound to the plasmid DNA, AcH3 would be detected in our ChIP experiments, as it was readily detected in the proximal promoter region of the GAPDH gene (Figure 4).
Figure 4.

Chromatin features characteristic for the undamaged GFP transgene. Quantification of histones with the indicated posttranslational modifications at the transiently transfected GFP gene (promoter, 3′UTR) and the promoter-proximal regions of three chromosomal genes (GAPDH, MYOD1 and AFM), and occupancy of the indicated gene fragments by RNA polymerase II (Pol2). Data of representative ChIP experiments with typical proportions in recoveries of the analysed DNA fragments (mean and data range of two determinations). Dotted line shows maximal recovery with non-immune IgG observed in the corresponding experiments.
Of the chromatin marks associated with gene silencing, dimethylation of lysine 9 of the histone H3 (H3K9Me2) was found associated with the non-damaged transfected plasmid DNA, as ChIP recoveries with the corresponding antibody were reproducibly higher than those of non-immune IgG in all three performed experiments. The amount of H3K9Me2 in the 3′UTR region of the plasmid GFP gene was reproducibly higher than in the promoter region (by factors between 1.7 and 3.0). Such pattern is in agreement with an elongation-dependent deposition of H3K9Me2 that has been recently reported for transcribed gene regions (28). The association of H3K9Me2 with the MYOD1 and AFM chromosomal regions was clearly and reproducibly detected, in accordance with the repressed status of the corresponding genes, while the GAPDH fragment showed low levels of H3K9Me2. Histone H1 was clearly present in all tested DNA fragments (judged from the comparison of the specific DNA recoveries with those of non-immune IgG). Average histone H1 levels were higher at the 3′UTR of the plasmid GFP gene, than at the promoter region (P = 0.004). Histone H1 levels were especially high at the MYOD1 and AFM fragments. Finally, association of trimethylated histone H3 (H3K27Me3) with plasmid DNA could not be reproducibly detected in three ChIP experiments. On average, recoveries of the 3′UTR region were marginally higher than those of the promoter region but still not significantly exceeding those of the non-immune IgG, though, MYOD1 and AFM (but not GAPDH) chromosomal fragments were reproducibly enriched in H3K27Me3 (Figure 4). We thus assume that this modification is not present in the plasmid chromatin, unless in minor amounts that could remain undetectable because of relatively low efficiencies of immunoprecipitations with this antibody.
We also tried to measure the amount of RNA polymerase II (Pol2) bound to the transfected DNA. Although the plasmid fragments were slightly enriched by immunoprecipitation in comparison to the MYOD1 fragment (Figure 4), the fragment recoveries relative to input were lower than in case of non-immune IgG. Due to the insufficient plasmid DNA recovery with Pol2 antibodies, we unfortunately could not further assess the recruitment of RNA polymerase II to the damaged DNA. Pol2 could be detected at the GAPDH fragment, in agreement with the reports that detection of Pol2 resting at the promoter-proximal posing sites is more efficient with respect to the elongating Pol2 (29,30).
We have thus found a transcription-activatory chromatin mark (AcH4) as well as features associated with gene repression (H3K9Me2 and histone H1) in the promoter of the transiently transfected gene. Since quantitative changes in each of the detected chromatin components could modulate the gene transcription, we aimed at the identification of chromatin alterations that would be characteristic for oxidatively damaged DNA.
Effects of DNA damage on the histone marks of the GFP transgene
To allow quantitative analyses of the chromatin components at the oxidatively damaged pZA plasmid in comparison to the undamaged plasmid, an internal reference DNA (pDsRed plasmid) was introduced to correct for the potentially variable DNA recoveries in ChIP. The reference plasmid DNA contains the same promoter sequence as the studied pZA plasmid and was thus expected to have the same chromatin structure as the corresponding pZA fragment. The reference plasmid (pDsRed) was co-transfected into the HeLa cells together with either undamaged or damaged pZA. Subsequently, non-transfected carrier cells were added, and the nuclei were isolated and used to produce soluble chromatin, suitable for immunoprecipitation (Figure 5A). The promoter DNA fragments derived from the two different plasmids can be distinguished by PCR due to a specific reverse primer annealing within the plasmid-specific sequences downstream from the transcription start site (Figure 5B and Supplementary Table S2). The amounts of the DNA fragments of the damaged or undamaged pZA in the ChIP samples were thus determined relative to the amount of pDsRed promoter (internal control).
Figure 5.

Quantitative comparison of the chromatin components in the undamaged and damaged plasmid DNA. (A) Experimental flowchart of transfection and chromatin preparation. The reference non-damaged pDsRed plasmid is introduced as an internal standard for chromatin immunoprecipitation. (B) Summary of DNA fragments analysed by real-time PCR. (C) DNA fragment size analyses after the reversal of cross-links in the samples transfected with an undamaged pZA (lane 1) or pZA containing three oxidized guanines (lane 2). M. molecular size marker. (D) ChIP enrichments of the undamaged pZA (oG = 0, white bars) and damaged pZA (oG = 3, black bars) DNA fragments, relative to the promoter fragment of the reference pDsRed plasmid (not plotted, equals to 1 in all graphs). Error bars indicate standard deviation of the mean for n ChIP experimens.
Since the size of chromatin fragments can strongly influence the quantitative recovery in ChIP, the samples transfected with different plasmids were carefully controlled to produce chromatin fragments of the same size (Figure 5C).
Histone H1 has been previously demonstrated to act as a general transcription repressor in transiently transfected DNA (25). We have therefore analysed the levels of histone H1 recruited to the damaged DNA. On average, histone H1 was enriched in the promoter region as a consequence of DNA damage, but the difference between the damaged and undamaged pZA was not highly significant (P = 0.16 in Student’s two-tailed t-test) (Figure 5D) and observable not in all experiments. We thus propose that the recruitment of histone H1 to the damaged DNA generally correlates with the decreased transcription but probably is not causal for repression of the damaged gene.
Acetylated histone H4 (AcH4) was observed in the same amounts at the promoter region of the undamaged pZA plasmid and at the promoter of the reference pDsRed plasmid (Figure 5D). At the same time, the recovery of the promoter region of the damaged pZA plasmid was decreased on average by 27% relative to the promoter of the reference undamaged pDsRed plasmid and by 25% in comparison to the same promoter region in non-damaged pZA (P < 0.0001). This effect was consistently detected in five ChIP experiments (Supplementary Figure S3). Interestingly, the decrease of AcH4 was reproducibly observed in the promoter region, but not in the other two DNA fragments. The levels of acetylated histone H3 (AcH3) were very low in the plasmid DNA and were not significantly influenced by the DNA damage (Figure 5D). In addition, there were no major differences in the levels of the methyl histone H3 marks (H3K9Me2 and H3K27Me3). However, since the chromatin immunoprecipitation data for H3K9Me2 and H3K27Me3 showed bigger interexperimental variations than those for the acetylated histones, small changes in their levels cannot be ruled out. For all antibodies employed, the recoveries of the corresponding promoter regions of non-damaged pZA and pDsRed plasmids were very similar (pDsRed promoter set to 1 in Figure 5).
Effect of trichostatin A on expression of the damaged plasmid DNA
The chromatin immunoprecipitation results described above show that among the analysed chromatin marks decrease of acetylation of the histone H4 could be relevant for transcriptional gene inactivation caused by the oxidative DNA damage. To test for a causal connection between the decrease of histone acetylation and silencing of the damaged GFP transgene, we treated transfected cells with the inhibitor of histone deacetylases trichostatin A (TSA). A dose-dependent recovery of expression of the damaged DNA was observed in the range of TSA concentrations between 111 and 333 nM (Figure 6). The dose-dependent effect of TSA was observed in three independent transfection experiments. TSA doses higher than 333 nM induced significant cytotoxicity and could not further improve the expression of the oxidatively damaged transgene (data not shown).
Figure 6.

Effect of trichostatin A on expression of the oxidatively damaged gene. Expression of the oxidatively damaged plasmid containing on average two oxidized guanines (oG = 2) relative to the undamaged plasmid (oG = 0) in presence of the indicated concentrations of trichostatin A. Cells were transfected with the same plasmid preparations for the whole range of trichostatin A concentrations. GFP expression for the treatments which were done in duplicates was measured by flow cytometry as median GFP fluorescence. Error bars indicate the data range.
DISCUSSION
Transfection of chemically modified plasmid DNA into undamaged cells is a useful strategy to study DNA repair and physiological effects of DNA damage, thanks to the defined chemical nature of the damage and localization of the damage sites within a restricted region of DNA. This method is widely used to study the repair of bulky DNA lesions, such as UV photoadducts (31), and proved valuable for investigation of mismatch repair (32) and translesion DNA synthesis (33). Since processing of the DNA lesions occurs in the nucleus of the host cell, it is supposed to be similar to the physiological processing of the damage occurring in the chromosomal DNA. The main concern about the plasmid based approach is the lack of comprehensive knowledge about the capacity of transiently transfected DNA to form a chromatin structure. It has often been assumed that transiently transfected plasmid DNA remains chromatin free. However, new evidence has been obtained that plasmid DNA is associated with histones shortly after being delivered to the nucleus and forms a nucleosomal structure stoichiometricaly similar to that of a chromosomal DNA fragment (25). Hence, DNA repair and transcription machineries operating on the plasmid DNA would likely have to deal with DNA wrapped around nucleosomes. Association of the transiently transfected plasmid DNA with histones is confirmed in the present study (Figures 3 and 4). Furthermore, the data presented here (Figure 4) indicate that posttranslational modifications of the nucleosome core histones, as well as linker histone H1, are present in the transiently transfected plasmid and thus can be relevant for regulation of the plasmid genes.
DNA quantification by real-time PCR indicated that a fraction of the damaged plasmids is lost during incubation in the host cells (Figure 2B). One possibility is that the nuclear import of plasmid DNA is handicapped by the presence of oxidative base damage. Since DNA transport through the nuclear pores requires binding of proteins (e.g. transcription factors) (34) and such binding can be inhibited by the presence of oxidized guanines (35,36), a fraction of the damaged DNA that failed to rapidly enter the nucleus could be subsequently degraded by cytoplasmic nucleases (37). Alternatively, incision of the oxidized DNA bases by specialized DNA repair endonucleases could promote degradation of the plasmid DNA by exonucleases in the nucleus. Whatever is the mechanism for DNA degradation, our data show that the remaining fraction of oxidatively damaged plasmid molecules in the cell nucleus (at least 40%) fails to sustain the reporter gene expression at a proportional level (Figure 2B). Concordantly, mRNA production was decreased several-fold in consequence of the DNA damage (Figure 2C), indicating a persistent transcriptional repression.
A major oxidative base lesion produced during DNA oxidation by methylene blue plus light is certainly 8-oxoG (38,15). There are several putative mechanisms by which oxidative DNA base lesions such as 8-oxoG could negatively influence the expression of a reporter protein. 8-OxoG does not significantly interfere with transcript elongation by RNA polymerase II (9–11), but it can interfere with the recruitment of transcription factors to the promoter region (35,36) possibly acting in the initiation phase of transcription. In addition, 8-oxoG is reported to cause misincorporation of adenine into messenger RNA in a process called ‘transcriptional mutagenesis’, leading to the synthesis of an aberrant protein (39). Nevertheless, several considerations make us believe that a direct effect of oxidative DNA base damage does not have a significant effect on gene expression under the studied conditions. Firstly, excision of 8-oxoG is very efficient in mammalian cell lines that express catalytically active OGG1. We observed a half-life of ∼3 h for Fpg-sensitive sites induced by photosensitisation in HeLa cells (Anderhub,S., unpublished data). If plasmid DNA is repaired with the same efficiency, we expect most of the DNA lesions to be repaired by the time when the decrease in gene expression takes place (between 8 and 32 h, Figure 1). Secondly, at the employed density of the DNA lesions (on average, 3 Fpg-sensitive DNA base modifications per plasmid molecule), the damage within the regulatory element of the promoter or in the coding region of the GFP gene would occur in only a minor fraction of the plasmid molecules. Finally, any direct effect of the DNA lesions would not explain the decrease of expression of the damaged gene with time. A more plausible scenario would be triggering of a gene silencing mechanism by DNA damage. Particularly intriguing in this case is the question which factor or group of factors mediate transcriptional repression at the damaged DNA, starting from the damage recognition event. Candidate factors could involve components of machineries dealing with chromatin maintenance, transcription, or DNA repair. For example, a change of the chromatin structure during DNA repair could modulate functional accessibility of the gene regulatory elements within the promoter region and potentially persist for a long time after removal of the lesion. Intriguingly, transcription factors Sp1 and CREB, which are most critical for regulation of the human CMV promoter (40), both require histone acetylation for accomplishment of their gene activatory functions (41,42).
To search for a chromatin signature marking the oxidatively damaged DNA, we have quantitatively analysed several histone modifications associated with gene activation or repression. Increased methylation of histone H3 lysines 9 (H3K9Me2) and 27 (H3K9Me3) are the best studied histone tail modifications that promote gene silencing in mammals via inducing formation of heterochromatin (26) and DNA methylation. H3K9Me2 and/or H3K27Me3 are robustly associated with gene silencing in human cancers (43) and therefore were initially regarded as candidate histone modifications relevant for silencing of the oxidatively damaged transgene. However, the ChIP data did not provide a convincing evidence for requirement of either H3K9Me2 or H3K27Me3 for silencing of the damaged transgene (Figure 5). At the same time, we cannot exclude that small changes in these histone modifications could escape detection due to overall low DNA recoveries and, in consequence, relatively broad experimental error margins. Among the histone modifications studied here, only acetylation of histone H4 was significantly different between the promoter regions of the damaged and undamaged plasmid genes. The capability of a HDAC inhibitor trichostatin A to partially restore expression of the damaged gene (Figure 6) indicates that histone deacetylation is involved in the gene silencing mechanism. However, in view of rather modest change in histone H4 acetylation levels (Figure 5), it is likely that other chromatin marks and gene silencing mechanisms contribute to the transcriptional inactivation of the oxidatively damaged DNA as well.
Transcriptional inactivation of the oxidatively damaged gene described here has common features with the silencing mechanism operating at a transgene integrated into the chromosomal DNA (44), which was found to be initiated by deacetylation of histones H3 and H4, followed by dimethylation of histone H3 K9 (after 17–19 days) and by massive methylation of CpG nucleotides in DNA (after 100 days). Interestingly, the gene silencing marks H3K9Me2 and H3K27Me3 were not found at the onset of transcriptional silencing (44). Possibly, these relatively late events of histone methylation are therefore not observable after 24 h in our study. In contrast to the integrated transgene (44), we found very low levels of histone H3 acetylation at the transiently transfected gene even under the conditions of active gene expression (Figure 4), indicating that histone H3 acetylation is not required for active expression of the transiently transfected DNA. This particularity can probably be explained by the expectation of a replacement variant H3.3 to be a major histone H3 species in the transiently transfected plasmid, as the conventional H3.1 variant requires DNA replication for its deposition (45,46).
In summary, we demonstrated that oxidative DNA base damage can cause transgene inactivation by triggering degradation of the damaged DNA, but primarily via induction of a persistent state of transcriptional repression. We have found that histone H4 acetylation was slightly but reproducibly decreased at the promoter region of the damaged gene, possibly contributing to a gene silencing mechanisms that act specifically on the oxidatively damaged DNA. Given that oxidative base damage is continuously induced in chromosomal DNA by endogenously generated reactive oxygen species under physiological conditions, it would be of great importance to establish whether such endogenously-generated damage could influence the expression of the adjacent genes in the cell.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by Deutsche Forschungsgemeinschaft (grant number EP 11/8-1).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Bernd Roβbach, Rosario Heck, and Ernesto Bockamp (Mainz) for making available their lab space and the real-time PCR equipment at the early stage of this work. We appreciate stimulating discussions with Barbara Tudek (Warsaw), Eugenia Dogliotti (Rome), and Thomas Lingg (Freiburg). We thank Uta Anderhub (Darmstadt) for carefully reading the manuscript.
REFERENCES
- 1.Hoeijmakers JH. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech. Ageing Dev. 2007;128:460–462. doi: 10.1016/j.mad.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Akbari M, Krokan HE. Cytotoxicity and mutagenicity of endogenous DNA base lesions as potential cause of human aging. Mech. Ageing Dev. 2008;129:353–365. doi: 10.1016/j.mad.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA. 1997;94:8010–8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjoras M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997;16:6314–6322. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl Acad. Sci. USA. 1997;94:7429–7434. doi: 10.1073/pnas.94.14.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klungland A, Rosewell I, Hollenbach S, Larsen E, Daly G, Epe B, Seeberg E, Lindahl T, Barnes DE. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl Acad. Sci. USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 8.Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- 9.Kathe SD, Shen GP, Wallace SS. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J. Biol. Chem. 2004;279:18511–18520. doi: 10.1074/jbc.M313598200. [DOI] [PubMed] [Google Scholar]
- 10.Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 2004;3:483–494. doi: 10.1016/j.dnarep.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Charlet-Berguerand N, Feuerhahn S, Kong SE, Ziserman H, Conaway JW, Conaway R, Egly JM. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 2006;25:5481–5491. doi: 10.1038/sj.emboj.7601403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen E, Kwon K, Coin F, Egly JM, Klungland A. Transcription activities at 8-oxoG lesions in DNA. DNA Repair (Amst) 2004;3:1457–1468. doi: 10.1016/j.dnarep.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Spivak G, Hanawalt PC. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 2006;5:13–22. doi: 10.1016/j.dnarep.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 14.Pastoriza-Gallego M, Armier J, Sarasin A. Transcription through 8-oxoguanine in DNA repair-proficient and Csb(-)/Ogg1(-) DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis. 2007;22:343–351. doi: 10.1093/mutage/gem024. [DOI] [PubMed] [Google Scholar]
- 15.Khobta A, Kitsera N, Speckmann B, Epe B. 8-Oxoguanine DNA glycosylase (Ogg1) causes a transcriptional inactivation of damaged DNA in the absence of functional Cockayne syndrome B (Csb) protein. DNA Repair (Amst) 2009;8:309–317. doi: 10.1016/j.dnarep.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 17.Polo SE, Roche D, Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127:481–493. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 18.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 19.Luijsterburg MS, Dinant C, Lans H, Stap J, Wiernasz E, Lagerwerf S, Warmerdam DO, Lindh M, Brink MC, Dobrucki JW, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 2009;185:577–586. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitsera N, Khobta A, Epe B. Destabilized green fluorescent protein detects rapid removal of transcription blocks after genotoxic exposure. Biotechniques. 2007;43:222–227. doi: 10.2144/000112479. [DOI] [PubMed] [Google Scholar]
- 22.Khobta A, Carlo-Stella C, Capranico G. Specific histone patterns and acetylase/deacetylase activity at the breakpoint-cluster region of the human MLL gene. Cancer Res. 2004;64:2656–2662. doi: 10.1158/0008-5472.can-03-1126. [DOI] [PubMed] [Google Scholar]
- 23.Epe B, Pflaum M, Boiteux S. DNA damage induced by photosensitizers in cellular and cell-free systems. Mutat. Res. 1993;299:135–145. doi: 10.1016/0165-1218(93)90091-q. [DOI] [PubMed] [Google Scholar]
- 24.Steenken S, Jovanovic SV. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997;119:617–618. [Google Scholar]
- 25.Hebbar PB, Archer TK. Altered histone H1 stoichiometry and an absence of nucleosome positioning on transfected DNA. J. Biol. Chem. 2008;283:4595–4601. doi: 10.1074/jbc.M709121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 27.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khobta A, Ferri F, Lotito L, Montecucco A, Rossi R, Capranico G. Early effects of topoisomerase I inhibition on RNA polymerase II along transcribed genes in human cells. J. Mol. Biol. 2006;357:127–138. doi: 10.1016/j.jmb.2005.12.069. [DOI] [PubMed] [Google Scholar]
- 31.Johnson JM, Latimer JJ. Analyses of DNA repair using transfection-based host cell reactivation. Methods Mol. Biol. 2005;291:321–335. doi: 10.1385/1-59259-840-4:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lei X, Zhu Y, Tomkinson A, Sun L. Measurement of DNA mismatch repair activity in live cells. Nucleic Acids Res. 2004;32:e100. doi: 10.1093/nar/gnh098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reissner T, Chaney S, Friedberg EC, Wang Z, Carell T, et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagstaff KM, Jans DA. Nucleocytoplasmic transport of DNA: enhancing non-viral gene transfer. Biochem. J. 2007;406:185–202. doi: 10.1042/BJ20070505. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh R, Mitchell DL. Effect of oxidative DNA damage in promoter elements on transcription factor binding. Nucleic Acids Res. 1999;27:3213–3218. doi: 10.1093/nar/27.15.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hailer-Morrison MK, Kotler JM, Martin BD, Sugden KD. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2′-deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry. 2003;42:9761–9770. doi: 10.1021/bi034546k. [DOI] [PubMed] [Google Scholar]
- 37.Lechardeur D, Sohn KJ, Haardt M, Joshi PB, Monck M, Graham RW, Beatty B, Squire J, O'Brodovich H, Lukacs GL. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 1999;6:482–497. doi: 10.1038/sj.gt.3300867. [DOI] [PubMed] [Google Scholar]
- 38.Schneider JE, Price S, Maidt L, Gutteridge JM, Floyd RA. Methylene blue plus light mediates 8-hydroxy 2′-deoxyguanosine formation in DNA preferentially over strand breakage. Nucleic Acids Res. 1990;18:631–635. doi: 10.1093/nar/18.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saxowsky TT, Meadows KL, Klungland A, Doetsch PW. 8-Oxoguanine-mediated transcriptional mutagenesis causes Ras activation in mammalian cells. Proc. Natl Acad. Sci. USA. 2008;105:18877–18882. doi: 10.1073/pnas.0806464105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stinski MF, Isomura H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 2008;197:223–231. doi: 10.1007/s00430-007-0069-7. [DOI] [PubMed] [Google Scholar]
- 41.Mottet D, Pirotte S, Lamour V, Hagedorn M, Javerzat S, Bikfalvi A, Bellahcene A, Verdin E, Castronovo V. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–256. doi: 10.1038/onc.2008.371. [DOI] [PubMed] [Google Scholar]
- 42.Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McInerney EM, Mullen TM, Glass CK, Rosenfeld MG. Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science. 1998;279:703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- 43.Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat. Genet. 2008;40:741–750. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 44.Mutskov V, Felsenfeld G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 2004;23:138–149. doi: 10.1038/sj.emboj.7600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmad K, Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell. 2002;9:1191–1200. doi: 10.1016/s1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- 46.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.