


Abstract
The retinoic acid receptor-related orphan receptor (ROR) alpha has been demonstrated to regulate lipid metabolism. We were interested in the RORα1 dependent physiological functions in skeletal muscle. This major mass organ accounts for ∼40% of the total body mass and significant levels of lipid catabolism, glucose disposal and energy expenditure. We utilized the strategy of targeted muscle-specific expression of a truncated (dominant negative) RORα1ΔDE in transgenic mice to investigate RORα1 signaling in this tissue. Expression profiling and pathway analysis indicated that RORα influenced genes involved in: (i) lipid and carbohydrate metabolism, cardiovascular and metabolic disease; (ii) LXR nuclear receptor signaling and (iii) Akt and AMPK signaling. This analysis was validated by quantitative PCR analysis using TaqMan low-density arrays, coupled to statistical analysis (with Empirical Bayes and Benjamini–Hochberg). Moreover, westerns and metabolic profiling were utilized to validate the genes, proteins and pathways (lipogenic, Akt, AMPK and fatty acid oxidation) involved in the regulation of metabolism by RORα1. The identified genes and pathways were in concordance with the demonstration of hyperglycemia, glucose intolerance, attenuated insulin-stimulated phosphorylation of Akt and impaired glucose uptake in the transgenic heterozygous Tg-RORα1ΔDE animals. In conclusion, we propose that RORα1 is involved in regulating the Akt2-AMPK signaling pathways in the context of lipid homeostasis in skeletal muscle.
INTRODUCTION
Retinoic acid receptor related orphan receptor alpha (RORα) is an orphan member of the nuclear receptor superfamily of transcription factors. Several in vitro and in vivo studies on RORα action and function (1–5) have suggested the involvement of this orphan nuclear receptor in lipid homeostasis (6) and hepatic phase I/II metabolism (7). RORα can be detected in many metabolic tissues including liver, kidney, adipose tissue and is highly expressed in skeletal muscle. In mice, RORα deficiency leads to profound metabolic disturbances. The homozygous staggerer (sg/sg) mice have a global RORα defect that results in decreased and dysfunctional expression of both mouse isoforms of RORα (1 and 4). These mice display hypoalphalipoproteinemia, dyslipidemia (decreased serum triglycerides and HDL-cholesterol) (5), susceptibility to atherosclerosis (2) and reduced adiposity and resistance to diet-induced obesity (8). The complex phenotype of the staggerer mice has been demonstrated to involve underlying changes in the expression of genes involved in fatty acid homeostasis, i.e. Apo-lipoprotein A1 (ApoA1) (5), Apo-lipoprotein C3 (ApoCIII) (1), sterol regulatory element-binding protein 1c (SREBP-1c), ATP-binding cassette transporter-binding proteins A1 and G1 (ABCA1 and G1), peroxisome proliferator-activated receptor gamma co-activator alpha/beta (PGC-1α/β), lipin1 and beta2-adrenergic receptor (8).
In the context of whole body metabolism, skeletal muscle has a vital contribution to the maintenance of energy balance. It is a major mass peripheral tissue with high energy demands. Consequently, multiple metabolic pathways converge in this tissue involving the utilization of both lipid and carbohydrate substrates. Skeletal muscle is considered a major site of glucose disposal and perturbation of insulin-mediated glucose uptake in this tissue is an important factor in the development of type 2 diabetes. In addition, the development of insulin resistance in skeletal muscle has been associated with increased intramuscular triglyceride accumulation, which can attenuate several insulin signaling pathways (9). As type 2 diabetes and associated complications are health issues that have global significance, it is therefore of considerable interest to investigate the regulatory machinery responsible for maintaining tight metabolic control in this tissue.
Previously, our analysis of skeletal muscle from staggerer mice (8) identified differential expression of a number of genes involved in fatty acid homeostasis. In this study, we aimed to investigate the role of RORα in skeletal muscle, without the complex interactions that result from the global defect. Therefore, we generated a transgenic mouse that overexpressed truncated human RORα1ΔDE (lacking the ligand-binding domain) in skeletal muscle to investigate the contribution of this energy demanding tissue to the RORα phenotype. We used a three-pronged genomic approach, utilizing llumina expression profiling, Ingenuity function and pathway analysis, and validation by rigorous quantitative PCR (qPCR) analysis on the TaqMan® low density array (TLDA) platform. This demonstrated that RORα has an important role in the regulation of carbohydrate and lipid metabolism in skeletal muscle. In particular, this animal model demonstrated that RORα1 also plays a critical role in skeletal muscle insulin signaling and glucose tolerance, via modulation of Akt2 and adenosine monophosphate kinase (AMPK) expression and activity. This correlated with increased expression of phospho ACC and genes involved in the regulation of fatty acid oxidation. In conclusion, we propose that in skeletal muscle RORα1 is an important metabolic regulator, contributing to both glucose and fatty acid homeostasis.
MATERIALS AND METHODS
Production of transgenic-mouse
The transgenic vector construct encodes a truncated version of human RORα1 (RORα1ΔDE). Amino acids 1–235 are present but the entire E region and part of the hinge/D region have been removed as previously described (3). To confer muscle specificity, the vector was placed under the control of the full-length human skeletal alpha-actin (HSA) promoter as described previously (10–12). Zygotes were generated by pronuclear microinjection of the transgene into oocytes from hybrid female donors (C57BL/6J X CBA) as described (13). Screening of founder mice and their offspring for stable germ line transmission was performed by real time PCR. The sequences of the specific mouse RORα and human RORα primers were as follows—hRORα: CAATGCCACCTACTCCTGTCC and CTACGGCAAGGCATTTCTGTAAT, mRORα: CAATGCCACCTACTCCTGTCC and GCCAGGCATTTCTGCAGC. Transgene copy number was determined using real time PCR as described (14). Two founders were selected for analysis, each of which carried two copies of the transgene. Both lines were then backcrossed with C57Bl/6J mice for a minimum of five generations. Experiments were performed on transgenic mice from the fifth generation onward, relative to wt littermates; therefore the genetic background of the animals in this study is >98.6% C57Bl/6J. The initial characterization was performed on both transgenic lines. No differences were observed in either phenotype or metabolic gene expression between the two lines, therefore we continued investigation on one line only.
Animal procedures
The mice were housed in the QBP vivarium (University of Queensland, St. Lucia, Queensland, Australia) with 12 h light–dark cycle and fed a standard diet containing 4.6% total fat (from Specialty Feeds, Glen Forrest, Western Australia). For the diet-induced obesity experiments, mice were transferred to a high-fat diet containing 34.9% fat (D12492, Research Diets, New Brunswick, NJ) from 4 weeks of age onward. Experimental animals were weighed weekly up to 14 weeks of age. Mice were fasted overnight by transferring to a new food-free holding cage with unrestricted access to water, prior to all experimental procedures. Care was taken to euthanize all animals at 9 am (i.e. 3 h into the light cycle, and at similar times), and excised tissues were immediately frozen in liquid nitrogen and then stored at −80°C. All aspects of animal experimentation were approved by The University of Queensland Animal Ethics Committee.
Microarray analysis
It is completely described in Supplementary Figure S1.
RNA extraction, cDNA synthesis and qPCR TLDA analysis
Total RNA extraction and cDNA synthesis were performed as described previously (4). We utilized custom designed ABI microfluidic TLDAs to analyze the expression of genes involved in metabolism (lipid, carbohydrate and energy homeostasis). Three control genes were utilized, including the mandatory control (18S rRNA) and four other controls: Gapdh, GusB, Hprt1 and 36B4. These controls span the relative abundance/Ct range of the genes on the card, and three (18S rRNA, GAPDH and 36b4) are approved real time PCR controls for NURSA supported nuclear receptor studies (15,16). The TLDAs were analyzed as described in Myers et al. (17). Briefly, significant changes in expression relative to wt littermate mice were analyzed using the ABI/integromics ‘StatMiner’ software package. Differentially expressed genes were identified by linear models (contained in the LIMMA package for bioconductor R embedded in StatMiner). Significance was assigned by the application of the Empirical Bayes statistic. B values represent the empirical bayes log odds of differential expression, and the t-value is the empirical bayes moderated t-statistic. Subsequently, we applied a more stringent/conservative data filtering (Benjamini–Hochberg) to control for false discovery rate, correct P values and further refine the subset of differentially expressed genes.
Primers and qPCR
Relative expression of genes was determined using the ABI 7500 real time PCR System (ABI, Singapore) as previously described (3). Primers for LXRα, GAAATGCCAGGAGTGTCGAC and GATCTGTTCTTCTGACAGCACACA; Akt1, GGCTGGCTGCACAAACG and GACTCTCGCTGATCCACATCCT; Akt3, CCTTCCAGACAAAAGACCGTTT and CGCTCTCTCGACAAATGGAAA; GLUT1, TACGCTGGAGGCGGTAGCT and AATGGGCGAATCCTAAAATGG.
Protein extraction
Total soluble protein was extracted from skeletal muscle (quadriceps) by the addition of lysis buffer (10 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X-100 and 5 mM EDTA) containing protease and phosphatase ‘cocktail’ inhibitors (Roche diagnostics GmbH, Mannheim, Germany). Lysates were passed through a 26-gauge needle and centrifuged at 10 000g for 20 min. The supernatant was collected and total protein concentration was determined by the bicinchoninic acid (BCA assay kit), as outlined by manufacturer’s instructions (Pierce Biotechnology Inc., Rockford, IL, USA).
Glucose and insulin tolerance tests and glucose uptake
Basal glucose measurements were obtained from the tail blood of overnight fasted animals (14–16 weeks of age). Mice were then administered a dose of either glucose solution (2 g/kg) or insulin (0.5 U/kg) by intraperitoneal injection. Blood glucose measurements were obtained at 10 or 15 min intervals for up to 90 min following challenge using the Accu-Chek Performa blood glucose testing system (Roche Diagnostics Australia, Castle Hill, NSW). Plasma insulin measurements were obtained using the insulin (mouse) Ultrasensitive EIA (ALPCO Diagnostics, Salem NH). Insulin-stimulated glucose uptake was performed on skeletal muscle (extensor digitorum longus, EDL) dissected from anesthetized mice and incubated in essential buffer (Krebs–Henseleit, pH 7.4) and the assay was performed as described previously (18,19).
Western blot analysis
Total soluble protein from the quadriceps of transgenic and their littermate wild-type mice were resolved on a 10% SDS–PAGE gel and transferred to a PVDF (Millipore Corporation, Billerica, MA, USA). The membranes were blocked for 1 h in 5% BSA in TBS–Tween 20, followed by an overnight incubation with primary antibody. The following antibodies were purchased from Cell Signaling Technology, Danvers, MA and used at 1:1000 dilution: AMPKα (#2532), pAMPK (Thr172) (#2535), Akt (#9272), pAKT (ser473) (#4058), ACC (#3662) and pACC(Ser79) (#3661). Anti-GAPDH (1:10000) was from R&D Systems, Minneapolis, MN. Following 3 × 10 min washes, the membrane was incubated with anti-rabbit horseradish peroxidase (HRP) (1:10000) for 1 h. Immunoreactive signals were detected using enhanced chemiluminescence Super Signal West Pico Substrate (Pierce) and visualized by autoradiography on an X-OMAT film developer (Kodak).
Chromatin immunoprecipitation analysis
C2C12 cells were differentiated for 4 days. Cells were harvested and subsequently washed twice in ice cold PBS and cross-linked in 1% formaldehyde solution. Chromatin immunoprecipitation (ChIP) was performed as described by Pearen et al. (20) using anti-RORα (Santa Cruz anti-RORα sc-6062) and IgG (Santa Cruz, CA). The following qPCR ChIP primers were used: RORRE Site 1 F—GCATGTGCTGCAAACATTCAG (−2897 to −2876), R—CTACACAGGGTCAGTGGCCA (−2866 to −2846). RORE Site 2 F—GAACATGAAGATTTGAACTTG (−2705 to −2684), R—CTGACCCAGAAACTCCTACA (−2668 to −2649). RORRE Site 3 F—CTGGGTCAGTAGAGTAGCAAGCC (−2657 to 2634), R—TAAGCATTCTGAGGTGACCTATGAA (−2631 to 2606). Downstream Negative control F—GTTCCCAAGTGAAGAACCGC (−278 to −258), R—CCGGCAATCAAAGGGCTT (−195 to −177).
Statistical analysis
The TLDA gene expression data were analyzed as described above. All other results were analyzed (and significance assigned) using a t-test, or ANOVA in the Graphpad Prism 4 software, unless otherwise indicated.
RESULTS
Overexpression of truncated RORα1ΔDE (lacking the ligand-binding domain) in skeletal muscle
As discussed, RORα has been demonstrated to regulate fat metabolism in several tissues. We were interested in identifying and validating the (physiologically relevant) in vivo functional role(s) and pathway(s) regulated by RORα action in skeletal muscle. This major mass peripheral tissue accounts for ∼40% of the total body mass, significant levels of fatty acid oxidation, glucose disposal and energy demand. We utilized the approach of attenuating RORα signaling, by the targeted skeletal muscle-specific expression of a truncated RORαΔDE (lacking the ligand-binding domain) in transgenic mice, to examine RORα action in this tissue. This approach was utilized in the absence of the availability of a reproducible and robust, native or synthetic agonist for the modulation of this orphan nuclear receptor.
We produced transgenic mice (by pronuclear injection) that selectively express a transgene encoding truncated human RORα1 in skeletal muscle [under the control of the human skeletal alpha actin promoter (10,11)]. The transgene (RORα1ΔDE) lacks the ligand-binding domain and part of the hinge region, and encoded amino acids 1–235 (6). McBroom et al. (21) reported that deletion of this segment preserved DNA recognition and binding, suppressed trans-activation and operated in a dominant negative manner. In addition, Hamilton et al. (22) reported the staggerer mutation is located in a similar position, and produces a non-functional ligand-binding domain. Moreover, we previously demonstrated in an in vitro myogenic cell culture model that this construct attenuated RORα mediated trans-activation, and ectopic expression suppressed endogenous RORα mRNA expression (3).
We observed that the heterozygous transgenic mice predominantly (and abundantly) expressed the ectopic transcript (transgene) in skeletal muscle relative to other organ/tissues (cerebellum, spleen, heart, white adipose, liver and pancreas; Figure 1A and B). Heart and brain expressed <2% of the ectopic transcript, relative to expression in skeletal muscle. This was consistent with several other studies that utilized this promoter in transgenic models (10,12,23). Quantitative real-time PCR (q-PCR) analysis of mRNA expression demonstrated that the ectopic truncated RORα transcript was expressed between 3- and 4-fold higher than the endogenous RORα transcript in the muscle of transgenic mice (Figure 1A). As expected, the ectopic RORα transcript was not detectable in wt littermates (data not shown). Concordantly, expression analysis demonstrated that total (endogenous plus ectopic) RORα transcript expression increased ∼3-fold in the muscle of transgenic, relative to wt littermate mice (Figure 1C). In this background we observed a decrease (that did not attain significance) in the levels of total endogenous RORα1 and 4 mRNA expression in the muscle of transgenic relative to wt littermate mice (Figure 1D). Surprisingly, isoform-specific q-PCR analysis of RORα1 and RORα4, revealed significant suppression of RORα1 (but not α4) mRNA expression (Figure 1E and F). Staels (1,5) and our group (3,8) amongst others have previously demonstrated that the α1 isoform has a physiological role in the regulation of lipid homeostasis. To further characterize the line of mice, we examined expression of a bona fide RORα (1 and 4) target gene, apolipoprotein A5, characterized by two independent groups (24,25), which was very significantly decreased in the Tg-RORα1ΔDE mice (Figure 1G). These data demonstrate the line of Tg-RORα1ΔDE mice, express the ectopic transcript, suppress endogenous RORα1 mRNA expression and attenuate RORα1-dependent gene expression.
Figure 1.

(A) qPCR of the ectopic transgene gene (hRORα1ΔDE) and endogenous RORα expression in various tissue/organs in heterozygous transgenic mice. Relative mRNA expression is normalized against 18S mRNA (n = 6/group, mean ± SEM, ***P < 0.001). (B) qPCR of the ectopic transgene gene (hRORα1ΔDE) in pancreas versus skeletal muscle from wild-type and heterozygous transgenic mice. Relative mRNA expression is normalized against 18S mRNA (n = 6/group). (C) qPCR of total RORα (ectopic and endogenous) in skeletal muscle of wild-type and transgenic mice. Relative fold change of (mRNA expression) is normalized against 18S mRNA (n = 6/group, mean ± SEM, ***P < 0.001). qPCR of (D) endogenous RORα1 and 4; (E) endogenous RORα1andF)endogenous RORα4 mRNA expression in skeletal muscle of wild-type and transgenic mice. Relative fold change of (mRNA expression) is normalized against 18S mRNA (n = 6/group, mean ± SEM), *P < 0.05. (G) qPCR of ApoA5 mRNA expression in skeletal muscle of wild-type and transgenic mice. Relative fold change of (mRNA expression) is normalized against 18S mRNA (n = 6/group, mean ± SEM, ***P < 0.001).
Expression profiling of skeletal muscle from the RORα1ΔDE mice: identification of functions and pathways
To rigorously identify the in vivo functional role(s), and pathway(s) regulated by RORα action in skeletal muscle, we carried out expression profiling coupled to functional and signaling characterization by Ingenuity pathway analysis (Supplementary Figures S2–S5). Using a P value cut-off of P < 0.05 and a fold change cut off of 1.3, the top 50 annotated genes that were differentially up- and down-regulated in a significant manner are shown in Tables 1 and 2, respectively [the complete list of genes (including the non-annotated genes) is shown in Supplementary Figure S2. In concordance with the overexpression of a dominant negative, the expression profiling, Genespring and Ingenuity analysis identifies a majority of genes in the down regulated category, and the bulk of highly ranked functions and pathways were attenuated/suppressed (This is evident in the complete list of genes in Supplementary Figure S2 and in the graphical representations of the data in Supplementary Figures S3–S5).
Table 1.
Downregulated annotated genes in RORα1ΔDE
| Symbol | Gene name | Fold change | P-value | Accession number |
|---|---|---|---|---|
| Ucp1 | Uncoupling protein 1 | 34.89 | 0.000 | NM_009463.2 |
| Fasn | Fatty acid synthase | 8.96 | 0.001 | NM_007988.3 |
| Cidea | Cell death-inducing DNA fragmentation factor, alpha subunit-like effector A | 8.58 | 0.000 | NM_007702.1 |
| Scd1 | Stearoyl-Coenzyme A desaturase 1 | 5.47 | 0.004 | NM_009127.3 |
| Hp | Haptoglobin | 4.22 | 0.004 | NM_017370.1 |
| Prkar2b | Protein kinase, camp dependent regulatory, type II beta | 4.06 | 0.004 | NM_011158.3 |
| Apoc1 | Apolipoprotein C-I | 3.11 | 0.006 | NM_007469.2 |
| Igh-6 | Immunoglobulin heavy chain 6 (heavy chain of IgM) | 2.93 | 0.014 | XM_354710.1 |
| Fah | Fumarylacetoacetate hydrolase | 2.72 | 0.008 | NM_010176.1 |
| Tmem45b | Transmembrane protein 45b | 2.62 | 0.000 | NM_144936.1 |
| Elovl6 | ELOVL family member 6, elongation of long chain fatty acids | 2.47 | 0.000 | NM_130450.2 |
| Myo1e | Myosin IE | 2.43 | 0.010 | NM_181072.3 |
| Ces3 | Carboxylesterase 3 | 2.21 | 0.001 | NM_053200.2 |
| Sfxn1 | Sideroflexin 1 | 2.11 | 0.013 | NM_027324.2 |
| Acaca | Acetyl-coenzyme A carboxylase alpha | 2.02 | 0.002 | NM_133360.2 |
| Rab15 | RAB15, member RAS oncogene family | 2.00 | 0.008 | NM_134050.2 |
| Cox6a1 | Cytochrome c oxidase, subunit VI a, polypeptide 1 | 2.00 | 0.000 | NM_007748.3 |
| Tkt | Transketolase | 1.97 | 0.004 | NM_009388.2 |
| Fgfbp1 | Fibroblast growth factor binding protein 1 | 1.90 | 0.006 | NM_008009.3 |
| Thrsp | Thyroid hormone responsive SPOT14 homolog | 1.90 | 0.006 | NM_009381.2 |
| Sfxn1 | Sideroflexin 1 | 1.90 | 0.010 | NM_027324.2 |
| Cox6a1 | Cytochrome c oxidase, subunit VI a, polypeptide 1 | 1.86 | 0.000 | NM_007748.3 |
| D12Ertd647e | DNA segment, Chr 12, ERATO Doi 647, expressed, transcript variant 4 | 1.85 | 0.006 | NM_194068.1 |
| Agpat2 | 1-Acylglycerol-3-phosphate O-acyltransferase 2 (lysophosphatidic acid acyltransferase, beta) | 1.84 | 0.000 | NM_026212.1 |
| Pygl | Liver glycogen phosphorylase | 1.84 | 0.000 | NM_133198.1 |
| Ehhadh | Enoyl-coenzyme A, hydratase/3-hydroxyacyl Coenzyme A dehydrogenase | 1.83 | 0.007 | NM_023737.2 |
| Aoc3 | Amine oxidase, copper containing 3 | 1.81 | 0.012 | NM_009675.1 |
| M6prbp1 | Mannose-6-phosphate receptor binding protein 1 | 1.81 | 0.003 | NM_025836.1 |
| Idh1 | Isocitrate dehydrogenase 1 (NADP+), soluble | 1.79 | 0.007 | NM_010497.2 |
| Atp1b4 | Atpase, (Na+)/K+ transporting, beta 4 polypeptide | 1.78 | 0.000 | NM_133690.2 |
| Exod1 | Exonuclease domain containing 1 | 1.77 | 0.012 | NM_027698.3 |
| Hsd17b12 | Hydroxysteroid (17-beta) dehydrogenase 12 | 1.76 | 0.002 | NM_019657.2 |
| Wnt4 | Wingless-related MMTV integration site 4 | 1.76 | 0.004 | NM_009523.1 |
| Cebpa | CCAAT/enhancer binding protein | 1.75 | 0.005 | NM_007678.2 |
| Slc27a2 | Solute carrier family 27 (fatty acid transporter), member 2 | 1.73 | 0.009 | NM_011978.2 |
| Apoc1 | Apolipoprotein C-I | 1.72 | 0.013 | NM_007469.2 |
| Tmcc3 | Transmembrane and coiled coil domains 3 | 1.71 | 0.004 | NM_172051.2 |
| Zfp691 | Zinc finger protein 691 | 1.71 | 0.000 | NM_183140.1 |
| Olfm1 | Olfactomedin 1, transcript variant 2 | 1.68 | 0.010 | NM_001038612.1 |
| Gnao1 | Guanine nucleotide binding protein, alpha o | 1.68 | 0.007 | NM_010308.3 |
| Gcat | Glycine C-acetyltransferase (2-amino-3-ketobutyrate coenzyme A ligase) | 1.67 | 0.012 | NM_013847 |
| Abhd8 | Abhydrolase domain containing 8 | 1.64 | 0.005 | NM_022419.1 |
| Pparg | Peroxisome proliferator activated receptor gamma | 1.64 | 0.001 | NM_011146.1 |
| Cox8a | Cytochrome c oxidase, subunit viiia | 1.63 | 0.001 | NM_007750.2 |
| Tspo | Translocator protein | 1.62 | 0.001 | NM_009775.2 |
| Lrtm1 | Leucine-rich repeats and transmembrane domains 1 | 1.62 | 0.001 | NM_176920.2 |
| Igh-1a | Immunoglobulin heavy chain 1a | 1.62 | 0.002 | XM_354704.1 |
| Slc16a10 | Solute carrier family 16 (monocarboxylic acid transporters), member 10 | 1.62 | 0.001 | NM_028247.1 |
| Tspo | Translocator protein | 1.60 | 0.003 | NM_009775.2 |
| Hsd11b1 | Hydroxysteroid 11-beta dehydrogenase 1, transcript variant 2 | 1.60 | 0.001 | NM_001044751.1 |
Table 2.
Upregulated annotated genes in RORα1ΔDE
| Symbol | Gene name | Fold change | P-value | Accession number |
|---|---|---|---|---|
| Zranb3 | Zinc finger, RAN-binding domain containing 3 | 2.64 | 0.000 | NM_172642.1 |
| Rom1 | Rod outer segment membrane protein 1 | 2.54 | 0.000 | NM_009073.2 |
| Tceal5 | Transcription elongation factor A (SII)-like 5 | 2.43 | 0.001 | NM_177919.1 |
| Col7a1 | Procollagen, type VII, alpha 1 | 2.21 | 0.010 | NM_007738.3 |
| Plekhb1 | Pleckstrin homology domain containing, family B (evectins) member 1 | 2.10 | 0.001 | NM_013746.1 |
| Tnfrsf22 | Tumor necrosis factor receptor superfamily, member 22 | 2.10 | 0.001 | NM_023680.2 |
| Tnfrsf22 | Tumor necrosis factor receptor superfamily, member 22 | 1.94 | 0.001 | NM_023680 |
| Gfer | Growth factor, erv1 (S. cerevisiae)-like (augmenter of liver regeneration) | 1.90 | 0.000 | NM_023040.3 |
| Tob1 | Transducer of erbb-2.1 | 1.79 | 0.011 | NM_009427.2 |
| Stab2 | Stabilin 2 | 1.79 | 0.001 | NM_138673.1 |
| Camk2n2 | PREDICTED: calcium/calmodulin-dependent protein kinase II inhibitor 2 | 1.77 | 0.003 | XM_993468.1 |
| Il17re | Interleukin 17 receptor E, transcript variant 2 | 1.71 | 0.001 | NM_001034029.1 |
| Lgals1 | Lectin, galactose binding, soluble 1 | 1.61 | 0.000 | NM_008495.2 |
| Slc22a4 | Solute carrier family 22 (organic cation transporter), member 4 | 1.61 | 0.007 | NM_019687.3 |
| Ush1c | Usher syndrome 1C homolog, transcript variant a1 | 1.59 | 0.008 | NM_023649.1 |
| Maged2 | Melanoma antigen, family D, 2 | 1.55 | 0.003 | NM_030700.1 |
| Gdf11 | Growth differentiation factor 11 | 1.52 | 0.007 | NM_010272.1 |
| Sfxn3 | Sideroflexin 3 | 1.50 | 0.010 | NM_053197.2 |
| Rab11fip5 | RAB11 family interacting protein 5 (class I), transcript variant 2 | 1.50 | 0.009 | NM_177466.4 |
| Synpo2l | Synaptopodin 2-like | 1.50 | 0.008 | NM_175132.3 |
| Hes1 | Hairy and enhancer of split 1 | 1.49 | 0.001 | NM_008235.2 |
| Actr1b | ARP1 actin-related protein 1 homolog B, centractin beta (yeast) | 1.47 | 0.001 | NM_146107 |
| Actr1b | ARP1 actin-related protein 1 homolog B | 1.47 | 0.007 | NM_146107.2 |
| Rala | V-ral simian leukemia viral oncogene homolog A (ras related) | 1.47 | 0.004 | NM_019491.5 |
| Sms | Spermine synthase | 1.46 | 0.000 | NM_009214.3 |
| C1qtnf4 | C1q and tumor necrosis factor related protein 4 | 1.44 | 0.006 | NM_026161.1 |
| Sorl1 | Sortilin-related receptor containing LDLR class A repeats | 1.42 | 0.009 | NM_011436 |
| Des | Desmin | 1.42 | 0.004 | NM_010043.1 |
| Nans | N-acetylneuraminic acid synthase (sialic acid synthase) | 1.40 | 0.002 | NM_053179.2 |
| Ccl25 | Chemokine (C-C motif) ligand 25 | 1.40 | 0.002 | NM_009138.1 |
| Rrm1 | Ribonucleotide reductase M1 | 1.39 | 0.002 | NM_009103 |
| Eif1ay | Eukaryotic translation initiation factor 1A, Y-linked | 1.39 | 0.012 | NM_025437 |
| Setd8 | SET domain containing (lysine methyltransferase) 8 | 1.38 | 0.007 | NM_030241.2 |
| Igfbp5 | Insulin-like growth factor binding protein 5 | 1.37 | 0.000 | NM_010518 |
| Mllt3 | Myeloid/lymphoid or mixed lineage-leukemia translocation to 3 homolog (Drosophila), transcript variant 2 | 1.36 | 0.008 | NM_029931.2 |
| Zbtb12 | Zinc finger and BTB domain containing 12 | 1.35 | 0.012 | NM_198886.2 |
| Smarcd3 | SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily d, member 3 | 1.35 | 0.006 | NM_025891.3 |
| Hdac4 | Histone deacetylase 4 | 1.35 | 0.000 | NM_207225.1 |
| Eif3s2 | Eukaryotic translation initiation factor 3, subunit 2 (beta) | 1.35 | 0.002 | NM_018799.1 |
| St3gal2 | ST3 beta-galactoside alpha-2,3-sialyltransferase 2, transcript variant 2 | 1.34 | 0.006 | NM_178048.2 |
| Slc25a15 | Solute carrier family 25 (mitochondrial carrier ornithine transporter), member 15 | 1.34 | 0.000 | NM_181325.2 |
| Klc2 | Kinesin light chain 2 | 1.34 | 0.007 | NM_008451 |
| Epdr1 | Ependymin-related protein 1 | 1.34 | 0.012 | NM_134065.2 |
| Slc11a2 | Solute carrier family 11 (proton-coupled divalent metal ion transporters), member 2 | 1.34 | 0.010 | AK049856 |
| Abca15 | ATP-binding cassette, subfamily A (ABC1), member 15 | 1.34 | 0.006 | NM_177213.3 |
| Pip5k2b | Predicted: phosphatidylinositol-4-phosphate 5-kinase, type II, beta, transcript variant 4 | 1.33 | 0.003 | XM_991639.1 |
| Ptp4a2 | Protein tyrosine phosphatase 4a2 | 1.33 | 0.003 | NM_008974.3 |
| Col11a2 | Procollagen, type XI, alpha 2 | 1.33 | 0.006 | NM_009926.1 |
| Prkcq | Protein kinase C, theta | 1.32 | 0.014 | NM_008859.2 |
| Ssbp2 | Single-stranded DNA-binding protein 2 | 1.32 | 0.012 | NM_024186.1 |
Interrogation of differentially expressed genes on the Ingenuity platform identified that a subset of differentially expressed genes in Tg-RORα1ΔDE mice were involved in/associated with lipid metabolism, small molecule transport and biochemistry, cardiovascular and metabolic disease, carbohydrate metabolism, endocrine system disorders etc (see Supplementary Figure S4), in concordance with the pathophysiological role of NRs. The majority of metabolic genes are down regulated in the major functional categories of lipid and carbohydrate metabolism (Supplementary Figure S4 and S5). This is consistent with several investigations in RORα deficient mouse models (2,8). In the framework of this investigation, the primary signaling pathways regulated in skeletal muscle were the glutathione metabolism (responsible for the tight control of ROS levels, which modulate insulin sensitivity), type 2 diabetic signaling and fatty acid biosynthesis. Notably, several NR signaling (and RXR-dependent) pathways including LXR, FXR VDR and PXR were identified in the analysis. The higher ranked NR signaling pathways control lipogenesis/fatty acid biosynthesis (LXR) and cholesterol homeostasis (LXR and FXR) in agreement with the functional annotation (see Supplementary Data S4 and S5). The majority of differentially expressed genes were associated with lipid/carbohydrate metabolism and metabolic disease (Supplementary Data S5A).
Expression of the truncated orphan nuclear receptor, RORα, affected the fatty acid biosynthetic pathway: decreased SREBP-1c, LXRα and downstream target genes in Tg-RORα1ΔDE mice
Previously we have demonstrated that staggerer mice (sg/sg), with decreased and dysfunctional RORα expression (in all organs) are resistant to (high fat) diet induced obesity (8). The genes, pathways and functional role identified (for RORα) from the interrogation of the data revealed that lipid metabolism (especially fatty acid biosynthesis/lipogenesis) functions and pathways were highly ranked. We have validated the illumina/genespring analysis by rigorous qPCR analysis utilizing a custom-designed ABI microfluidic TLDA, that encoded taqman primer sets for critical metabolic genes involved in the control of lipid homeostasis. Specifically, in the context of lipogenic gene expression, TLDA analysis revealed significant differential expression (normalized against 18S rRNA) of the mRNAs encoding acyl-CoA synthetase long chain family member 4 (ACSL4), fatty acid translocase/cluster of differentiation 36 (FAT/CD36), fatty acid synthase (FAS), stearyl-coA dehydrogenase 1 (SCD-1), SREBP-1c and many other genes in the skeletal muscle of male Tg-RORα1ΔDE mice relative to wild-type littermates (assigned by the Empirical Bayes statistic, see Table 3, Supplementary Data S6—Supplementary Figure S6 shows the complete list). Stringent filtering of data using the Benjamini–Hochberg method to control P value false discovery rate (FDR) refined the significant subset of differentially expressed genes to SREBP-1c, FAS, SCD-1, ACSL4, CD36 and HIF1α (Figure 2A and Table 3).
Table 3.
Relative quantification of gene expression in skeletal muscle of transgenic compared to wt littermate controls
| Entrez gene symbol- TaqMan assay ID | ΔΔCt | P-value | B-value | t-value | RQ. Log10 | RQ. Linear | Significance | Adj. P-value | Significance FDR |
|---|---|---|---|---|---|---|---|---|---|
| Lipogenesisa | |||||||||
| Abca1-Mm00442646_m1 | 0.628 | 0.038 | −4.081 | 2.283 | −0.189 | 0.647 | Significant | 0.080 | NS |
| Acsl4-Mm00490331_m1 | 1.396 | 0.001 | −0.313 | 4.273 | −0.420 | 0.380 | Significant | 0.012 | Significant |
| Cav3-Mm01182632_m1 | 0.452 | 0.043 | −4.190 | 2.220 | −0.136 | 0.731 | Significant | 0.081 | NS |
| Cd36-Mm00432403_m1 | 1.115 | 0.013 | −3.121 | 2.814 | −0.335 | 0.462 | Significant | 0.042 | Significant |
| Cebpb-Mm00843434_s1 | 0.908 | 0.023 | −3.614 | 2.546 | −0.273 | 0.533 | Significant | 0.061 | NS |
| Cebpd-Mm00786711_s1 | 1.086 | 0.029 | −3.827 | 2.428 | −0.327 | 0.471 | Significant | 0.068 | NS |
| Fasn-Mm00662319_m1 | 1.844 | 0.005 | −2.163 | 3.316 | −0.555 | 0.279 | Significant | 0.023 | Significant |
| Hif1a-Mm00468875_m1 | 0.797 | 0.013 | −3.082 | 2.834 | −0.240 | 0.575 | Significant | 0.042 | Significant |
| Scd1-Mm00772290_m1 | 2.099 | 0.001 | −0.881 | 3.978 | −0.632 | 0.233 | Significant | 0.012 | Significant |
| Srebf1-Mm00550338_m1 | 0.764 | 0.004 | −2.034 | 3.383 | −0.230 | 0.589 | Significant | 0.023 | Significant |
| Nuclear receptorsb | |||||||||
| Nr1h3-Mm00443454_m1 | 0.814 | 0.039 | −3.531 | 2.522 | −0.245 | 0.569 | Significant | 0.393 | NS |
| Nr2c1-Mm00449123_m1 | 0.601 | 0.032 | −3.353 | 2.661 | −0.181 | 0.659 | Significant | 0.393 | NS |
| Nr3c1-Mm00433832_m1 | 0.912 | 0.005 | −1.734 | 4.037 | −0.275 | 0.531 | Significant | 0.234 | NS |
| Ppard-Mm00803186_g1 | 0.926 | 0.041 | −3.564 | 2.496 | −0.279 | 0.526 | Significant | 0.393 | NS |
| Rorc-Mm00441139_m1 | 0.860 | 0.019 | −2.911 | 3.013 | −0.259 | 0.551 | Significant | 0.393 | NS |
| Glucose homeostasisa | |||||||||
| Akt2-Mm00545827_m1 | 1.684 | 0.001 | −0.621 | 4.337 | −0.507 | 0.311 | Significant | 0.037 | Significant |
| Cs- Mm00466043_m1 | 0.515 | 0.011 | −2.781 | 3.061 | −0.155 | 0.700 | Significant | 0.087 | NS |
| Foxo1-Mm00490672_m1 | 1.229 | 0.042 | −4.074 | 2.295 | −0.370 | 0.427 | Significant | 0.173 | NS |
| Il15-Mm00434210_m1 | 1.076 | 0.026 | −3.634 | 2.561 | −0.324 | 0.474 | Significant | 0.132 | NS |
| Lepr-Mm00440181_m1 | 1.173 | 0.009 | −2.664 | 3.129 | −0.353 | 0.443 | Significant | 0.087 | NS |
| Pdk3-Mm00455220_m1 | 1.071 | 0.028 | −3.703 | 2.520 | −0.323 | 0.476 | Significant | 0.132 | NS |
| Pdk4-Mm00443325_m1 | 0.887 | 0.018 | −3.293 | 2.763 | −0.267 | 0.541 | Significant | 0.119 | NS |
| Stat5b-Mm00839889_m1 | 0.892 | 0.003 | −1.646 | 3.723 | −0.269 | 0.539 | Significant | 0.053 | NS |
| Myogenesisa | |||||||||
| Acvr2a-Mm00431657_m1 | 0.616 | 0.029 | −3.719 | 2.502 | −0.185 | 0.653 | Significant | 0.123 | NS |
| Gdf8-Mm00440328_m1 | 0.888 | 0.047 | −4.155 | 2.234 | −0.267 | 0.540 | Significant | 0.141 | NS |
| Hdac5-Mm00515917_m1 | 0.599 | 0.001 | −0.721 | 4.283 | −0.180 | 0.660 | Significant | 0.026 | Significant |
| Mef2a-Mm00488969_m1 | 0.397 | 0.042 | −4.061 | 2.293 | −0.119 | 0.760 | Significant | 0.140 | NS |
| Mef2d-Mm00504929_m1 | 0.499 | 0.021 | −3.398 | 2.693 | −0.150 | 0.708 | Significant | 0.114 | NS |
| Smad1-Mm00484721_m1 | 0.503 | 0.034 | −3.847 | 2.424 | −0.151 | 0.706 | Significant | 0.123 | NS |
| Smad2-Mm00487530_m1 | 0.701 | 0.011 | −2.793 | 3.050 | −0.211 | 0.615 | Significant | 0.087 | NS |
| Tgfbr1-Mm00436971_m1 | 1.296 | 0.002 | −0.925 | 4.157 | −0.390 | 0.407 | Significant | 0.026 | Significant |
| Tgfbr2-Mm00436978_m1 | 0.675 | 0.013 | −2.977 | 2.941 | −0.203 | 0.626 | Significant | 0.087 | NS |
| Tnni2-Mm00437157_g1 | 0.510 | 0.009 | −2.561 | 3.185 | −0.154 | 0.702 | Significant | 0.087 | NS |
Significance was assigned by the application of the Empirical Bayes statistic. RQ, relative quantification; FDR, false detection rate; NS, not significant. Bold text denotes significant changes in gene expression after more conservative data filtering (Benjamini-Hochberg) to control for false discovery rate, and correct P-values.
aTarget genes normalized to 18S RNA.
bTarget genes normalized to gusB, hprt1 and gapdh.
Figure 2.

Graphs showing significant changes in expression of lipogenic genes in the skeletal muscle of transgenic mice. (A) Data derived from Table 3 are expressed as fold change (log10) normalized to 18S mRNA, following application of Benjamini–Hochberg false detection rate algorithm. qPCR of (B) LXRα, (C) Tip47 and (D) Dgat2 in skeletal muscle of transgenic and wt littermate control mice. Relative fold change is normalized against 18S mRNA (n = 6/group, mean ± SEM. *P < 0.05, **P ≤ 0.01 ***P ≤ 0.001). (E) Graphs showing significant changes in expression of the entire NR gene superfamily in the skeletal muscle of transgenic mice. Data derived from Table 3 and S7 are expressed as fold change (log10) after normalization against the median of three genorm-selected controls GAPDH, gusB and Hprt1 following application of Benjamini–Hochberg false detection rate algorithm.
Subsequently, we used (manual) qPCR to examine (and validate) the expression of several other genes in the lipogenic pathway that were identified in the illumina analysis as significant, differentially expressed targets. The Tg-RORα1ΔDE mice also displayed significantly reduced expression of the mRNAs encoding the nuclear hormone receptor, LXRα (Figure 2B), a critical transcriptional regulator of SREBP-1c and the genetic program that regulates lipogenesis. Interestingly, the expression of both FAS and SCD-1 (responsible for the synthesis of de novo fatty acids and monounsaturated fatty acids, respectively) was also decreased. These genes are downstream targets of LXRα and SREBP1c (the master transcriptional regulators of the genetic program that modulates lipogenesis). Moreover, these changes in gene expression were not observed in either liver or adipose tissue (data not shown).
In addition, we analyzed several genes that are involved in intramuscular triglyceride (IMTG) accumulation. We identified that the expression of the two mRNAs encoding tail interacting protein (Tip47) and di-acyl glycerol acetyl transferase 2 (Dgat2) were significantly reduced in the male Tg-RORα1ΔDE transgenic mice relative to wild-type littermate pairs (Figure 2C and D). In summary, we observed qPCR validation of the master transcriptional regulators of fatty acid biosynthesis (LXR and SREBP-1c) and several important downstream target genes (including FAS and SCD-1).
The observation of attenuated LXR expression and decreased downstream target gene expression was consistent with the identification of this NR signaling cascade in the pathway analysis (Supplementary Figures S4 and S5). We explored the NR signaling pathways more rigorously by performing qPCR analysis utilizing a custom-designed ABI microfluidic TLDA that encoded taqman primer sets targeting all 48 mouse NRs. The analysis revealed small, but significantly reduced (1.5–2.5-fold) expression of the mRNAs encoding the nuclear hormone receptors: LXRα, TR2, PPARδ, GR and RORγ in the Tg-RORα1ΔDE mice, relative to the wt littermates (Figure 2E, Table 3 and Supplementary Figure S7 shows the complete list of nuclear hormone receptors). Please note increased RORα expression in the Tg-RORαΔDE was not detected because the TLDA was mouse specific and the transgenic line expressed the truncated human transcript.
In summary, parallel analysis by illumina, ingenuity and qPCR on the TLDA platform have demonstrated that RORα in skeletal muscle leads regulates the lipogenic/fatty acid biosynthetic pathway.
Expression of truncated RORα attenuated Akt (mRNA and protein) expression in Tg-RORα1ΔDE mice
Pathway analysis revealed the involvement of RORα expression in carbohydrate metabolism and type 2 diabetic signaling, and that truncated RORα expression effected Akt2 mRNA levels (Supplementary Figures S2, S4 and S5A). This led us to further examine the expression of critical genes involved in carbohydrate metabolism using the custom-designed ABI microfluidic TLDA to perform qPCR analysis. We identified statistically significant differential expression of several important genes that control insulin signaling and glucose uptake (Table 3, Supplementary Figure S8 shows the complete list of genes analyzed) in the Tg-RORα1ΔDE mice, relative to wild-type littermates. TLDA analysis revealed the expression of the mRNAs encoding Akt2, pyruvate dehydrogenase kinase isozyme 3 and 4 (Pdk3 and 4), and several other genes were significantly reduced in the skeletal muscle of male Tg-RORα1ΔDE mice relative to wild-type littermates (see Table 3, Supplementary Figure S8. Subsequently, after correction/adjustment of P values according to the Benjamini–Hochberg FDR method, Akt2 remained the only transcript that was significantly and differentially expressed (and repressed) in the Tg-RORα1ΔDE line (see Table 3, Supplementary Figure S8 and Figure 3A). It should be noted that the data (prior to FDR filtering) has been derived from sensitive qPCR TLDA analysis from six littermate pairs of mice focused on metabolic genes. Assignment of significance prior to application of FDR analysis is relatively robust.
Figure 3.

(A) Graph showing a significant fold repression of Akt2 (RQ_Log10) in insulin-stimulated glucose uptake after stringent filtering of data by the application of Benjamini–Hochberg false detection rate algorithm.In the skeletal muscle of transgenic mice relative wild-type mice. Data are derived from Table 3 and is expressed as fold change (log10) normalized to 18S mRNA, following application of Benjamini–Hochberg false detection rate algorithm (n = 6/group) (B) western blot analysis of Akt and pAkt in skeletal muscle of male transgenic mice and wt littermate control mice. Densitometry analysis of western blots (C) Akt and (D) pAkt (n = 3/group, mean ± SEM. *P < 0.05, **P ≤ 0.01). (E) Body weight development in transgenic mice versus wt littermate controls over 14 weeks on normal chow diet (n = 19 male and n = 6 female, mean ± SEM. *P < 0.05). (F) Plasma glucose levels of overnight fasted male transgenic and wt littermate control mice (n = 6/group, mean ± SEM. *P < 0.05). (G) Blood glucose concentrations measured at various times after I/P administration of glucose (t = 0) to overnight fasted male transgenic and wt littermate control mice (n = 7/group, mean ± SEM. *P < 0.05, **P ≤ 0.01). (H) Blood glucose concentrations measured at various times after IP administration of insulin (t = 0) to overnight fasted male transgenic and wt littermate control mice. Data are presented as percentage of starting blood glucose concentration at time = 0 (I = 7/group, mean ± SEM).
Subsequently, we also assessed the expression of the other Akt family members. Although Akt2/PKB was significantly decreased in the skeletal muscle of male Tg-RORα1ΔDE mice relative to wild-type littermates, we observed no change in the expression of the mRNAs encoding, Akt1 or 3 (data not shown). Notably, no significant changes in gene expression were observed in either liver or adipose tissue after TLDA analysis (data not shown). In addition, we investigated whether attenuation in Akt2 mRNA expression correlated with changes in the protein levels of total Akt and phosphorylated (ser473) Akt/PKB using western analysis (Figure 3B). We observed a significant decrease in total Akt/PKB protein in the Tg-RORα1ΔDE mice relative to wild-type littermate pairs (Figure 3B and C), corresponding with the mRNA expression data. Moreover, the levels of the active phosphorylated (ser473) Akt species were also significantly decreased (Figure 3B and D). Akt/PKB is a critical target in the regulation of GLUT4-[solute carrier family 2a (facilitated glucose transporter), member 4] mediated glucose uptake. In summation, corresponding investigation by illumina, qPCR (on the TLDA platform) and western analysis have demonstrated that RORα in skeletal muscle regulates Akt2 mRNA, protein and phosphorylation. This is consistent with the high ranking of carbohydrate metabolism and type 2 diabetic signaling by the ingenuity platform.
Expression of truncated RORα in skeletal muscle induced mild hyperglycemia and glucose intolerance: attenuated insulin-mediated phosphorylation of Akt in the Tg-RORα1ΔDE mice
Suppression of (total and phospho) Akt in skeletal muscle suggests the mouse model may display increased plasma glucose and impaired glucose tolerance. We conducted several experiments to characterize the phenotypic (and metabolic effects) of aberrant RORα expression in skeletal muscle to validate the results of the illumina/ingenuity analysis.
The transgenic mice presented with no gross or histological phenotypic abnormalities, although they did appear to be slightly smaller than their wt littermates. However, growth curve analysis from 4 to 14 weeks indicates that both male and female Tg-RORα1ΔDE mice on a regular chow diet showed no significant reductions in body weight, relative to wild-type littermate pairs (Figure 3E). All subsequent studies were performed on male mice.
We subsequently measured blood glucose and observed mild hyperglycemia in the transgenic mice, with increased fasting glucose levels in male transgenic mice relative to wild-type littermate pairs (Figure 3F). This is consistent with decreased (total and phospho) Akt (mRNA and protein) levels in skeletal muscle. We further examined systemic glucose metabolism, and performed intraperitoneal glucose and insulin tolerance tests. In the Tg-RORα1ΔDE mice, glucose clearance was significantly delayed following a glucose challenge (Figure 3G). No significant differences in plasma insulin levels between the transgenic and wt mice (on regular chow diets) were detected (data not shown). Furthermore, the glucose excursions displayed by Tg-RORα1ΔDE compared to wild-type mice during an insulin tolerance test were comparable (Figure 3H). Moreover, no differences in plasma insulin concentration were observed either at baseline or ten minutes subsequent to glucose administration (data not shown).
We subsequently examined whether skeletal muscle-specific expression of Tg-RORα1ΔDE mice played a role in insulin signaling. Protein extracts were isolated from saline and insulin injected (littermate pairs of) wt and Tg mice and subsequently analyzed by immunoblot analysis using Ab’s specific to Akt2 and phospho- Akt2.
Quantification of the western blots (Figure 4E and F) demonstrated that there were no significant differences in (basal) total Akt2 levels in between saline and insulin-treated wild-type and Tg animals. As expected, insulin treatment in wild-type mice significantly stimulated the levels of phosphoS473Akt2 relative to saline treated mice (∼3-fold Figure 4A and B). However, we observed that insulin treatment did not significantly increase phosphorylation of Akt Ser473 in the male Tg-RORα1ΔDE mice (Figure 4C and D, respectively).
Figure 4.

(A) and (B) Western blot and densitometry analysis of pAkt, total Akt and Gapdh in skeletal muscle of saline and insulin treated wild-type (wt) mice, respectively. (C) and (D) Western blot and densitometry analysis of pAkt, total Akt and Gapdh in skeletal muscle of saline and insulin-treated heterozygous transgenic-RORα1ΔDE mice, respectively. Briefly, fasted mice were IP injected with either saline or insulin (0.5 U/kg). Mice treated with saline (n = 4 wt and 4 tg) or insulin (n = 3 wt and 3 tg) was sacrificed after 10 min and skeletal muscle (quadricep) was excised and snap frozen. Densitometry expressed as the mean +/− SEM, ***P < 0.001 (E) Ex-vivo insulin-stimulated glucose uptake in type II glycolytic muscle of wt and transgenic-RORα1ΔDE mice (n = 7–8). Insulin-stimulated glucose uptake was performed from skeletal muscle (extensor digitorum longus, EDL) dissected from anesthetized mice and incubated in essential buffer (Krebs–Henseleit, pH 7.4) and the assay was performed as described previously (18,19). *P < 0.05, NS, not significant.
Furthermore, impaired glucose tolerance and insulin stimulation of Akt phosphorylation did not involve (or result in) significant changes in GLUT 2, 4 or 8 mRNA expression (determined by qPCR-microfluidic TLDA analysis, see Supplementary Figure S7). GLUT1 mRNA expression was analyzed independently by qPCR, and data presented as a footnote in Supplementary Figure S8. The expression of the mRNAs encoding Glut1, 4 and 8 was decreased by ∼20%, however, after Bayes and FDR analysis, these changes did not attain significance (Supplementary Figure S8).
In the context of the hyperglycemia, impaired glucose tolerance and insulin stimulation of Akt phosphorylation, we investigated whether ex vivo glucose uptake was affected by transgenic RORΔDE expression. (Figure 4E). In wild-type type 2 (fast twitch glycolytic EDL, Figure 4E) skeletal muscle, glucose uptake was increased ∼2-fold by insulin treatment. Similar results were observed in soleus muscle (data not shown). In contrast, in the Tg-RORα1ΔDE mice, glucose uptake did not significantly respond to insulin treatment (Figure 4E).
To further explore the molecular basis of aberrant Akt2 mRNA, protein and phospho AKt expression in the transgenic Tg-RORα1ΔDE mice, we examined the mouse Akt2 promoter sequence (as reported on the Ensembl site) for putative RORE elements. Interestingly, we identified three putative ROR response elements (RORREs) between nucleotide positions, −2900 to −2600 bp upstream of the transcription start site that were concordant with the optimal RORα1 binding site, 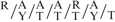 RGGTCA as described by Giguere et al., (26). (Figure 5A and B). In order to determine whether RORα was selectively recruited to the putative ROR responsive elements (NR1, 2 and 3) within the mouse Akt2 promoter, we designed three sets of primers and one set of negative primers downstream of the putative sites within the promoter (Figure 5) for ChIP in skeletal muscle cells. It was observed that the RORα antibody [using anti-RORalpha (Santa Cruz anti-RORα sc-6062)] effectively immunoprecipitated the NR1 (−2859/−2853) and NR2 (−2655/−2649 [and to a lesser extent the NR3 (−2626/2620)] motifs relative to IgG, and the no antibody controls (Figure 5C). The specificity of RORalpha recruitment to these NR half sites was underscored by the lack of recruitment, i.e. the failure to immunoprecipitate the negative control region further downstream between nucleotide positions −278/−177 [∼1.6-kb downstream of these putative RORE sites, (Figure 5C)]. In summary, these data clearly demonstrate that RORα is recruited directly to the mouse Akt2 promoter, and suggest the direct functional involvement of RORα1 in the regulation of AKt2 expression.
RGGTCA as described by Giguere et al., (26). (Figure 5A and B). In order to determine whether RORα was selectively recruited to the putative ROR responsive elements (NR1, 2 and 3) within the mouse Akt2 promoter, we designed three sets of primers and one set of negative primers downstream of the putative sites within the promoter (Figure 5) for ChIP in skeletal muscle cells. It was observed that the RORα antibody [using anti-RORalpha (Santa Cruz anti-RORα sc-6062)] effectively immunoprecipitated the NR1 (−2859/−2853) and NR2 (−2655/−2649 [and to a lesser extent the NR3 (−2626/2620)] motifs relative to IgG, and the no antibody controls (Figure 5C). The specificity of RORalpha recruitment to these NR half sites was underscored by the lack of recruitment, i.e. the failure to immunoprecipitate the negative control region further downstream between nucleotide positions −278/−177 [∼1.6-kb downstream of these putative RORE sites, (Figure 5C)]. In summary, these data clearly demonstrate that RORα is recruited directly to the mouse Akt2 promoter, and suggest the direct functional involvement of RORα1 in the regulation of AKt2 expression.
Figure 5.

(A) and (B) RORα1 is recruited to the Akt2 promoter. Diagrammatic representation of predicted RORalpha response elements in [concordance with the optimal RORα1 binding site, (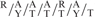 RGGTCA) as described by Giguere et al. (26)] on the promoter of mouse Akt2. Nucleotide numbering is based on the sequence and start site as reported on the ensemble web site (http://www.ensembl.org/Mus_musculus/Gene/Sequence?g=ENSMUSG00000004056). The nucleotide positions and sequence for NR sites 1–3 for is shown on Figure 5A. The downstream negative control was located between nucleotide positions –278/–177 (∼1.6 kb downstream of these putative RORE sites). (C) The recruitment of RORαl onto the AKt2 promoter in C2C12 myotubes by ChIP assay (representative assay) using anti-RORα (Santa Cruz anti-RORα sc-6062). Triplicate real-time PCR analysis was performed and the results are expressed, as the mean ± SD. Results are representative of two independent experiments.
RGGTCA) as described by Giguere et al. (26)] on the promoter of mouse Akt2. Nucleotide numbering is based on the sequence and start site as reported on the ensemble web site (http://www.ensembl.org/Mus_musculus/Gene/Sequence?g=ENSMUSG00000004056). The nucleotide positions and sequence for NR sites 1–3 for is shown on Figure 5A. The downstream negative control was located between nucleotide positions –278/–177 (∼1.6 kb downstream of these putative RORE sites). (C) The recruitment of RORαl onto the AKt2 promoter in C2C12 myotubes by ChIP assay (representative assay) using anti-RORα (Santa Cruz anti-RORα sc-6062). Triplicate real-time PCR analysis was performed and the results are expressed, as the mean ± SD. Results are representative of two independent experiments.
In summary, parallel analysis by illumina, ingenuity, qPCR and western and metabolic analysis has demonstrated that RORα expression in skeletal muscle regulates Akt expression (and phosphorylation state), blood glucose levels, glucose tolerance and insulin-stimulated Akt2 phosphorylation and glucose uptake.
Reduced Akt correlates with increased levels of phospho (thr172)-adenosine monophosphate kinase in Tg-RORα1ΔDE mice
Recent research has demonstrated that Akt2/PKB is a potent negative regulator of AMPK activity (Figure 6 A) in murine cardiac muscle and fibroblasts (27,28). Secondly, ingenuity interrogation of the illumina data identified AMPK signaling as a significantly regulated pathway. Therefore, we undertook western analysis of the basal and phosphorylated AMPK species (Figure 6B). We detected no change in total AMPK protein levels (Figure 6B and C). However, phosphorylated T172 AMPK levels were significantly elevated in skeletal muscle from Tg-RORα1ΔDE mice (Figure 6B and C). The increased levels of phospho-AMPK are entirely consistent with reduced Akt2 activity.
Figure 6.

(A) A pictorial representation of the cross-talk between Akt2 and AMPK pathways, highlighting the implications for lipogenesis in skeletal muscle. (B) and (C) Western blot and densitometry analysis of AMPK and pAMPK in skeletal muscle of transgenic RORα1ΔDE and wt littermate control mice. (D) and (E) Western blot and densitometry analysis of AMPK and pAMPK in skeletal muscle of wt and staggerer (sg/sg) littermate control mice. (n = 3–4/group, mean ± SEM, *P < 0.05, **P ≤ 0.01).
We further investigated the levels of the basal and phosphorylated AMPK species AMPK in homozygous staggerer (sg/sg) mice that lack RORα in all tissues (in contrast, to the muscle-specific overexpression of the dominant negative). We detected a slight but significant ∼1.5-fold increase in total AMPK protein levels (Figure 6D and E) in sg/sg mice relative to wt littermates. However, the levels of phosphorylated T172 AMPK levels did not change in the skeletal muscle tissue from the sg/sg mice relative to wt littermates (Figure 6D and E). In summary, transgenic and muscle-specific overexpression of RORα1ΔDE leads to elevated levels of phospho-AMPK.
Elevated phospho-AMPK in Tg-RORα1ΔDE mice leads to increased phosphoACC Levels, and increased PGC-1α and CPT-1b mRNA expression
The literature reports that the positive effect of activated AMPK on fatty acid oxidation is mediated by phosphorylation of Acetyl CoA carboxylase ACC (and decreased production of malonyl CoA) (see Figure 7A). Moreover, AMPK activators increase the expression of PGC-1α and PPARα target genes, for example CPT-1 in skeletal muscle (29,30). Consequently, we were particularly interested to assess whether increased expression of activated AMPK resulted in the induction of phosphoACC (Ser79) and two critical genes associated with fatty acid oxidation in the skeletal muscle of the Tg-RORα1ΔDE mice.
Figure 7.

(A) A pictorial representation of the cross-talk between the AMPK and fatty acid oxidation pathways, highlighting the implications for fatty acid oxidation in skeletal muscle. (B) Western blot analysis of ACC and pACC in skeletal muscle of transgenic and wt littermate control mice. Densitometry analysis of western blots (n = 3/group, mean ± SEM, **P ≤ 0.01). (C) densitometry analysis of ACC and pACC in skeletal muscle of transgenic RORα1ΔDE and wt littermate control mice. (D) qPCR of PGC-1α and (E) CPT1b in skeletal muscle of transgenic and wt littermate control mice. Relative fold change is normalized against 18S mRNA (n = 6/group, mean ± SEM. *P < 0.05, **P ≤ 0.01).
We undertook analysis of the basal and phosphorylated ACC species (Figure 7B). We detected a significant change in total ACC protein levels (Figure 7B). More importantly, phosphorylated ACC levels (detected by western blot) were significantly elevated in skeletal muscle from Tg-RORα1ΔDE mice (Figure 7B and C). qPCR analysis revealed the significant (differential) expression of the mRNAs encoding PGC-1α (Figure 7D) and CPT-1b (Figure 7E) in the skeletal muscle of male Tg-RORα1ΔDE mice relative to wild-type littermates. The elevated levels of pACC and PGC-1α and CPT-1 mRNA expression in the Tg mice are consistent with increased levels of pAMPK and pACC.
Expression of the truncated orphan nuclear receptor, RORα1 had minimal effects on important skeletal muscle markers of fiber type and muscle mass
We utilized qPCR-TLDA analysis to assess whether the expression of the truncated receptor affect markers of contractile function, fiber type and muscle mass (see Table 3, Supplementary Figure S9). Specifically, in this context, TLDA analysis revealed significant differential expression of the mRNAs encoding histone deacetylase 5 (HDAC5), transforming growth factor beta receptor 1 (Tgfbr1) and several other genes (including Mef2a/d, Tnni2 and Acvr2A) in the skeletal muscle of male Tg-RORα1ΔDE mice relative to wild-type littermates (see Table 3, Supplementary Figure S9) when normalized against 18S rRNA. However, following the application of conservative data filtering using the FDR–Benjamini–Hochberg algorithm, only HDAC5 (∼1.25-fold) and Tgfbr1 (2.5-fold) survived as significant (but weak) decreases in expression (Table 3, and Supplementary Figure S9). This suggested that the changes in glucose tolerance, insulin signaling and lipogenesis were not indirect effects of changes in muscle mass and/or contractile function.
DISCUSSION
The biochemical and molecular characterization of RORs in cell culture and animal models has revealed that RORs play a critical role in the modulation of lipid homeostasis in a tissue-specific manner (1–5,31,32). Our previous studies reported that homozygous (male and female) staggerer mice (with decreased and dysfunctional expression of RORα in all organs) display reduced adiposity and are resistant to high-fat diet-induced obesity. The lean phenotype of staggerer mice was associated with significantly increased expression of genes involved in fatty acid oxidation (including PGC-1, lipin1, etc.), and significantly reduced expression of SREBP-1c (and several lipogenic genes) in all major metabolic tissues.
Surprisingly, studies investigating the role of RORα in the regulation of glucose homeostasis have not been reported. In this context, we were particularly interested in exploring the role of RORα1 in skeletal muscle, a major mass peripheral tissue that accounts for the majority of glucose disposal and lipid catabolism. We probed the role of RORα signaling in this major mass peripheral tissue, by the skeletal muscle-specific overexpression of truncated RORα1ΔDE (lacking the ligand-binding domain) to investigate the contribution of this peripheral tissue to the RORα phenotype. This construct has been described to operate in a dominant negative manner in several reports (21,22). This strategy also allows the probing of RORα-dependent gene expression in the absence of an identified native and/or synthetic ligand.
The human skeletal α-actin promoter was utilized to construct the muscle-specific vector. This promoter has been demonstrated to function in a cell/tissue-specific manner (10,11). As expected the transgene, RORα1ΔDE, was efficiently and specifically expressed in skeletal muscle of transgenic mice relative to other organ/tissues. Second, our RORα1ΔDE expression vector retains the native RORα1 amino-terminal AF-1 domain (i.e. AB region) and the native zinc finger region; these properties ensure that the NR targets the native RORα1 response elements in the skeletal muscle of transgenic mice. Giguere et al. (26) demonstrated that each ROR isoform displays isoform-specific DNA binding, and the amino-terminal domain and the zinc finger region work in parallel to confer sequence-specific DNA recognition and binding.
Expression profiling, coupled to analysis on the ingenuity platform identified a RORα1-regulated subset of genes that were involved in or associated with lipid metabolism, small molecule transport and biochemistry, cardiovascular and metabolic disease, carbohydrate metabolism, endocrine system disorders etc. This is in concordance with investigations in RORα-deficient mouse models and in vitro promoter/cell culture investigations that have demonstrated the involvement of RORα in a number of physiological process including atherosclerosis, lipid homeostasis, fat deposition, obesity, inflammation, immunity, etc (6). In the framework of this investigation, the primary signaling pathways regulated in skeletal muscle were glutathione metabolism (that tightly controls ROS levels and modulates insulin sensitivity), type 2 diabetic signallng and fatty acid biosynthesis. Notably, the LXR signaling (and RXR dependent) pathway was identified in the analysis, which controls lipogenesis and cholesterol homeostasis.
In concordance with the array analysis, qPCR analysis on the TLDA platform revealed significantly attenuated expression of the mRNAs (encoding the hierarchical regulators of lipogenesis), LXRα and SREBP-1c and several other downstream target genes involved in fatty acid biosynthesis in skeletal muscle. For example, we observed decreased expression of the downstream target genes, SCD1 and FAS. Moreover, in the context of lipogenesis, we observed the suppression of Ascl4, Dgat2, Tip47, HIF-1 and Cd36. Acsl4, the long chain acyl CoA synthetase, plays an important role in lipid metabolism by routing fatty acids to several different metabolic pools. Decreased Acsl4 expression is associated with decreased triglycerides and fatty acids partitioning to di- and tri-acyl glycerol (33,34) and consistent with reduced Dgat2 (that catalyzes the final step in triglyceride production) and Tip47 (a perilipin family member that co-ordinates the storage of triacylglycerol (35). Furthermore, it has been reported that decreased Dgat2 expression is associated with reduced expression of SREBP-1c and SCD-1 and increased CPT-1 expression (see below), in concordance with our observations (36). Finally, suppression of HIF-1alpha is associated with reduced fatty acid uptake and biosynthesis, and in concordance with the suppression of the genetic program controlling lipogenesis and the decrease in the fatty acid translocase, CD36 (37).
In the context of the decrease in LXR and other NR signaling pathways, we completed TLDA analysis of the entire NR supergene family. This analysis identified decreases in the expression of the mRNAs encoding GR, PPARδ, TR2 and RORγ. These nuclear receptors have been implicated in the regulation of lipid homeostasis. We did not observe decreases in THR(α / β), FXR, VDR, etc. that were identified by ingenuity. We suggest that the detection of these pathways by ingenuity reflects secondary consequences of RORα-mediated dysregulation and the aberrant expression of NRs identified above.
As discussed, ingenuity analysis of the array data identified carbohydrate metabolism as a significantly regulated function. We observed the male tg-RORα1ΔDE mice display significantly increased fasting plasma glucose levels, impaired glucose tolerance and attenuated insulin-stimulated glucose uptake relative to wild-type littermates. Our qPCR TLDA profiling identified the significantly decreased expression of the mRNA encoding Akt2/PKB in skeletal muscle, after Bayes assignment of significance and stringent FDR filtering. Furthermore, the total amount of Akt and phospho-Akt protein was significantly reduced. In this context, several studies have demonstrated that, Akt2 plays a critical role in insulin-mediated glucose disposal in skeletal muscle. For example, Cho et al. (38) demonstrated that disruption of the Akt2 gene expression in mice was associated with insulin resistance and type2 diabetes-like syndrome. Subsequently, Garofalo et al. (39) showed mice lacking Akt2/PKB gene displayed a diabetic phenotype and age-dependant lipodystrophy.
Interestingly, the mild hyperglycemia (in the fasted state), glucose intolerance and impaired glucose uptake occur without any significant change in whole body insulin sensitivity (i.e. the insulin tolerance test) and/or change in GLUT1 and four expression. First, this does not appear to involve an insulin secretion defect as the transgene is not expressed in the pancreas and the mice develop hyperinsulinemia after a high-fat challenge (data not shown). It should be noted that muscle-specific deletion of an essential component (rictor) of the mTOR complex 2 leads to the attenuation of insulin-induced phosphorylation of Akt2 at Ser473. Moreover, these mice displayed glucose intolerance, however, insulin sensitivity remained unchanged (40). Similarly, our Tg-RORα1ΔDE mice displayed an attenuated insulin-mediated Ser473 phosphorylation of Akt and glucose uptake. In this context, Gonzalez and McGraw (41) demonstrated that ‘insulin signalling diverges into Akt-dependent and independent signals’. James and colleagues (42) recently stated that several studies underscore the pivotal role of Akt2 in GLUT4 function; however, many gaps remain in understanding the signaling cascades involved. For example, similar inconsistencies (between impaired glucose uptake and/or normal glucose tolerance and insulin sensitivity) have been described in other animal models that have perturbed expression and/or knockout in critical genes that regulate glucose homeostasis.
ChIP analysis revealed RORαl was directly involved in the regulation of Akt2. We identified several potential RORa1 response elements in the mouse Akt2 promoter between −2900 and −2600 nt upstream of the transcription start site. These sites were accommodated by the motif 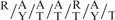 RGGTCA described by Giguere et al. (26), as an optimal RORα1-binding site. ChIP identified functional RORα1 recruitment to three sites (in close proximity) within this region. Further characterization of the Akt2 promoter is required to elucidate the mechanism mediating regulation by RORα1.
RGGTCA described by Giguere et al. (26), as an optimal RORα1-binding site. ChIP identified functional RORα1 recruitment to three sites (in close proximity) within this region. Further characterization of the Akt2 promoter is required to elucidate the mechanism mediating regulation by RORα1.
Ingenuity identified AMPK signaling as a regulated pathway in the Tg mice overexpressing the dominant negative RORα1. It has been well established that AMPK is master regulator of energy homeostasis through the suppression of ATP-consuming lipogenic pathways and by the enhancement of ATP producing catabolic pathways, including fatty acid oxidation in skeletal muscle tissue (43,44 and references therein). Moreover, Akt2/PKB is a potent modulator of AMPK activity. For example, Hahn-Windgassen et al. (28) showed that Akt/PKB regulates intracellular ATP levels through regulating AMPK activity. Furthermore, Planavila et al. (45) showed that mice treated with troglitazone (PPARγ agonist) in skeletal muscle were associated with increased Akt2/PKB and decreased AMPK activity. Moreover, recent investigations on Akt2/PKB and AMPK pathways in heart tissue showed that Akt2/PKB is a negative regulator of AMPK activity (27). Finally, the association between increased AMPK activity and the reduction of lipogenic gene expression and genes regulating fatty acid oxidation is well documented (46–49). Furthermore, the inhibitory effect of potent AMPK activators such as aminoimidazole carboxamide ribonucleotide (AICAR) or Metformin on lipogenesis in skeletal muscle has been described in both human and rodents (29,30). Consequently, in the context of our studies, we were particularly interested in examining the cross-talk between lipid homeostasis (lipogenesis and fatty acid oxidation), Akt2 signaling (and glucose tolerance) and AMPK signaling in a background of aberrant RORα1 (NR) signaling.
Interestingly, the hyperglycemia and significantly decreased Akt2 (mRNA and protein) and phosphoAKT levels in the Tg-RORα1ΔDE mice are associated with increased phosphoAMPK levels, in concordance with the studies discussed above. This is consistent with studies demonstrating type 2 diabetic patients also display normal AMPK signaling (50,51). Paradoxically, some studies report that increased AMPK activity mediates (only exercise induced) glucose transport. However, AMPK null mice exhibit typical exercise stimulated glucose uptake in skeletal muscle and AICAR does not induce glucose transport in muscle (52). Interestingly, we did not observe increases in pAMPK in the skeletal muscle of staggerer (sg/sg) mice that lack RORα expression in all tissues (although we observed mild elevation in the levels of basal/total AMPK). Differential AMPK activity in these lines is not completely unexpected for several reasons. Firstly, we are comparing a line of mice overexpressing dominant negative RORa1 (in skeletal muscle), against a line of mice lacking RORα1 expression. Second, in mice lacking NR expression, genes (and/or phenotypes) maybe silenced or derepressed depending on cofactor requirements, expression and/or promoter characteristics (53).
The increased AMPK activity in the Tg-RORα1ΔDE mice did not only correlate with decreased Akt activity, but was also consistent with (i) attenuated mRNA expression of LXRα and SREBP-1c, and several other genes involved in fatty acid biosynthesis in skeletal muscle (44,54,55) and (ii) induction of the genes/pathways increasing fatty acid oxidation (56). For example, SCD1 deficiency has been reported to be associated with increased AMPK activity, fatty acid oxidation and reduced ceramide synthesis (41,42,57). In addition, in this interconnected regulatory milieu, we observed increased expression of two critical regulators of fatty acid oxidation, PGC-1α and CPT1b, concomitant with elevated levels of phospho-Ser79 ACC in the Tg-RORα1ΔDE mice, relative to the wt littermate mice. This is also consistent with the observations (47–49,56 and references therein) that activated AMPK and AMPK agonists regulate PGC-1 and pACC expression.
Finally, overexpression of truncated RORα in skeletal muscle resulted in very minor changes in the expression of genes regulating muscle mass, proliferation and differentiation. Interestingly, the analysis did identify HDAC5 as a differentially expressed gene. A recent study has demonstrated the link between AMPK, energy balance and transcriptional regulation of GLUT4 expression mediated by HDAC5. That study indicated that increased AMPK activity induced HDAC phosphorylation that reduces HDAC5 association with the GLUT4 promoter. Interestingly, in our mouse model, although insulin mediated phosphorylation of Akt2, and glucose uptake was perturbed; the association between increased AMPK activity and HDAC5 was maintained.
In conclusion our investigation reveals that the orphan nuclear receptor RORα operates at the nexus of pathways controlling the association between lipid homeostasis (lipogenesis, and fatty acid oxidation), Akt2 signaling (glucose tolerance and uptake) and AMPK signaling.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research project grant from the National Health and Medical Research Council (NHMRC) of Australia, and the Diabetes Australia Research Trust (DART). GEOM is a Principal Research Fellow of the NHMRC, and Suryaprakash Raichur was a recipient of an International Postgraduate Research Scholarship (IPRS).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Raspe E, Duez H, Gervois P, Fievet C, Fruchart JC, Besnard S, Mariani J, Tedgui A, Staels B. Transcriptional regulation of apolipoprotein C-III gene expression by the orphan nuclear receptor RORalpha. J. Biol. Chem. 2001;276:2865–2871. doi: 10.1074/jbc.M004982200. [DOI] [PubMed] [Google Scholar]
- 2.Mamontova A, Seguret-Mace S, Esposito B, Chaniale C, Bouly M, Delhaye-Bouchaud N, Luc G, Staels B, Duverger N, Mariani J, et al. Severe atherosclerosis and hypoalphalipoproteinemia in the staggerer mouse, a mutant of the nuclear receptor RORalpha. Circulation. 1998;98:2738–2743. doi: 10.1161/01.cir.98.24.2738. [DOI] [PubMed] [Google Scholar]
- 3.Lau P, Nixon SJ, Parton RG, Muscat GE. RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J. Biol. Chem. 2004;279:36828–36840. doi: 10.1074/jbc.M404927200. [DOI] [PubMed] [Google Scholar]
- 4.Raichur S, Lau P, Staels B, Muscat GE. Retinoid-related orphan receptor gamma regulates several genes that control metabolism in skeletal muscle cells: links to modulation of reactive oxygen species production. J. Mol. Endocrinol. 2007;39:29–44. doi: 10.1677/jme.1.00010. [DOI] [PubMed] [Google Scholar]
- 5.Vu-Dac N, Gervois P, Grotzinger T, De Vos P, Schoonjans K, Fruchart JC, Auwerx J, Mariani J, Tedgui A, Staels B. Transcriptional regulation of apolipoprotein A-I gene expression by the nuclear receptor RORalpha. J Biol. Chem. 1997;272:22401–22404. doi: 10.1074/jbc.272.36.22401. [DOI] [PubMed] [Google Scholar]
- 6.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang HS, Angers M, Beak JY, Wu X, Gimble JM, Wada T, Xie W, Collins JB, Grissom SF, Jetten AM. Gene expression profiling reveals a regulatory role for ROR alpha and ROR gamma in phase I and phase II metabolism. Physiol. Genomics. 2007;31:281–294. doi: 10.1152/physiolgenomics.00098.2007. [DOI] [PubMed] [Google Scholar]
- 8.Lau P, Fitzsimmons RL, Raichur S, Wang SC, Lechtken A, Muscat GE. The orphan nuclear receptor, ROR{alpha}, regulates gene expression that controls lipid metabolism: STAGGERER (SG/SG) mice are resistant to diet-induced obesity. J. Biol. Chem. 2008;283:18411–18421. doi: 10.1074/jbc.M710526200. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz-Peiffer C. Signalling aspects of insulin resistance in skeletal muscle: mechanisms induced by lipid oversupply. Cell Signal. 2000;12:583–594. doi: 10.1016/s0898-6568(00)00110-8. [DOI] [PubMed] [Google Scholar]
- 10.Brennan KJ, Hardeman EC. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J. Biol. Chem. 1993;268:719–725. [PubMed] [Google Scholar]
- 11.Muscat GE, Kedes L. Multiple 5′-flanking regions of the human alpha-skeletal actin gene synergistically modulate muscle-specific expression. Mol. Cell Biol. 1987;7:4089–4099. doi: 10.1128/mcb.7.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaenisch R. Transgenic animals. Science. 1988;240:1468–1474. doi: 10.1126/science.3287623. [DOI] [PubMed] [Google Scholar]
- 14.Mitrecic D, Huzak M, Curlin M, Gajovic S. An improved method for determination of gene copy numbers in transgenic mice by serial dilution curves obtained by real-time quantitative PCR assay. J. Biochem. Biophys. Methods. 2005;64:83–98. doi: 10.1016/j.jbbm.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang X, Downes M, Yu RT, Bookout AL, He W, Straume M, Mangelsdorf DJ, Evans RM. Nuclear receptor expression links the circadian clock to metabolism. Cell. 2006;126:801–810. doi: 10.1016/j.cell.2006.06.050. [DOI] [PubMed] [Google Scholar]
- 17.Myers SA, Eriksson N, Burow R, Wang SC, Muscat GE. Beta-adrenergic signaling regulates NR4A nuclear receptor and metabolic gene expression in multiple tissues. Mol. Cell Endocrinol. 2009;309:101–108. doi: 10.1016/j.mce.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Brozinick JT, Jr, Birnbaum MJ. Insulin, but not contraction, activates Akt/PKB in isolated rat skeletal muscle. J. Biol. Chem. 1998;273:14679–14682. doi: 10.1074/jbc.273.24.14679. [DOI] [PubMed] [Google Scholar]
- 19.Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, Kemp BE, Febbraio MA, Steinberg GR. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat. Med. 2006;12:541–548. doi: 10.1038/nm1383. [DOI] [PubMed] [Google Scholar]
- 20.Pearen MA, Myers SA, Raichur S, Ryall JG, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology. 2008;149:2853–2865. doi: 10.1210/en.2007-1202. [DOI] [PubMed] [Google Scholar]
- 21.McBroom LD, Flock G, Giguere V. The nonconserved hinge region and distinct amino-terminal domains of the ROR alpha orphan nuclear receptor isoforms are required for proper DNA bending and ROR alpha-DNA interactions. Mol. Cell Biol. 1995;15:796–808. doi: 10.1128/mcb.15.2.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- 23.Clapham JC, Arch JR, Chapman H, Haynes A, Lister C, Moore GB, Piercy V, Carter SA, Lehner I, Smith SA, et al. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature. 2000;406:415–418. doi: 10.1038/35019082. [DOI] [PubMed] [Google Scholar]
- 24.Genoux A, Dehondt H, Helleboid-Chapman A, Duhem C, Hum DW, Martin G, Pennacchio LA, Staels B, Fruchart-Najib J, Fruchart JC. Transcriptional regulation of apolipoprotein A5 gene expression by the nuclear receptor RORalpha. Arterioscler. Thromb. Vasc. Biol. 2005;25:1186–1192. doi: 10.1161/01.ATV.0000163841.85333.83. [DOI] [PubMed] [Google Scholar]
- 25.Lind U, Nilsson T, McPheat J, Stromstedt PE, Bamberg K, Balendran C, Kang D. Identification of the human ApoAV gene as a novel RORalpha target gene. Biochem. Biophys. Res. Commun. 2005;330:233–241. doi: 10.1016/j.bbrc.2005.02.151. [DOI] [PubMed] [Google Scholar]
- 26.Giguere V, Tini M, Flock G, Ong E, Evans RM, Otulakowski G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994;8:538–553. doi: 10.1101/gad.8.5.538. [DOI] [PubMed] [Google Scholar]
- 27.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 28.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 29.Kola B, Boscaro M, Rutter GA, Grossman AB, Korbonits M. Expanding role of AMPK in endocrinology. Trends Endocrinol. Metab. 2006;17:205–215. doi: 10.1016/j.tem.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Osler ME, Zierath JR. Adenosine 5′-monophosphate-activated protein kinase regulation of fatty acid oxidation in skeletal muscle. Endocrinology. 2008;149:935–941. doi: 10.1210/en.2007-1441. [DOI] [PubMed] [Google Scholar]
- 31.Wada T, Kang HS, Jetten AM, Xie W. The emerging role of nuclear receptor ROR{alpha} and its crosstalk with LXR in Xeno- and endobiotic gene regulation. Exp. Biol. Med. (Maywood) 2008;233:1191–1201. doi: 10.3181/0802-MR-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wada T, Kang HS, Angers M, Gong H, Bhatia S, Khadem S, Ren S, Ellis E, Strom SC, Jetten AM, et al. Identification of oxysterol 7alpha-hydroxylase (Cyp7b1) as a novel retinoid-related orphan receptor alpha (RORalpha) (NR1F1) target gene and a functional cross-talk between RORalpha and liver X receptor (NR1H3) Mol. Pharmacol. 2008;73:891–899. doi: 10.1124/mol.107.040741. [DOI] [PubMed] [Google Scholar]
- 33.Askari B, Kanter JE, Sherrid AM, Golej DL, Bender AT, Liu J, Hsueh WA, Beavo JA, Coleman RA, Bornfeldt KE. Rosiglitazone inhibits acyl-CoA synthetase activity and fatty acid partitioning to diacylglycerol and triacylglycerol via a peroxisome proliferator-activated receptor-gamma-independent mechanism in human arterial smooth muscle cells and macrophages. Diabetes. 2007;56:1143–1152. doi: 10.2337/db06-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudo T, Tamagawa T, Kawashima M, Mito N, Shibata S. Attenuating effect of clock mutation on triglyceride contents in the ICR mouse liver under a high-fat diet. J. Biol. Rhythms. 2007;22:312–323. doi: 10.1177/0748730407302625. [DOI] [PubMed] [Google Scholar]
- 35.Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149:942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- 36.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J. Biol. Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 37.Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw E.B., 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 39.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J. Clin. Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence J.C., Jr Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol. Cell Biol. 2008;28:61–70. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez E, McGraw TE. Insulin signaling diverges into Akt-dependent and -independent signals to regulate the recruitment/docking and the fusion of GLUT4 vesicles to the plasma membrane. Mol. Biol. Cell. 2006;17:4484–4493. doi: 10.1091/mbc.E06-07-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larance M, Ramm G, James DE. The GLUT4 code. Mol. Endocrinol. 2008;22:226–233. doi: 10.1210/me.2007-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hegarty BD, Turner N, Cooney GJ, Kraegen EW. Insulin resistance and fuel homeostasis: the role of AMP-activated protein kinase. Acta. Physiol. (Oxf) 2009;196:129–145. doi: 10.1111/j.1748-1716.2009.01968.x. [DOI] [PubMed] [Google Scholar]
- 44.Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, et al. AMPK: lessons from transgenic and knockout animals. Front. Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Planavila A, Alegret M, Sanchez RM, Rodriguez-Calvo R, Laguna JC, Vazquez-Carrera M. Increased Akt protein expression is associated with decreased ceramide content in skeletal muscle of troglitazone-treated mice. Biochem. Pharmacol. 2005;69:1195–1204. doi: 10.1016/j.bcp.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Rutter GA, Da Silva Xavier G, Leclerc I. Roles of 5′-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem. J. 2003;375:1–16. doi: 10.1042/BJ20030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J. Biol. Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 48.Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, et al. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- 49.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Musi N, Fujii N, Hirshman MF, Ekberg I, Froberg S, Ljungqvist O, Thorell A, Goodyear LJ. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes. 2001;50:921–927. doi: 10.2337/diabetes.50.5.921. [DOI] [PubMed] [Google Scholar]
- 51.Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001;280:E677–684. doi: 10.1152/ajpendo.2001.280.5.E677. [DOI] [PubMed] [Google Scholar]
- 52.Fujii N, Aschenbach WG, Musi N, Hirshman MF, Goodyear LJ. Regulation of glucose transport by the AMP-activated protein kinase. Proc. Nutr. Soc. 2004;63:205–210. doi: 10.1079/PNS2004340. [DOI] [PubMed] [Google Scholar]
- 53.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, et al. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol. Cell Biol. 2003;23:5780–5789. doi: 10.1128/MCB.23.16.5780-5789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006;574:41–53. doi: 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Craddock L, Hong S, Liu ZM. AMP-activated protein kinase suppresses LXR-dependent sterol regulatory element-binding protein-1c transcription in rat hepatoma McA-RH7777 cells. J. Cell Biochem. 2009;106:414–426. doi: 10.1002/jcb.22024. [DOI] [PubMed] [Google Scholar]
- 56.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu. Rev. Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 57.Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Cohen P, Asilmaz E, Hardie DG, Friedman JM, Ntambi JM. Stearoyl-CoA desaturase-1 deficiency reduces ceramide synthesis by downregulating serine palmitoyltransferase and increasing beta-oxidation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005;288:E599–E607. doi: 10.1152/ajpendo.00439.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.