

Abstract
FoxM1 is a member of the Forkhead family of transcription factors and is implicated in inducing cell proliferation and some forms of tumorigenesis. It binds promoter regions with a preference for tandem repeats of a consensus ‘TAAACA’ recognition sequence. The affinity of the isolated FoxM1 DNA-binding domain for this site is in the micromolar range, lower than observed for other Forkhead proteins. To explain these FoxM1 features, we determined the crystal structure of its DNA-binding domain in complex with a tandem recognition sequence. FoxM1 adopts the winged-helix fold, typical of the Forkhead family. Neither ‘wing’ of the fold however, makes significant contacts with the DNA, while the second, C-terminal, wing adopts an unusual ordered conformation across the back of the molecule. The lack of standard DNA–‘wing’ interactions may be a reason for FoxM1’s relatively low affinity. The role of the ‘wings’ is possibly undertaken by other FoxM1 regions outside the DBD, that could interact with the target DNA directly or mediate interactions with other binding partners. Finally, we were unable to show a clear preference for tandem consensus site recognition in DNA-binding, transcription activation or bioinformatics analysis; FoxM1's moniker, ‘Trident’, is not supported by our data.
INTRODUCTION
In the field of cancer biology, intense interest has recently focused upon the Forkhead transcription factor FoxM1. This is because its expression is tightly linked to a cell’s proliferative potential (1). FoxM1 regulates the transcription of a number of proteins required for cell-cycle progression (e.g. Cdc25C, Plk1, cyclin B2 and securin) (2) as well as those that alleviate the oxidative burden incurred by proliferating cells (3). As such, FoxM1 is expressed in cycling cells, but becomes barely detectable when a cell is quiescent or terminally differentiated. However, high levels of FoxM1 may again be observed in states of uncontrolled cellular growth, as has been documented in a number of cancerous cell-lineages (4–13). Moreover, some tumors are known to become dependent upon FoxM1 over-expression for survival (3). This ‘addiction’ of certain diseased states to overexpressed FoxM1 has led to an effort to characterize its potential as a therapeutic chemotherapy target and several inhibitory compounds are now being investigated (14–17).
FoxM1 is a member of the Forkhead Box (Fox) family of transcription factors (18). Each Fox protein contains a conserved stretch of 90 amino acids that comprise a DNA-binding Forkhead box, a domain that serves to target the proteins to genomic promoter regions. Humans have over 40 Fox proteins, which are grouped into subfamilies with consecutive alphabetic designations (18). These Fox-subfamilies are varied and share little sequence similarity or global domain-architecture, their only unifying link being the conserved DNA-binding domain. Across the super-family, the residues within this domain have levels of sequence identity that approach 60%. Unsurprisingly, this results in the different Fox proteins having somewhat similar sequence-specificities. Despite this overlap in specificity, each family member regulates a distinct subset of genes during cell division and development.
The transcriptional activity of FoxM1 is regulated in part through dynamic interactions between its different domains. The full-length protein has three distinct components: a central DNA-binding Forkhead box, an N-terminal auto-inhibitory domain and a C-terminal transcriptional transactivation domain. Cell-cycle dependence of FoxM1's transcriptional activity arises through control mechanisms that influence inter-domain interactions by controlling their phosphorylation status at defined cell-cycle stages. Expression of FoxM1 peaks in late G1 and early S-phase, but activity is only induced later in two separate stages that initiate different transcriptional pathways, the first in S/G2-phase and the second around the G2/M transition. When first expressed, the transactivating domain of FoxM1 is kept inactive through an ‘intra’-molecular auto-inhibitory interaction with the N-terminal domain (19,20). From S-phase to the G2/M transition the protein is then phosphorylated at several sites by CyclinA/Cdk, disrupting the FoxM1 auto-inhibitory interaction and leading to the transcription of the first set of target genes (21,22). Full initiation of FoxM1’s mitotic program requires a second round of phosphorylation that further enhances transcriptional activity. This occurs when the mitotic kinase Plk-1 recognizes pre-primed FoxM1 as a substrate, resulting in further phosphorylation of the C-terminal domain (23), thus allowing the initiation of the FoxM1 transcriptional pathways necessary for mitotic progression.
The conserved Forkhead box of FoxM1 adopts a winged-helix-type fold commonly found in DNA-binding proteins (24). The DNA specificity of this domain has been previously characterized using coupled pull-down and gel-shift assays (25,26). Such assays screen a pool of random oligonucleotides identifying those that bind to the FoxM1 DNA-binding domain (FoxM1-DBD, residues 207–348) with high affinity (1), and led to the establishment of a core consensus site, TAAACA. This site is similar to that known for other Forkhead proteins: (G/A)(T/C)(A/C)AA(C/T)A. When the FoxM1 consensus sequence was first identified in the gel-shift assays it was also observed that the DBD preferentially bound to tandem copies of the site (1). This was intriguing as the Forkhead proteins are well conserved; if one member of the family has a unique avidity for repeated sequences this could represent a physiological difference in the way it regulates transcription. We were thus interested in the mechanism by which FoxM1 might induce an allosteric increase in its affinity for tandem consensus sites. In this paper, we present the first crystal structure of the human FoxM1 DBD, in complex with a tandem promoter-like recognition sequence. We then combine the structural information with DBD affinity measurements for FoxM1 recognition site variants, transcription activation assays and bioinformatics searches, in an effort to understand the mechanism of action of the protein.
METHODS
Protein production
The FoxM1C construct (residues 222–360) was cloned into a pET-28-based vector with the N-terminal tag MAHHHHHHSAALEVLFQGPG. This was transformed into Escherichia coli BL21 (DE3) and grown at 37°C until reaching an Abs600nm of 0.8–1.0, protein was induced at 15°C for 16 h by adding 0.5 mM IPTG. Bacteria were harvested by centrifugation and resuspended in 100 ml of buffer A (20 mM Tris pH 8.0, 500 mM NaCl, 2 mM Imidazole, 0.5 mM TCEP). After which they were lysed and then cleared by high-speed centrifugation before loading on a Ni–NTA metal affinity column (Qiagen Valencia, CA, USA). After washing the protein was eluted in buffer A containing 400 mM imidazole and the first 17 tag residues removed by overnight cleavage using human rhinovirus 3C protease. The product was then loaded on a Superdex G75 16/60 HiLoad size exclusion column (GE Healhcare Life Sciences) equilibrated in buffer A. FoxM1C.222.360 eluted as a monomer with the main peak pooled and concentrated to 15–20 mg/ml, flash-frozen in liquid nitrogen and stored at −80°C until required. Protein concentration was determined through 280 nm absorbance measurements in the presence of 6 M guanidinium hydrochloride (Abs 1g/l = 1.78).
Crystallization
The oligonucleotides aaattgtttataaacagcccg and ttcgggctgtttataaacaat were dissolved in water and heat-annealed. For crystallization protein, DNA and buffer A were mixed to give a final concentration of 12 mg/ml protein (0.72 mM) and 0.5 mM Fm1-2x-19nt-aa/tt oligonucleotide. The mixture was incubated for 30 min on ice prior to being set in a 96-well sitting-drop format using 75 µl reservoirs and drops consisting of 400 nl protein/DNA mixed with 400 nl of reservoir. Crystal grew in 24% w/v PEG 3350 and 0.2 M sodium malonate and was serially transferred into the same solution with an additional 25% v/v Ethylene Glycol for cryoprotectant. Diffraction data were collected at 100K at the European Synchrotron Radiation Facility on beamline ID23-1. The data thus obtained were integrated and scaled at 2.2 Å using Mosflm and Scala.
Phases were obtained by Molecular Replacement using AMoRe (27) and a single protein monomer of FoxK1 [pdb: 2C6Y (28)]. Two molecules of FoxM1 and one DNA duplex were present in the asymmetric unit. The resulting electron density was clear allowing iterative model building in Coot (29) and refinement with Refmac5 (27,30). The final data reduction and refinement statistics are shown in Table 1.
Table 1.
Data collection and refinement statistics
| pH 7.5 | |
|---|---|
| Data collection statistics | |
| Number of crystals | 1 |
| Space group | C2221 |
| Unit cell: a, b, c (Å) | 63.1, 119.8, 153.0 |
| Resolution Limits (High) (Å) | 38.1–2.2 (2.3–2.2) |
| Completeness (%) | 91.9 (61.3) |
| Number of reflections | 25 545 |
| Multiplicity | 5.0 (4.4) |
| Rpim | 0.030 (0.213) |
| <I/σ(I)> | 17.2 (3.2) |
| Wilson's B-factor (Å2) | 48.1 |
| Model refinement statistics | |
| Rfactora | 0.203 |
| Rfreea | 0.234 |
| Number of protein atoms | 1644 |
| Number of DNA atoms | 945 |
| Number of waters (ions) | 163 (2) |
| Ramachandran plotb | |
| – favored (%) | 96.1 |
| – outliers (%) | 0.0 |
| RMS bond, Z-score (Å)a | 0.009, 0.370 |
| RMS angle, Z-score (°)a | 1.45, 0.538 |
Fluorescence anisotropy assay
The dissociation constants and IC50 values of the FoxM1-DBD for different consensus site DNA sequences were measured by fluorescence anisotropy (FP) assay. Measurements were performed on a EnVision 2101 multilabel reader (Perkin Elmer) using 96-well optiplates (Perkin Elmer). The excitation filter was a Perkin Elmer X531 with a CWL of 531 nm, while the P and S emission filters were M579p with a CWL of 579 nm, all measurements were performed at 19°C. Each 5′ TAMRA-labeled oligonucleotide (Sigma Genosys) was dissolved in water was heat annealed with its counterpart, the resulting duplex was purified over a Superdex-75 10/300 Hi-Load gel filtration column (GE Healthcare). Dye concentration was determined through UV–Vis measurements. Assays were performed as either serial DBD-dilutions or in a competition format to calculate EC50 values for the longer oligonucleotides. The labeled DNA concentration was 1 nM, binding reactions were carried out in the buffer 20 mM HEPES pH 7.5, 140 mM NaCl, 1 g/l chicken ovalbumin, 0.05 mM TCEP. Three measurements were collected and averaged for each binding isotherm. The competition oligonucleotides used were ERS6 (gggggaatCAAACAgaaag), ERS7 (aaagagagaCAAACAgaga), ERS6/7 (ggaatCAAACAgaaagagagaCAAACA), Ran20 (ccgatccgctcgcccgggcc), 2x-1 (ccgatGTTTTAAACAtgcc), 2x + 1 (ccgaTGTTTAgTAAACAgcc), 2x (ggcTGTTTATAAACAatcgg) and 1x (ccgatTGTTTAgcccgtgcc).
Luciferase reporter assays
A mammalian expression plasmid for the full-length wild-type FoxM1c protein had been previously made (21). The mutation L291Y within this plasmid was generated by site-directed mutagenesis. Plasmids encoding FoxM1 ΔC (aa 1–600) and FoxM1 DNA binding domain (DBD) (aa 210–378) were subcloned as N-terminal flag-tagged or myc-tagged versions in pCDNA-3 (Invitrogen) by using standard techniques. U2OS cells (osteosarcoma human cells) were maintained in DMEM medium with 6% fetal calf serum and antibiotics. Cells were transfected with plasmid DNA using the standard calcium phosphate transfection protocol. After transfection, cells were synchronized in G1/S with thymidine (2.5 mM, 24 h) and in G2/M by releasing the cells from the thymidine block in fresh medium for 14 h. For competition experiments, non-synchronized cells were collected 48 h after transfection. Luciferase activity was determined using the dual luciferase kit (Promega) according to the manufacturer’s instructions. Relative luciferase was expressed as the ratio of firefly luciferase activity to control Renilla luciferase activity. For western blot analysis, protein concentration in the luciferase lysates was determined by Bradford assay (Bio-Rad). Equivalent amounts were subjected to SDS-PAGE and western blotting was carried out with the indicated antibodies. FoxM1 (C-20) and Actin (I-19) were from Santa Cruz. Anti-flag (M2) and anti-myc (9E10) were from Sigma and Covance, respectively.
Bioinformatics
The 3000 bp upstream of transcripts from human or mammalian promoter sequences (Ensembl version 51) were searched for: the Forkhead consensus motif TAACA or ATTTGT; the palindrome TGTTTA.{0,5}TAAACA; and the palindrome TAAACA.{0,5}TGTTTA. Motif conservation is computed as the fraction of promoters of vertebrate orthologs that contain a hit to this motif in a similar position to that of the human motif hit, applying a 1% window to allow for small shifts.
RESULTS
The FoxM1-DBD recognizes its consensus and natural promoter sequences with micromolar affinity
To characterize the DNA-binding behavior of FoxM1, we used fluorescence polarization anisotropy experiments to measure the affinity of the DBD for its consensus sequence. The FoxM1-DBD (residues 222–360) was expressed in bacteria and purified to homogeneity. Affinity measurements were performed using a TAMRA-labeled 11 bp DNA containing a consensus ccaTAAACAac sequence. In the conditions used, wild-type FoxM1-DBD binds to this sequence with an apparent dissociation constant (KD) of 7000 ± 300 nM (Figure 1A). The FoxM1-DBD thus has a micromolar affinity for the classical Forkhead consensus site. This is at least an order of magnitude lower than the affinity reported for the FoxO3a-DBD [KD of 300 nM for a 26 bp oligonucleotide (31)] and four orders of magnitude lower than that reported for the FoxD3-DBD [KD of 0.3 nM for a 25 bp oligonucleotide (32)].
Figure 1.

DNA-binding properties of FoxM1-DBD as measured by fluorescence anisotropy. (A) Anisotropy measurements of 1 nM of TAMRA-labeled consensus sequence binding to different concentrations of FoxM1-DBD (residues 222–360). Error bars represent standard deviations of three different experimental measurements. (B) Competition assays in which increasing concentrations of non-labeled oligonucleotides derived from estrogen receptor alpha promoter elements are added allowing the determination of relative affinities expressed as EC50 values. (C) Anisotropy measurements using the TAMRA labeled consensus sequence as in (A), a sequence without the A(i + 4) consensus base and a non-consensus (random) sequence. (D) Competition assays in which increasing concentrations of various non-labeled palindromic oligonucleotides are added allowing the determination of relative affinities expressed as EC50 values.
The previously described consensus sequences are merely the best derived from past in vitro experiments. The physiological promoters recognized by FoxM1 will be related, but not necessarily identical. To examine if natural promoter sequences transactivated by FoxM1 have different properties, we used 11 bp and 19 bp sections of the S6 and S7 response elements of the estrogen receptor (ERα) promoter(6), as well as a 21 bp sequence containing the combined S6/S7 element. For this experiment, we had to use competition fluorescence anisotropy assays to compensate for the reduced tumbling times of the longer DNA. The competition assay was set up containing 20 µM of the FoxM1-DBD, 1 nM of labeled 11 bp DNA and a dilution series of different oligonucleotides to obtain EC50 values (Figure 1B). We find that the EC50 values between the control 11 bp consensus site (61 000 nM), the 11 bp S6 element (80 000 nM), and the 11 bp S7 element (71 000 nM) to be the same within their 99% confidence level. The EC50 values for the longer, 19 bp sequences of the S6 element (29 000 nM), the S7 element (12 200 nM) as well as the S6/S7 element (16 800 nM) are significantly lower than these for their 11 bp counterparts, taking into account the 99% confidence levels. From these experiments, we conclude that FoxM1-DBD has similar affinity to the core recognitions sites of the natural promoters and the consensus site. The affinity for longer promoter sequences, appears tighter, but at most by a factor of two, which would be still much less than what has been reported for other Fox family members for similar size recognition sequences.
Structure determination of the FoxM1-DBD in complex with its consensus sequence
To understand why the FoxM1-DBD has a lower affinity for its consensus sequence compared to other Forkhead proteins we sought further details about the mechanism through which it recognizes DNA. We were able to obtain a 2.2 Å X-ray crystal structure of the DBD (residues 222–360) in complex with a DNA duplex containing two consensus sites (see Table 1 for data collection and refinement statistics). This 19-bp duplex contained two FoxM1 recognition sites in a palindromic orientation (atTGTTTA-TAAACAgcccg) and cohesive ends to aid self-association during crystallization. Phases were obtained by molecular replacement of FoxK1 [pdb code 2C6Y (28)] yielding electron density that allowed a model to be built and refined (Figure 2A and B). Due to the palindromic sites two FoxM1 protein molecules are bound per DNA duplex, with the asymmetric unit containing one of these protein:DNA complexes. Our model consists of: the entire double-stranded DNA molecule; 2 magnesium ions; 163 water molecules; FoxM1 residues 232–321 in the A molecule and residues 235–327 in the B molecule (Figure 2A and B). The two protein molecules are nearly identical in structure [RMSD of 0.73 Å over 88 Cα atoms as calculated by RAPIDO (33)]. Neither molecule displays electron density for the first 10 N-terminal residues or the 25–30 C-terminal ones, indicating a degree of disorder in these regions.
Figure 2.
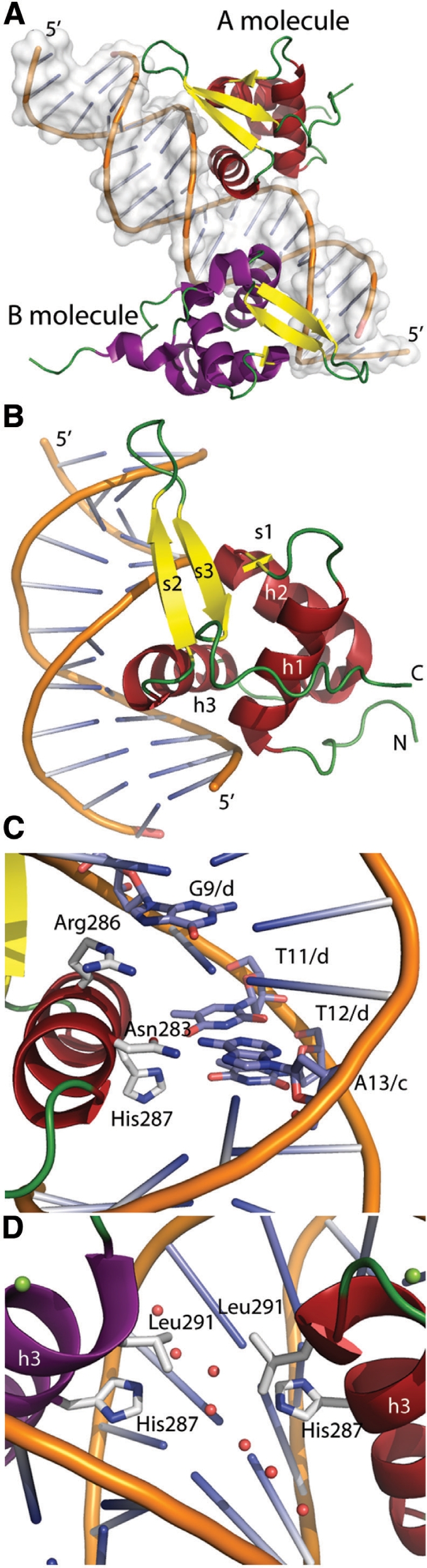
Crystal structure of the FoxM1 DNA binding domain. (A) cartoon representation of the macromolecules present in the asymmetric unit. The FoxM1 A molecule is shown with helices in red and β-strands in yellow and loop regions in green. The B molecule has the same coloring but with helices in purple. (B) A magnified representation of the FoxM1 A subunit bound to the major groove of the DNA. Secondary structural elements and the Nand C-termini are labeled. (C) The residues and bases involved in sequence-specific DNA-protein interactions are shown in stick-representation. Each residue and base is labeled, the chain identifiers of the DNA bases are also shown. (D) Close-up view of the protein-protein interface at the site of the palindromic psuedo 2-fold axis. Water molecules are shown in pink and magnesium ions in green.
The winged-helix fold
The FoxM1 DBD adopts a Forkhead winged-helix fold. This is constructed from three α-helices and three β-strands with the topology αβααββ (Figure 2B). The α-helices make up the center of the fold and pack tightly into a squat-cylindrical volume that sits perpendicularly alongside the DNA; an arrangement that allows helix h3 to insert into the major groove where it can make sequence-specific DNA contacts. The mixed three-stranded β-sheet lies at one end of this helical bundle with strands s2 and s3 running parallel to the DNA-helix, contacting the phosphate-backbone at several points. The first ‘wing’ of a winged-helix-fold describes the loop between these s2 and s3 strands while the second ‘wing’ refers to the residues immediately C-terminal to s3. An additional small 310-helix lies within the loop connecting h2 to h3, a structure also present in FoxD3 (34), FoxC2 (35), FoxO4 (36) and FoxQ1 (37).
Protein–DNA recognition
FoxM1 makes both sequence-specific and sequence-independent protein–DNA contacts. Sequence-independent contacts are in the majority and are primarily made between the DNA backbone (Figure 3A) and three regions of the protein: the N-terminus of h1; residues within strands s2 and s3; and the N-terminus of h3. This last set of interactions is mediated through the hydration sphere of a coordinated magnesium ion also present in some other Fox-DBD structures (28,38). To describe the sequence-specific FoxM1 interactions we label the consensus site as iTAAACAi+5 where i is the first base of the two palindromic sites (base 11 in chain C and base 14 in chain D of the DNA, Figures 2C and 3).
Figure 3.

Interactions made between FoxM1 and its cognate DNA. A two-dimensional representation is shown that highlights the interactions made between FoxM1 and the DNA. The phosphate-sugar backbone for the oligonucleotide C-chain is on the left and Dchain on the right. Next to each backbone sugar is the single letter code of the respective base. Chemical moieties that are seen to be the correct distance in the crystal structure to form hydrogen bonds (solid line) or van-der-Waals interactions (brackets) with residues in the A protein subunit are indicated. Similar interactions are observed for the palandromicly-equivalent residues of the DNA and the B protein subunit.
Direct contact with the DNA bases is mainly made through protein residues from within h3 (Figures 2C and 3). As in other Fox proteins, the major contributors to specificity are three invariantly conserved residues: Asn-283, Arg-286 and His-287. Specificity for the A(i + 2) base derives from two hydrogen bonds with the side chain of Asn-283 (Figure 3), while the A(i + 1) specificity arises from a hydrogen bond between its complementary base and His-287 (Figure 2). The reported specificity for the other bases in the FoxM1 consensus sequence are harder to determine from the crystal structure alone; indirect water mediated interactions with the complimentary bases possibly yield a preference for the A(i + 3) position (via Asn-286) and the C(i + 4) position (via Arg-286). Additionally a van der Waals contact between Ser-290 and the T(i) base could promote selectivity at this position. We observe no contact between the A(i + 5) base and the protein.
Consensus sequence specificity
In light of our findings, we proceeded to examine the affinity of the FoxM-DBD for its consensus site as well as its ability to discriminate against other DNA sequences. We used the same fluorescence anisotropy experiments as above, with different oligonucleotides. The experiment shown in Figure 1C gave KD values for the FoxM1-DBD of: 7500 ± 800 nM for the standard consensus sequence ccaTAAACAac similar to previous measurements (Figure 1A); 20 500 ± 1500 nM when the specifically recognized A(i + 2) base is mutated to a G (ccaTAgACAac); and 26 000 ± 1700 nM for the random non-consensus sequence gcaggtcgacg. Thus, the FoxM1-DBD has only a 3–4-fold higher affinity for its consensus site over random DNA. Moreover, altering the A(i + 2) base, which based on the structure is the major sequence-specific readout within the consensus site, is sufficient to considerably abrogate this weak discrimination. One reason the FoxM1-DBD may bind so weakly is its lack of ‘wing’-interactions. It is noteworthy that truncation of the wing-2 residues 240–253 in FoxO3a resulted in a 5-fold drop in affinity [Kd of 1500 nM (27)], yielding binding with a similar order of magnitude to what we observe for FoxM1.
FoxM1 had previously been shown to have an increased affinity for tandem consensus recognition sites, even being named Trident to showcase an observed preferential binding of triple-consensus sequences (1). Given that in our structure the two molecules binding the tandem consensus sites are in close proximity, we wished to examine if there was an obvious means by which the FoxM1-DBD could preferentially bind such tandem sequences in solution.
Tandem sequence affinity
To determine whether tandem sites could enhance the affinity of the FoxM1-DBD for its consensus sequence, we measured the affinities of longer DNA sequences, using the competition fluorescence anisotropy described above. We have used oligonucleotides containing one consensus site (x); two consensus sites (2x); two consensus sites separated by 1 base (2x + 1); two consensus sites one base closer (2x-1) or non-consensus DNA (Ran). As shown in Figure 1D, the competition curves for each of these experiments show that they bind with similar affinities. The respective EC50 values are 7000 nM, 9000 nM, 6000 nM, 9000 nM for the tandem variants, and finally 18 000 nM for the non-consensus site. From these data, we can conclude that the FoxM1-DBD does not display a noticeably higher affinity for DNA with two-tandem Forkhead consensus binding sites.
Next, we wished to examine if the contacts between the two protein molecules bound to the palindromic DNA in our structure are likely to represent a true interaction interface that could facilitate increased affinity for tandem recognition sites.
Protein ‘dimerization’ interface analysis
We looked for evidence of interaction between the FoxM1 A and B molecules that were bound to the tandem palindromic repeats of the DNA in our structure. The palindromic inter-molecule interface shows minimal contacts, the only instance of a direct interaction being between the two Leu-291 side chains. Notably, this residue is conserved in almost all Fox-DBDs. A network of ordered water molecules also bridges the two His-287 side chains (Figure 2D). PISA (39) assigns this interface a buried surface area of 41.2 Å2 and a relatively minor solvation free energy gain upon formation (−0.9 kcal/mol). In addition, there is no obvious protein–protein interface between symmetry-related subunits, consistent with the model of the isolated DBD being monomeric as reported (40).
We established no obvious mechanism from our structure that would favor tandem repeat recognition in the FoxM1-DBD, but such an arrangement could still be physiologically relevant. Thus, we next wished to determine if the full-length protein could still utilize such an oligomerization mechanism like that seen in the arrangement of molecules in the crystal structure as mediated through a Leu-291 interaction.
Mutation of the ‘dimerization’ interface does not affect transcription assays for the full-length FoxM1 protein
To determine if a change in transcriptional activity is observed when the DNA-induced FoxM1 dimer is prevented from forming, we expressed the full-length FoxM1c protein and an L291Y mutant. To prevent tandem repeat recognition we used this mutation as our structure suggests a large residue at that position would result in steric hindrances that inhibit dimer formation (Figure 2D). A luciferase reporter assay was used to monitor the transcriptional activity of the wild-type protein and the L291Y mutant in U2OS cells (Figure 5A). Protein was expressed in transfected cells synchronized in either G1/S-phase or G2/M-phase, and transcriptional activity was then measured from a responsive reporter bearing six tandem canonical Forkhead-binding elements [6xDBE (41)]. As expected, cells transfected with wild-type FoxM1c showed increased transcription of the luciferase gene compared to controls. Moreover, transcriptional activity increased when cells entered mitosis, which is when further FoxM1 activation is expected following its phosphorylation by upstream kinases. The FoxM1c L291Y mutant also expressed abundantly and resulted in considerable luciferase activity. When transcriptional activity was normalized by protein expression level, the L291Y mutant and wild-type FoxM1c are seen to be equally able to promote transcription from tandem Forkhead consensus sites.
Figure 5.

Transcription activation assays and genomic search for Fox consensus sequences. (A) Luciferase reporter assays showing the FoxM1-mediated transcription rates from a Forkhead consensus promoter for wild type full-length protein or the L291Y mutant. As a control the western blot showing the cell-lysates expression levels of each protein is at the bottom of the figure. (B)The left panel shows a histogram of the number of forkhead consensus sites in either orientation found within the 3000 bp upstream of each gene in human genome colored in grey. Overlaid upon this are similar histograms that display the degree of conservation of such sequences within vertebrate genomes. The right panel depicts the results of an identical search made for tandem or palindromic forkhead consensus sequences and their degree of conservation. (C) Luciferase reporter assays where the FoxM1-mediated transcription rates from a forkhead consensus promoter for wild type full-length protein are competed with FoxM1 DC (1–600) or FoxM1-DBD (210–378). As a control the western blot showing the cell-lysates expression levels of each protein and a loading control is at the bottom of the figure. (D) Luciferase reporter assays where the FoxM1-mediated transcription rates from a forkhead consensus promoter for wild type full-length protein are competed with FoxM1-DBD or FoxO3a-DBD. Both the actual luciferase readout (blue bars) and the same reading normalized for relative protein expression (cyan bars) are shown for clarity. As a control the western blot showing the cell-lysates expression levels of each protein and a loading control is at the bottom of the figure.
Tandem Forkhead consensus sequences are rare and poorly conserved in the genome
Although we were unable to detect an obvious preference of FoxM1 for tandem consensus sites, our inability to detect enhanced affinity may be due to missing experimental factors that are present in vivo and promote enhanced recognition in the full-length protein. Therefore, we wished to examine whether tandem Forkhead consensus sequences were conserved in the promoter regions of vertebrate genomes. To ascertain whether this was the case we performed a computational search of the 3000 bp upstream of each transcript in the human genome to determine whether such sites exist and whether they are conserved. We first searched for the single-site minimal consensus sequence TAAACA or its complement within promoter regions (Figure 5B). As multiple Forkhead proteins are expected to bind to this or similar sites, it is not surprising that in many genes one or more conserved copies of the sequence can be found. In contrast, if the palindromic sequence TGTTTA. TAAACA (Figure 5B) or the same sequence with up to 5 bp of separation between the repeats (TGTTTA.{0,5}.TAAACA) are searched for, a statistically insignificant number of hits are observed (only four promoters have a palindromic sequence with a conservation cutoff of 0.1). Thus, tandem arrays of Forkhead consensus sequences do not appear to be highly conserved within vertebrate genomes. It must be noted that such a search is complicated by both the small size of the consensus site and the apparent promiscuity of some isolated Forkhead domains as evidenced by the relatively low discrimination of FoxM1.
The FoxM1 ‘wing’ regions adopt unique conformations that are not involved in DNA binding
The FoxM1 DBD has fewer features defining specificity than other Forkhead proteins, and the two ‘wings’ of the winged-helix fold make minimal contacts with the DNA. The first wing of the fold, consisting of the loop between s2 and s3, is one of the less-conserved regions of the DBD and varies in both length and sequence in different Fox proteins. In FoxD3 wing1 is 11-residue long (Figure 4) and NMR-measurements (42) showed that it associates dynamically with the minor groove of the DNA influencing binding. Moreover, in the crystal structure of the DNA-bound complex between FoxP2 and NFAT (43) (Figure 4) wing1 is seen to be a primary site of interaction between the two transcription factors. These facets of this loop imply it may be an important region within Forkhead proteins that acts as a transcriptional cofactor recognition site and due to its variability may lend a degree of ‘specificity’ to the Forkhead regulatory networks by coupling the relatively conserved Forkhead consensus site with those from different transcription factors.
Figure 4.

DNA bound Forkhead domains. Cartoon representation of the Fox proteins for which structures in complex with DNA are known. The first wing loop is highlighted in pink and the second in black. In Fox K2 and FoxP2 the fourth helix preceding wing2 is also highlighted. In the lower right a superposition between FoxM1 (coloured as in Figure 2) and FoxO3 is shown detailing the different conformations of the proteins Ctermini. The FoxM1 C-terminus is colored teal.
The first wing of FoxM1, which is only 6-residue long, is short and reminiscent of the FoxP2 structure in which the loop is so abridged it consists of only a tight β-turn (38) (Figure 4). In the asymmetric unit, the two FoxM1 molecules have their wing1 loops in slightly different conformations; the A-molecule wing1 crosses the unligated phosphate backbone of the DNA cohesive end and is therefore less likely to represent a physiologically relevant state. However, in both subunits, the loop diverges away from the DNA and does not contact the minor groove, as for example in FoxD3 (44) or FoxK1a (28). Consequently, like in the FoxP2 structure, this ‘wing’ in FoxM1 is unable to directly affect protein–DNA interactions.
The second wing of the winged-helix fold describes a loosely structured loop at the extreme C-terminus of the DBD (Figure 3B). In other Fox proteins, 10–20 amino acids after s3 are stretches of charged positive rich sequences that can interact with the DNA. These positive residues follow a solvent exposed 4th helix within the wing, a helix that may dynamically form upon DNA binding. The charged wing2 residues may interact with the DNA through diverse means, contacting the phosphate backbone in FoxO1 (45) and FoxO4, contacting bases via the minor groove of the DNA in FoxK1 (28) and FoxD3 (42); or reconnecting with the major groove in the case of FoxO3a (31). The C-terminus of our FoxM1 structure differs significantly from these DNA-bound Forkhead structures. We see no 4th helix C-terminal to s3, although a helical turn does encompass residues 311–314 (see C-terminus in Figures 2B and 4). The residues C-terminal to the FoxM1-DBD instead adopt a unique structure that consists of a sharp turn leading into an extended loop packed perpendicularly across h1. This loop diverges away from the DNA so is not merely a different conformation of the second wing loop.
We next wanted to examine whether the lack of wing regions in the FoxM1-DBD is a reason for the low DNA-binding affinity compared to other Forkhead family members.
Low DNA biding affinity is likely due to the lack of ‘wing’ regions in FoxM1-DBD
To test this, we performed further transcriptional assays. Here, we out-compete the full-length protein’s ability to transactivate its consensus promoter, by increasing concentrations of various other protein constructs which lack transactivation domains. We expect that the construct with higher affinity for DNA target sequences, will out-compete the wild-type protein more effectively from these sites, and will result in lower transcriptional activity, as measured by the luciferase reporter.
For this experiment we used our FoxM1-DBD and the FoxO3-DBD, which has typical wings (Figure 3B) and 10 times higher DNA-binding affinity in vitro. Under identical plasmid concentrations, the expressed FoxO3-DBD out-competed full-length FoxM1 for promoter binding at a level approximately equal to that of FoxM1-DBD (Figure 5D, blue bars). However, FoxM1-DBD is much higher expressed than FoxO3-DBD (see lower panel in Figure 5D), indicating that, in fact, FoxO3-DBD inhibits FoxM1 transactivation more efficiently than FoxM1-DBD, as expected from its binding affinity in vitro. To emphasize this effect, we normalized the transcriptional activity to protein expression levels (Figure 5D, light blue bars). This result indicates that the ‘winged’ FoxO3-DBD has greater ability to act as a dominant negative, when compared to the ‘wingless’ FoxM1-DBD. This is consistent with the hypothesis that the wing regions missing in FoxM1-DBD result in the lower consensus site affinity.
Next, we wanted to examine if the missing wings of FoxM1-DBD are likely ‘substituted’ by other regions in the FoxM1 full-length protein, that display additional interactions contributing to binding specificity.
Regions outside the FoxM1-DBD are likely involved in target DNA interaction
To examine this hypothesis, we wanted to see if a FoxM1 construct with additional elements, but still lacking the C-terminal transactivation domain, would be a better competitor of full-length FoxM1, compared to FoxM1-DBD alone. We used the competition transcriptional assay described above, with FoxM1-DBD and a construct lacking only the C-terminal transactivating domain, ΔC(1–600), extending both upstream and downstream of the FoxM1-DBD. The ΔC(1–600) construct was able to reduce luciferase production much more effectively than FoxM1-DBD (Figure 5C). This suggests that ΔC(1–600) binds promoter sites with higher affinity than FoxM1-DBD, and supports the concept that elements additional to the FoxM1-DBD, either upstream or downstream of it, are important for DNA recognition, taking the role of the ‘wing’ regions present in other Forkhead family members.
DISCUSSION
We found that the FoxM1-DBD has an unusually low affinity for single copies of its Forkhead consensus sequence. The low affinities observed for the isolated domain are anomalous and it seems highly unlikely that the full-length FoxM1 transcription factor would be able to exert any coordinated response with such poor discrimination.
To facilitate our understanding of FoxM1 promoter recruitment we determined the structure of the FoxM1-DBD in complex with DNA. This new structure was both informative and puzzling. Surprisingly we observed that the FoxM1-DBD lacks contacts between the DNA and the loop regions described as the ‘wings’ of the winged-helix fold. Moreover, while the first wing loop does not contact the DNA the second wing loop is entirely absent. The C-terminal residues of the DBD instead form an extended structure distal from the DNA unique to FoxM1. The loss of wing–DNA contacts presumably in part explains the low affinity of FoxM1 for single copies of its consensus sequence. Comparing the ability of the (‘wingless’) FoxM1-DBD to the ‘winged’ FoxO3-DBDs to limit transcription in a reporter assay supports this hypothesis. Moreover, we show indirectly that a FoxM1 construct containing elements both upstream and downstream of the FoxM1-DBD, has a higher apparent affinity for consensus sites in our transcription reporter assay.
These observations argue that additional regions in FoxM1, outside the DBD, increase the avidity of the protein for its promoter regions. These sequences are likely to directly interact with DNA promoter sequences, increasing the affinity and target site recognition specificity of the full-length FoxM1 protein. Alternatively, these additional regions maybe required to mediate post-translational modifications or other interactions of the FoxM1 protein that result in higher apparent DNA binding affinity. Further analysis will become possible when structural information about the full-length protein is available.
Despite crystallizing a construct that extended to residue 360, any additional C–terminal residues that could make up a DNA-interacting wing appear to remain distant from the DNA and disordered. This could imply two possibilities: that FoxM1 does not have a 2nd wing-like loop, perhaps transferring this function to another part of the full-length protein; or that this wing does not bind to the minor groove but instead the major groove of the DNA as in FoxO3a (31), in which case our palindromic sequence may perturb its binding.
A major motivation to study the FoxM1 transcription factor, apart from its biological interest and its profound implication in cell proliferation and cancer, was its reported preference for multiple Forkhead binding sites occurring in tandem in either a back-to-forward (e.g. TGTTTA-TAAACA) or forward-to-back (TAAACA-TGTTTA) orientation (1). We wished to examine the structural means by which a preference for tandem sequences could uniquely arise within the FoxM1 family member. This question is especially intriguing as the Forkhead DBDs are well conserved and any behavior distinguishing FoxM1 from the other Forkhead transcription factors may be related to its role in regulating cellular proliferation.
To that end we used a forward-to-back tandem repeat DNA for crystallization. This resulted in an arrangement of DBDs with few inter-protein contacts and none that could be described as being specific to FoxM1. Indeed, we note that the FoxP2 structure of Stroud et al. (38) displays an arrangement of DBDs similar to FoxM1 in its asymmetric unit’s F- and J-molecules; This is despite the fact that the oligonucleotide within this structure lacks a palindrome-like sequence (i.e. the F-molecule is not bound to a Forkhead consensus site) as well as the fact that one of the molecules exists in a domain-swapped form. That both FoxP2 and FoxM1 can form these weakly interacting dimers when bound to DNA is intriguing, and due to the conserved nature of the residues involved could reflect a means of interaction by which multiple Forkhead DBDs could associate. However, two points should be held in mind: the first is that such an arrangement may also be promoted artificially through the requirements of crystallization; the second is that all current Fox DBD structures are of truncated constructs and not full-length proteins. DNA-induced dimers could be either enhanced or entirely prevented by the missing parts of the molecule. For FoxM1c this possibility was tested indirectly by comparing the transcription rate of a reporter gene in cells expressing wild-type protein or a mutant unable to form the arrangement of molecules seen in our crystal structure. No fundamental changes in transcriptional activity were observed, thus indicating that this dimerization mechanism is either not a part of all FoxM1-promoter recognition events, or plays a relatively minor role in regulating transcription. We can draw one firm conclusion from the structural data, which is that we do not detail any means by which the isolated FoxM1-DBD could have a unique affinity for tandem consensus sequences.
In the absence of further experiments, we conclude it is unlikely that FoxM1 mediated transcription relies on the direct recognition of tandem consensus sites. This conclusion is strengthened since we were unable to observe conserved tandem Forkhead sequences in genomic promoter regions. The initial gel-shift assays that established the preference for tandem sequence recognition were possibly misled by the low affinity of the isolated FoxM1-DBD for its consensus sequence thus leading to preferential identification of multi-site oligonucleotides. This scenario raises doubt about the physiological significance of such poorly conserved multi-site sequences.
FoxM1 contains a central domain located C-terminal to its DBD that plays a contextual role, inhibiting FoxM1c mediated transcription from consensus Forkhead promoters and activating promoters with c-myc like TATA boxes (2). It could be hypothesized that this central domain influences recognition of tandem consensus sequences by providing additional interactions to those observed across the palindromic interface in the crystal structure. This hypothesis is also supported by our indirect observation that the construct ΔC(1–600) binds tighter to FoxM1 recognition sequences. Unfortunately, the DBD construct used in our biophysical assay lacked this domain and it proved difficult to express a well-behaved version due to degradation. In the present structure, the residues immediately prior to the central domain are ordered and adopt a unique strand-like conformation that extends away from the DNA. It is difficult to extend any conclusions from this substructure about where the central domain would be positioned with respect to the remainder of the molecule.
CONCLUSIONS
We have obtained the structure of the FoxM1-DBD and measured its affinity for a number of DNA sequences. Unlike other DNA bound winged helix structures FoxM1 displays a relatively short first wing loop that does not interact with the DNA. In addition, our structure shows no evidence for the presence of the second wing loop found within other Fox proteins, indicating that this DNA-binding element is missing or migrated to a distal segment of the full-length protein. As compared to other family members, these subtle changes appear to result in a lower affinity and selectivity of FoxM1 for its consensus recognition sequence. We also show that sequences outside the DBD are likely directly or indirectly involved to infer higher affinity and selectivity toward promoter regions. Finally, structural, functional and bioinformatics data show no preference of FoxM1 for tandem recognition sequences. Should FoxM1 maintain its moniker, ‘Trident’, it is important that the consensus site be derived from the full-length protein and not the isolated DBD, in a physiological context.
ACCESSION NUMBER
3G73.
FUNDING
The European Commission under the Grant Agreements SPINE2-Complexes (LSH-2004-1.1.2-1 to A.P.), AntiPathoGN (223101 to P.A.); Spanish Ministerio de Innovación y Ciencia (BIO2007-62426, PSS-010000-2009 to P.A.). Funding for open access charge: Netherlands Cancer Institute.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Evangelos Christodoulou for providing the pET-28 ligation-independent vector and advice during cloning. The myc-FoxO3a DBD expression plasmid was provided by Boudewijn Burgering.
REFERENCES
- 1.Korver W, Roose J, Clevers H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997;25:1715–1719. doi: 10.1093/nar/25.9.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wierstra I, Alves J. The central domain of transcription factor FOXM1c directly interacts with itself in vivo and switches from an essential to an inhibitory domain depending on the FOXM1c binding site. Biol. Chem. 2007;388:805–818. doi: 10.1515/BC.2007.094. [DOI] [PubMed] [Google Scholar]
- 3.Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, Lau LF, Costa RH, Raychaudhuri P. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28:2908–2918. doi: 10.1038/emboj.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, Aldape KD, Xie TX, Pelloski CE, Xie K, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–3602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- 5.van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, Sommer C, Reifenberger G, Hanash SM. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am. J. Pathol. 2003;163:1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madureira PA, Varshochi R, Constantinidou D, Francis RE, Coombes RC, Yao KM, Lam EW. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J. Biol. Chem. 2006;281:25167–25176. doi: 10.1074/jbc.M603906200. [DOI] [PubMed] [Google Scholar]
- 7.Wonsey DR, Follettie MT. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005;65:5181–5189. doi: 10.1158/0008-5472.CAN-04-4059. [DOI] [PubMed] [Google Scholar]
- 8.Kim IM, Ackerson T, Ramakrishna S, Tretiakova M, Wang IC, Kalin TV, Major ML, Gusarova GA, Yoder HM, Costa RH, et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–2161. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 9.Wang IC, Meliton L, Tretiakova M, Costa RH, Kalinichenko VV, Kalin TV. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene. 2008;27:4137–4149. doi: 10.1038/onc.2008.60. [DOI] [PubMed] [Google Scholar]
- 10.Chan DW, Yu SY, Chiu PM, Yao KM, Liu VW, Cheung AN, Ngan HY. Over-expression of FOXM1 transcription factor is associated with cervical cancer progression and pathogenesis. J. Pathol. 2008;215:245–252. doi: 10.1002/path.2355. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–8300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 12.Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, Monzon FA. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalin TV, Wang IC, Meliton L, Zhang Y, Wert SE, Ren X, Snyder J, Bell SM, Graf L., Jr, Whitsett JA, et al. Forkhead Box m1 transcription factor is required for perinatal lung function. Proc. Natl Acad. Sci. USA. 2008;105:19330–19335. doi: 10.1073/pnas.0806748105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhat UG, Zipfel PA, Tyler DS, Gartel AL. Novel anticancer compounds induce apoptosis in melanoma cells. Cell Cycle. 2008;7:1851–1855. doi: 10.4161/cc.7.12.6032. [DOI] [PubMed] [Google Scholar]
- 15.Gartel AL. FoxM1 inhibitors as potential anticancer drugs. Expert Opin. Ther. Targets. 2008;12:663–665. doi: 10.1517/14728222.12.6.663. [DOI] [PubMed] [Google Scholar]
- 16.Gusarova GA, Wang IC, Major ML, Kalinichenko VV, Ackerson T, Petrovic V, Costa RH. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J. Clin. Invest. 2007;117:99–111. doi: 10.1172/JCI27527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kwok JM, Myatt SS, Marson CM, Coombes RC, Constantinidou D, Lam EW. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 2008;7:2022–2032. doi: 10.1158/1535-7163.MCT-08-0188. [DOI] [PubMed] [Google Scholar]
- 18.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer. 2007;7:847–859. doi: 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- 19.Park HJ, Wang Z, Costa RH, Tyner A, Lau LF, Raychaudhuri P. An N-terminal inhibitory domain modulates activity of FoxM1 during cell cycle. Oncogene. 2008;27:1696–1704. doi: 10.1038/sj.onc.1210814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wierstra I, Alves J. Despite its strong transactivation domain, transcription factor FOXM1c is kept almost inactive by two different inhibitory domains. Biol. Chem. 2006;387:963–976. doi: 10.1515/BC.2006.120. [DOI] [PubMed] [Google Scholar]
- 21.Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, Medema RH. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell. Biol. 2008;28:3076–3087. doi: 10.1128/MCB.01710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen YJ, Dominguez-Brauer C, Wang Z, Asara JM, Costa RH, Tyner AL, Lau LF, Raychaudhuri P. A conserved phosphorylation site within the forkhead domain of FoxM1b is required for its activation by cyclin-Cdk1. J. Biol. Chem. 2009;284:30695–30706. doi: 10.1074/jbc.M109.007997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ, Chen J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell. Biol. 2008;10:1076–1082. doi: 10.1038/ncb1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gajiwala KS, Burley SK. Winged helix proteins. Curr. Opin. Struct. Biol. 2000;10:110–116. doi: 10.1016/s0959-440x(99)00057-3. [DOI] [PubMed] [Google Scholar]
- 25.Korver W, Roose J, Wilson A, Clevers H. The winged-helix transcription factor Trident is expressed in actively dividing lymphocytes. Immunobiology. 1997;198:157–161. doi: 10.1016/S0171-2985(97)80036-8. [DOI] [PubMed] [Google Scholar]
- 26.Ye H, Kelly TF, Samadani U, Lim L, Rubio S, Overdier DG, Roebuck KA, Costa RH. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 1997;17:1626–1641. doi: 10.1128/mcb.17.3.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Tsai KL, Huang CY, Chang CH, Sun YJ, Chuang WJ, Hsiao CD. Crystal structure of the human FOXK1a-DNA complex and its implications on the diverse binding specificity of winged helix/forkhead proteins. J. Biol. Chem. 2006;281:17400–17409. doi: 10.1074/jbc.M600478200. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Tsai KL, Sun YJ, Huang CY, Yang JY, Hung MC, Hsiao CD. Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 2007;35:6984–6994. doi: 10.1093/nar/gkm703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiyanova T, Liao X. The dissociation rate of a winged helix protein-DNA complex is influenced by non-DNA contact residues. Arch. Biochem. Biophys. 1999;362:356–362. doi: 10.1006/abbi.1998.1040. [DOI] [PubMed] [Google Scholar]
- 33.Mosca R, Schneider TR. RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Res. 2008;36:W42–W46. doi: 10.1093/nar/gkn197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsden I, Jin C, Liao X. Structural changes in the region directly adjacent to the DNA-binding helix highlight a possible mechanism to explain the observed changes in the sequence-specific binding of winged helix proteins. J. Mol. Biol. 1998;278:293–299. doi: 10.1006/jmbi.1998.1703. [DOI] [PubMed] [Google Scholar]
- 35.van Dongen MJ, Cederberg A, Carlsson P, Enerback S, Wikstrom M. Solution structure and dynamics of the DNA-binding domain of the adipocyte-transcription factor FREAC-11. J. Mol. Biol. 2000;296:351–359. doi: 10.1006/jmbi.1999.3476. [DOI] [PubMed] [Google Scholar]
- 36.Weigelt J, Climent I, Dahlman-Wright K, Wikstrom M. Solution structure of the DNA binding domain of the human forkhead transcription factor AFX (FOXO4) Biochemistry. 2001;40:5861–5869. doi: 10.1021/bi001663w. [DOI] [PubMed] [Google Scholar]
- 37.Sheng W, Rance M, Liao X. Structure comparison of two conserved HNF-3/fkh proteins HFH-1 and genesis indicates the existence of folding differences in their complexes with a DNA binding sequence. Biochemistry. 2002;41:3286–3293. doi: 10.1021/bi011908k. [DOI] [PubMed] [Google Scholar]
- 38.Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, Chen L. Structure of the forkhead domain of FOXP2 bound to DNA. Structure. 2006;14:159–166. doi: 10.1016/j.str.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 39.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Wierstra I, Alves J. FOXM1c and Sp1 transactivate the P1 and P2 promoters of human c-myc synergistically. Biochem. Biophys. Res. Commun. 2007;352:61–68. doi: 10.1016/j.bbrc.2006.10.151. [DOI] [PubMed] [Google Scholar]
- 41.Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 2000;349:629–634. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin C, Marsden I, Chen X, Liao X. Dynamic DNA contacts observed in the NMR structure of winged helix protein-DNA complex. J. Mol. Biol. 1999;289:683–690. doi: 10.1006/jmbi.1999.2819. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Borde M, Heissmeyer V, Feuerer M, Lapan AD, Stroud JC, Bates DL, Guo L, Han A, Ziegler SF, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 44.Jin C, Liao X. Backbone dynamics of a winged helix protein and its DNA complex at different temperatures: changes of internal motions in genesis upon binding to DNA. J. Mol. Biol. 1999;292:641–651. doi: 10.1006/jmbi.1999.3106. [DOI] [PubMed] [Google Scholar]
- 45.Brent MM, Anand R, Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure. 2008;16:1407–1416. doi: 10.1016/j.str.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lovell SC, Davis IW, Arendall WB, III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]