

Abstract
Human β-hexosaminidase A (Hex A) (αβ) is composed of two subunits whose primary structures are ~60% identical. Deficiency of either subunit results in severe neurological disease due to the storage of GM2 ganglioside; Tay–Sachs disease, α deficiency, and Sandhoff disease, β deficiency. Whereas both subunits contain active sites only the α-site can efficiently bind negatively charged 6-sulfated hexosamine substrates and GM2 ganglioside. We have recently identified the αArg424 as playing a critical role in the binding of 6-sulfate-containing substrates, and βAsp452 as actively inhibiting their binding. To determine if these same residues affect the binding of the sialic acid moiety of GM2 ganglioside, an αArg424Gln form of Hex A was expressed and its kinetics analyzed using the GM2 activator protein:[3H]-GM2 ganglioside complex as a substrate. The mutant showed a ~3-fold increase in its Km for the complex. Next a form of Hex B (ββ) containing a double mutation, βAspLeu453 AsnArg (duplicating the α-aligning sequences), was expressed. As compared to the wild type (WT), the mutant exhibited a >30-fold increase in its ability to hydrolyze a 6-sulfated substrate and was now able to hydrolyze GM2 ganglioside when the GM2 activator protein was replaced by sodium taurocholate. Thus, this α-site is critical for binding both types of negatively charge substrates.
Keywords: Tay–Sachs disease, Sandhoff disease, GM2 Gangliosidosis, Structure–function
The β-hexosaminidases (Hex) are lysosomal hydrolases that catalyze the cleavage of terminal β-N-acetylglucosamine or β-N-acetylgalactosamine from a broad range of glycoconjugated substrates. In normal human tissues, two major Hex isozymes predominate, Hex A (αβ) and Hex B (ββ). Hex A is a heterodimer composed of an α and a βsubunit. The α subunit is encoded by the HEXA gene mapped to chromosome 15q23–q24 [1]. The β subunit is encoded by the HEXB gene which maps to chromosome 5q13 [2]. In addition to these, a very small amount of an unstable acidic isozyme, Hex S (αα), can be found in cells from patients deficient in the β protein (Sandhoff patients, see below) (reviewed in Ref. [3]).
The primary structure of the α and β subunits are approximately 60% identical and therefore must share very similar three dimensional structures with conserved functional domains. Whereas no previous data have identified an active monomeric subunit the existence of the three Hex isozymes, containing all possible dimeric combinations of the two subunits, necessitates each subunit to contain all the residues necessary to form an active site (reviewed in Refs. [4,5]). It is likely that the dimer interface occurs at or near enough to the active sites that some residues in the opposing subunit either complete or stabilize the active site of the other. Whereas all three isozymes can cleave terminal non-reducing β1–4 linked glycosidic bonds of either amino sugar (GlcNAc or GalNAc), contained on substrates such as Asn-linked oligosaccharides on proteins or on neutral glycolipids, only α subunit-containing isozymes (Hex A or Hex S) can cleave β1–4 linked GlcNAc-6-sulfate residues from keratan sulfate, and only heterodimeric Hex A can cleave the β1–4 linked GalNAc from the sialic acid-containing GM2 ganglioside. Artificial substrates have been developed that mimic these natural groupings. 4-Methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside (MUG) is cleaved by all three isozymes and therefore is used to measure “total” Hex activity. 4-Methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside-6-sulfate (MUGS) is cleaved primarily by the α-subunit containing isozymes (Hex A and S). However, the β active site can slowly hydrolyze this substrate with MUG/MUGS ratios of 150–300: 1 for Hex B, 3–4: 1 for Hex A, and 1–1.5: 1 for Hex S [6]. The only substrate completely unique to one of the Hex isozymes, Hex A, is GM2 ganglioside. The hydrolysis of GM2 ganglioside to free N-acetylgalactosamine and GM3 ganglioside also requires the assistance of a small thermostable protein known as the GM2 activator protein (Activator). This protein binds GM2 ganglioside, and presents it to the Hex A isozyme for hydrolysis. In vivo, therefore, it is the Activator:GM2 complex that is the true Hex A-specific substrate. Although the Activator interacts primarily with the α subunit and thus can bind both Hex A and Hex S, elements of the β subunit are needed to allow the β-GalNAc residue of GM2 to be properly orientated in the α – active site for hydrolysis [8], i.e. Hex S may be able to bind the complex, but it cannot hydrolyze GM2 [9]. Thus, genetic defects in any one of three gene products (α subunit, β subunit, and Activator) can result in impaired hydrolysis and intra-lysosomal accumulation of GM2. This accumulation is seen most dramatically in neuronal tissues where synthesis of this important brain lipid is greatest. Collectively these diseases are known as the GM2 gangliosidoses, which consist of Tay–Sachs disease (HEXA mutations), Sandhoff disease (HEXB mutations), and the AB-variant form of the disease (GM2A mutations) [3,4].
Human Hex A and B are members of the family 20 glycosidases. This group of proteins from many different organisms are believed to be evolutionarily and, thus, structurally related. Based on the crystal structure for one member of this family of proteins, a bacterial chitobiase, a model for human Hex was developed [11]. However, the bacterial enzyme shares sequence identity with the human enzyme only in its active site region and then only at a level of 26%. Additionally, it exists as a monomer. Previous work from our lab [12–14] and others [15,16] has demonstrated experimentally that this model of the active site is at least partially accurate. Recently using this model, we [12] and Kaplan et al. [17] identified αArg424 as the residue most likely to be involved in the direct binding of the 6-sulfate group of the artificial substrate MUGS by Hex A and S. In our previous report Hex S was purified from CHO cells transfected with an α subunit cDNA which carried various site specific mutations, one of which was αArg424Gln. This mutation raised the Km of Hex S for MUGS ninefold. Tranfection of CHO cells with a human [18–20] αcDNA produces only Hex S, because few, if any, inter-species dimers form [6,13]. In this report we produce Hex A containing the mutant [or wild-type (WT)] α-subunit by permanently transfecting a transformed glial cell line from a Tay–Sachs patient with the appropriate αcDNA [15]. After ion-exchange separation of the isozymes, the mutant and WT Hex A could be tested using [3H]-GM2 and purified human Activator. In our previous report we also attempted to analyze Hex B containing βAspLeu453 AsnArg substitutions (βAspLeu453 aligns with αAsnArg424). Although mutant Hex B levels were low, possibly due to its partial recognition by the endoplasmic reticulum’s quality control system [8], we were able to document a large increase in its hydrolytic efficiency towards MUGS. In this report we express this mutation along with additional sequences encoding a C-terminal His6-tag for easy purification [13,14] and re-analyze its MUG/MUGS hydrolysis ratio. More importantly, we assess its ability to hydrolyze GM2 in an assay where the function of the Hex A-specific Activator is replaced by a nonspecific detergent (reviewed in Ref. [10]).
1. Materials and methods
1.1. DNA construction and mutagenesis
As previously described [12], site-directed mutagenesis was used to create the Arg424Gln α-subunit mutation carried in the pREP4 expression vector. The βAspLeu453 AsnArg double mutation was created as described previously [12]. A section of the βcDNA containing the mutations was excised using BstX1 and then subcloned into a similarly digested C-terminal His6-tag encoding β-construct in the pcDNA3.1 vector [13] using standard techniques.
1.2. Hex A and Hex B from human placenta
Human Hex A and B were purified from human placenta as previously reported [21].
1.3. Cell culture and DNA transfections
Transformed Tay–Sachs neural glial cells (TSD-NG, from Fernandes et al. [15]) were grown in α-minimum essential medium (α-MEM) with 15% fetal calf serum (FCS) and antibiotics at 37° in 5% CO2. Transfections were performed according to the Superfect reference manual from Qiagen. The neural glial cells were seeded overnight in 100-mm tissue culture dishes and grown until they were about 80% confluent (overnight). For each dish of cells being transfected, 10 μg of DNA was mixed with 60 μl of Superfect in 300 μl of serum-free α-MEM. The mixture was allowed to incubate for 10 min at room temperature to form the DNA–Superfect complexes. Three milliliters of α-MEM containing serum was added prior to being added dropwise to the culture dishes. The dishes were incubated at 37° for 3 h, after which the cells were washed and then fed with α-MEM containing 15% FCS for 48 h. Following this, the cells were fed α-MEM plus 15% FCS and 1 μl/ml of hygromycin B (Calbiochem) every 2 days. After drug selection for 1 month, the mixed colonies were trypsinized and the cells were passaged into larger dishes for further growth and analyses. Hygromycin B was present in the growth media of all stable transfectants. The mutant Hex B construct in pcDNA 3.1 was transfected into CHO cells essentially as described as above except that the media used for growth contained 10% FCS and with drug selection being performed with 400 μg/ml neomycin.
1.4. Separation of Hex A by diethylaminoethyl (DEAE) ion-exchange chromatography
Cell pellets collected from the stably transfected neural glial cells containing either the WT or αArg424Gln Hex A mutant (Q mutant) construct cells were kept at −20 °C until use. Cells were lysed by multiple freeze–thaw cycles in 10 mM phosphate buffer pH 6. Following centrifugation, the lysate (10-mg total protein for WT, 20-mg total protein for Q mutant) was applied directly onto a 2-ml column of DEAE Sepharose CL-6B (Pharmacia) pre-equilibrated with 10 mM phosphate buffer pH 6. The column was washed with this phosphate buffer and then fractions eluted using a NaCl gradient (from 0 to 300 mM NaCl) in 10 mM phosphate buffer pH 6 [6]. Fractions were assayed for MUG and MUGS specific activity as described below. Appropriate fractions were pooled and concentrated using BSA-pretreated Centricon YM-10 spin concentrator.
1.5. Separation of the mutant Hex B by metal affinity chromatography
Large plates (Falcon P150) of transfected CHO cells were initially subcultured using α-MEM containing 10% FCS. When the cells were 80% confluent, the cells were washed well with PBS and then the media was changed to serum-free media (CHO SFMII, Gibco). After 4 days, the media was collected and replaced. This procedure was repeated twice more. The media was pooled, centrifuged at 3000 rpm, and the pellet discarded. The cleared media (200 ml) was concentrated to 50 ml using a centrifugal concentrator device (Millipore Ultracell 80, 10 kDa cut-off). The concentrated media was then dialyzed against buffer (20 mM imidazole, 500 mM NaCl, 50 mM NaH2PO4, 1% Tween 20, and 10% glycerol, pH 8.0). This dialyzed media was then transferred into a tube containing 400 μl of washed QIAGEN Ni–NTA resin and rotated at 4 °C overnight. The resin was packed into a column and then washed with 20 ml of the same buffer. The column was washed further with 20 ml of same buffer containing 100 mM imidazole. The mutant Hex B was eluted with 3 ml of this buffer containing 250 mM imidazole, collecting 0.5-ml fractions. MUG and MUGS specific activity was determined. Fractions containing the highest activity were combined and then concentrated using a BSA-pre-treated Centricon YM-10 spin concentrator.
1.6. Hex activity assay
Hex activity was measured using MUG and MUGS substrates as previously reported [22] using 1.6 mM MUG or MUGS substrate incubated at pH 4.2 in citrate-phosphate buffer at 37 °C for 15 min or 1 h, respectively. Fluorescence measurements were performed on an LS-50B Perkin Elmer Fluorescence Spectrometer with sipper attachment.
1.7. Preparation of GM2 ganglioside-containing liposomes
The hydrolysis of glycolipids in general has been shown to be enhanced by their presence in liposomes containing other anionic lipids [23]. Thus, liposomes containing tritiated GM2 ganglioside and other lipid components were prepared essentially as previously described [7]. Briefly, [3H]-GM2 ganglioside (4680 cpm/nmol, 10 mol%), cholesterol (20 mol%), phosphatidyl inositol (20 mol%), and phosphatidyl choline (50 mol%) were dried down under high vacuum so that upon rehydration a final lipid concentration of either 2 or 4 mM could be achieved. The lipid mixture thus obtained was rehydrated in 2 mM Tris/HCl buffer pH 7.4 and then freeze–thawed 10 times to ensure solute equilibration between bulk and trapped solutions. Unilamellar vesicles were prepared by successive passage of the rehydrated lipid suspension through polycarbonate filters of various pore size (Avestin, 400, 200, and 100 nm) mounted in a mini-extruder (Liposo-Fast, Avestin), with 19 passes as recommended by the manufacturer’s literature.
1.8. GM2 ganglioside hydrolysis kinetics
The standard incubation mixture for the hydrolysis reactions using GM2 ganglioside-containing liposomes contained the following components with a final volume of 100 μl; citrate-phosphate buffer (pH 4.1, 20 mM), bovine serum albumin (50 μg/ml), GM2 activator protein (isolated and refolded from E. coli as previously described [7,24], 2 μg), Hex A constructs (various amounts), and GM2-containing liposomes (as prepared above, 50 μl, 0–120 μM of 3H-GM2). After an 18-h incubation, the reaction was stopped by the addition of 1 ml of chloroform and 1 ml of 12 mM citrate-phosphate buffer containing 13.6 mM GlcNAc. The hydrolyzed product from GM2, [3H]-GalNAc, was separated from the unreacted substrate by passage through a positively charged ion-exchange 0.6 ml mini-column of AG3X4 (acetate form) resin (BioRad). The unbound fraction containing [3H]-GalNAc was determined by liquid scintillation counting. Less than 10% of the total input substrate counts was hydrolyzed and released during the reaction.
Experiments to establish the kinetics of the WT Hex A and mutant construct with respect to amount GM2 ganglioside added were performed by varying the amount of the substrate liposomes containing GM2 ganglioside added to the reaction mixture. Kinetics with respect to amount of GM2 added were performed essentially as described above except that liposomes were prepared at a higher concentration (4 mM) prior to addition into the final assay volume. Kinetic constants were calculated using a computerized nonlinear least squares curve-fitting program for the Macintosh, KaleidaGraph 3.0. This method provided the best estimation of the kinetic parameters [25]. Standard errors were calculated from the best-fit curve generated by the computer program.
1.9. GM2 hydrolysis by Hex B mutants
This assay does not utilize liposomes or the Activator; their functions being replaced by sodium taurocholate [18,20,26]. The incubation mixture for these assays contained the following components in a final volume of 100 μl; citrate-phosphate buffer (pH 4.1, 20 mM), bovine serum albumin (50 μg/ml), Hex enzyme (Hex B WT or the double mutant βAsp452Asn; βLeu453Arg Hex B), sodium taurocholate (final concentration 10 mM), and [3H]-GM2 ganglioside (6 nmol, 17,000 cpm/nmol). After a 4-h incubation at 37 °C, the samples were processed as above and the unbound fraction containing [3H]-GalNAc was counted using liquid scintillation counting.
2. Results
To examine whether the amino acid residue involved in sulfate binding of the MUGS substrate might also be involved in binding the sialic acid residue of GM2 ganglioside, constructs encoding the conservative substitution αArg424Gln (Q Hex) and WT α were permanently transfected into a transformed Tay–Sachs neural glial cell line. Hex A, WT, and Q Hex A were analyzed after removal of small amounts of residual Hex B and S by ion-exchange chromatography (data not shown). Kinetic examination of the partially purified Hex A forms was then performed using 50 μl of liposomes containing various amount of GM2 ganglioside (0–120 μM). This enhanced assay system was critical in that it allowed us to assay very small amounts of the partially purified Hex A forms. As well, using this system produced a typical Michaelis–Menten saturation curve, where we have previously assayed constant amounts of Hex A and Activator with various amounts of GM2 alone, i.e. in a micellar assay, up to 200 μM, without observing any indication of substrate saturation, making Km and Vmax calculations impossible (unpublished data). When each form of Hex was assayed using the liposome based assay conditions, differences between WT Hex A and the Q Hex A were observed (Fig. 1). Pure placental Hex A and WT Hex A were found to have Km values for the Activator:GM2 complex which were not significantly different (Table 1). Their Vmax values were slightly different, but this likely reflected an estimated 1% contamination by Hex B in the WT and presumably mutant Hex A preparations (as calculated from the slightly higher MUG/MUGS ratio of the WT Hex A as compared to the pure placental Hex A (Table 1)). However, due to the fact that Hex B is inactive towards the GM2/Activator complex this would have no effect on the resulting Km’s. Significantly, the Km of Q Hex A for the complex was increased by approximately ~3-fold while its Vmax was increased by ~10-fold. In addition, the MUG/MUGS hydrolysis ratio for Q Hex A, as compared to pure Hex A or WT Hex A, was increased approximately ~10 fold.
Fig. 1.
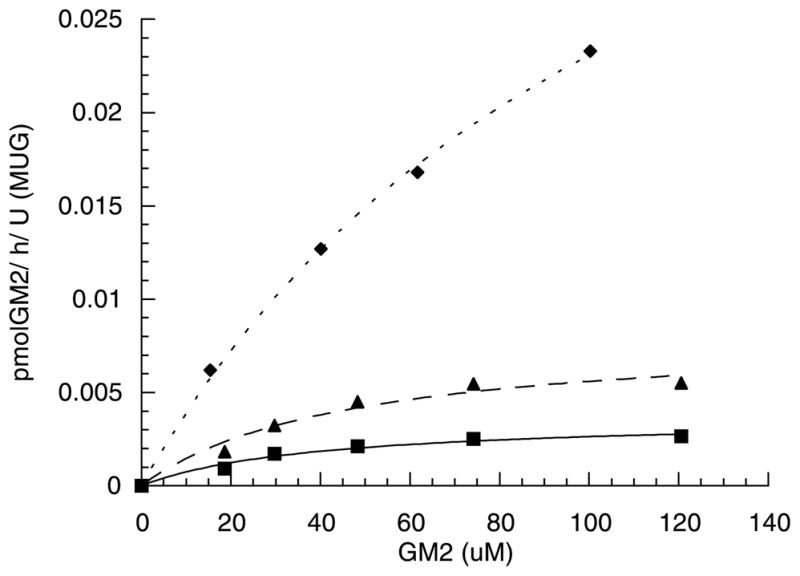
Kinetic examination of pure placental WT Hex A (closed triangles), WT Hex A derived from transfected glial cells (closed squares), and Q Hex A (αArg424Gln) also derived from transfected glial cells (closed diamonds), using varying amounts of [3H]-GM2 ganglioside contained in 50 μl of liposomes and saturating amounts of Activator protein as a substrate. Experimental data points are shown with the computer generated best-fit curve overlaid. Velocity values are given in nmol [3H]-GM2 ganglioside hydrolyzed/h/μg Hex A (estimated from its known SA towards MUG).
Table 1.
Characterization of the artificial substrate specificity and Km and Vmax for the Activator:GM2 complex of each isozyme
| Forms of Hex | MUG/MUGSa | Km(μM GM2) | Vmax [pmol GM2 h−1 U−1 (MUG)]b |
|---|---|---|---|
| Pure Hex Ac | 3.5 | 46 ± 15c | 0.008 ± 0.001 |
| WT Hex Ad | 5.4 | 39 ± 11 | 0.004 ± 0.001 |
| NQ Hex Ae | 59 | 120 ± 15 | 0.051 ± 0.004 |
| Pure Hex Bc | 190 | NDf | ND |
| WT Hex Bg | 150 | ND | ND |
| NR Hex Bh | 4.5 | ND | ND |
The ratio of units of MUG to MUGS hydrolyzed by the indicated forms of Hex at pH 4.2 with substrate concentration of 1.6 mM.
Vmax was normalized using the total units (nmol/h) of MUG activity added to the assay.
Hex A and Hex B purified from human placenta [21].
Hex A was isolated from transformed Tay–Sachs neural glial cells permanently transfected with the wild-type encoding αcDNA.
Hex A was isolated from transformed Tay– Sachs neural glial cells permanently transfected with an Arg424Gln encoding αcDNA.
Not determined.
Hex B was isolated through a His6-tag from the media of CHO cells transfected with the wild-type encoding βcDNA.
Hex B was isolated through a His6-tag from the media of CHO cells transfected with an βAsp452Asn; βLeu453Arg encoding βcDNA.
We have previously identified residues in both subunits that control the efficiency of MUGS binding. The αArg424 (aligns with βLeu453) enhances, whereas βAsp452 (aligns with αAsn423) actively inhibits binding of MUGS [12]. Thus, substitutions for these residues were made in the βcDNA to make it more α-like, i.e. βAspLeu453 AsnArg (NR Hex B). The constructs encoding WT Hex B and NR Hex B also encoded a C-terminal His6-tag which allowed purification of each isozyme from the media of the transfected cells using metal-affinity chromatography. The MUG/MUGS hydrolysis ratio for NR Hex B was decreased >30-fold (Table 1), but was still three- to fourfold higher than WT Hex S [12]. When the abilities of WT Hex A and Hex B to hydrolyze [3H]-GM2 in the presence of detergent [18,20,26], were compared to that of NR Hex B, a ~ 4-fold increase over WT Hex A levels was observed, whereas WT Hex B had no detectable activity (Table 2). Taken together these data indicate that the same residues affect both the binding of 6-sulfate-containing hexosamines and the sialic acid moiety of GM2.
Table 2.
Pure Hex A and WT and NR forms of Hex B assayed using GM2 and a nonspecific detergent, i.e. a taurocholate:[3H]-GM2 complex, as the substrate
| Form of Hex | MUG units added (nmol/h) | SA [pmol GM2 h−1 U−1 (MUG)]a |
|---|---|---|
| WT Hex Bb | 48 | 0.000 |
| NR Hex Bb | 48 | 0.098 |
| Pure Hex Ac | 96 | 0.021 |
Units (nmol/h) of MUG activity were used to calculate the specific activity.
Hex B was isolated from CHO cells transfected with either wild-type or βAsp452Asn; βLeu453Arg encoding βcDNAs.
Hex A purified from human placenta [17].
3. Discussion
Of the three Hex isozymes only heterodimeric Hex A (αβ) appears to be essential for life, as it can hydrolyze all known Hex substrates. Although Hex B (ββ) and Hex S (αα) are functional toward many of the same substrates, only Hex A can utilize the Activator:GM2 complex as a substrate. There are at least two reasons for this; first, Activator-docking requires interaction with portions of both the α and β subunit and second, only the α-active site can efficiently bind GM2. Thus, both Hex A and S can hydrolyze GM2 when the Activator’s function is replaced by a detergent. However, even in the presence of detergent, Hex B cannot efficiently hydrolyze GM2. That this α-specificity is caused by the presence of the negatively charged sialic acid residue of GM2 has been extrapolated from the observation that Hex B can hydrolyze the asialo-derivative of GM2, GA2, in the presence of detergent better than Hex A. Similarly the presence of a negative charge from a 6-sulfate group on β-linked GlcNAc substrates also appeared to convey α-specificity to the substrate (reviewed in Refs. [3,10]). Two questions remained to be answered concerning the mechanism by which the α-subunit obtains its unique substrate-specificity: What are the differences in the α-sequence as compared to β that allow it to bind these negatively charged substrates? Are these two apparently different negatively charged groups bound by the same site? The first question was partially answered by us [12] and by Kaplan et. al. [17], who identified the aligned sequence αAsnArg424 and βAspLeu453 as promoting and inhibiting the binding of the 6-sulfate group, respectively. However, neither in these previous reports nor in the present study (Table 1) has it been possible to completely convert the specificity of Hex B for MUGS into that of Hex S by substituting these two α-residues for their β-companions. These data indicate that there is some as yet unidentified α-residue(s) not conserved in the β-subunit, also involved in MUGS-binding, that likely hydrogen bonds with αArg424.
The data presented in this report do clarify the second question. Despite their apparent lack of structural similarity, the sialic acid moiety of GM2 and the 6-sulfate group of MUGS are bound by the same site in the α subunit; the major residue involved in this binding being αArg424. Conservative substitution of this residue with a neutral Gln increases the Km’s of both MUGS binding to Hex S (~10-fold) [12] and Activator:GM2 complex binding to Hex A (~3-fold). Interestingly, whereas the Vmax of Q Hex S for MUGS was slightly reduced, the Vmax of Q Hex A for the complex was increased ~ 10-fold (Table 1). The difference in the two results likely reflects the role of the Activator, which participates in the binding of the complex to Hex A. When Hex A and GM2 concentrations are held in excess and the concentration of the Activator varied, the Km of the reaction is 0.17 μM of Activator [27]. Thus, release of the product, GM3, must be slow and perhaps rate-limiting. The decreased affinity of Q Hex A for sialic acid would affect its affinity for both GM2 and GM3, which may facilitate the disassociation of the Activator:GM3 complex and increase the rate of GM2 turn over. This hypothesis could also explain why the rate of GM2 hydrolysis is 10-fold higher using taurocholate instead of the Activator.
To confirm the role of α AsnArg424 in sialic acid binding, we tested the ability of β AspLeu453 AsnArg substituted Hex B to hydrolyze GM2 in the presence of detergent. A significant increase in hydrolysis from 0.00 to 0.10 pmol GM2 h−1 U−1 (MUG) was noted (Table 2). Surprisingly, this rate was about fivefold that of purified placental Hex A. Some of this increase can be explained by the presence of two functional active sites for GM2 in the mutant Hex B, as compared to only one, the α-site, in Hex A.
Acknowledgments
We would like to thank Amy Leung for her technical assistance with the work presented in the report. This work was supported by a Canadian Institutes of Health Research (CIHR) operating grant to D.M.
Abbreviations
- Hex
β-hexosaminidase
- GM2
GM2 ganglioside, GalNAc β (1–4)-[NANAα (2–3)-]-Galβ (1–4)-Glc-ceramide
- GM2 activator protein
Activator
- MU
4-methylumbelliferone
- MUG
4-methylumbelliferyl-β-N-acetylglucosamine
- MUGS
4-methylumbelliferyl-β-N-acetylglucosamine-6-sulfate
- FCS
fetal calf serum
- MEM
minimum essential medium
- Q Hex A
Hex A containing an αArg242Gln substitution
- WT
wild type
- SA
specific activity
References
- 1.Nakai H, Byers MG, Nowak NJ, Shows TB. Cytogenet Cell Genet. 1991;56:164. doi: 10.1159/000133077. [DOI] [PubMed] [Google Scholar]
- 2.Bikker H, Meyer MF, Merk AC, deVijlder JJ, Bolhuis PA. Nucleic Acids Res. 1988;16:8198. doi: 10.1093/nar/16.16.8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahuran DJ. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 4.Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill; New York: 1995. pp. 2839–2879. [Google Scholar]
- 5.Mahuran D, Novak A, Lowden JA. Isozymes, Curr Top Biol Med Res. 1985;12:229–288. [PubMed] [Google Scholar]
- 6.Hou Y, Tse R, Mahuran DJ. Biochemistry. 1996;35:3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- 7.Smiljanic-Georgijev N, Rigat B, Xie B, Wang W, Mahuran DJ. Biochim Biophys Acta. 1997;1339:192–202. doi: 10.1016/s0167-4838(97)00002-2. [DOI] [PubMed] [Google Scholar]
- 8.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. J Biol Chem. 1998;273:21386–21392. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
- 9.Kytzia HJ, Sandhoff K. J Biol Chem. 1985;260:7568–7572. [PubMed] [Google Scholar]
- 10.Mahuran DJ. Biochim Biophys Acta. 1998;1393:1–18. doi: 10.1016/s0005-2760(98)00057-5. [DOI] [PubMed] [Google Scholar]
- 11.Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. Nat Struct Biol. 1996;3:638–648. doi: 10.1038/nsb0796-638. [DOI] [PubMed] [Google Scholar]
- 12.Sharma R, Deng H, Leung A, Mahuran D. Biochemistry. 2001;40:5440–5446. doi: 10.1021/bi0029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou Y, Vocadlo D, Withers S, Mahuran D. Biochemistry. 2000;39:6219–6227. doi: 10.1021/bi992464j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou Y, Vocadlo D, Leung A, Withers S, Mahuran D. Biochemistry. 2001;40:2201–2209. doi: 10.1021/bi002018s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandes MJG, Yew S, Leclerc D, Henrissat B, Vorgias CE, Gravel RA, Hechtman P, Kaplan F. J Biol Chem. 1997;272:814–820. doi: 10.1074/jbc.272.2.814. [DOI] [PubMed] [Google Scholar]
- 16.Mark BL, Wasney GA, Salo TJS, Khan AR, Cao ZM, Robbins PW, James MNG, Triggs-Raine BL. J Biol Chem. 1998;273:19618–19624. doi: 10.1074/jbc.273.31.19618. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan F, Boulay B, Cordeiro P, Hechtman P. Gene Funct Dis. 2001;1:38–45. [Google Scholar]
- 18.Bach G, Suzuki K. J Biol Chem. 1975;250:1328–1332. [PubMed] [Google Scholar]
- 19.Conzelmann E, Sandhoff K. Proc Natl Acad Sci. 1978;75:3979–3983. doi: 10.1073/pnas.75.8.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandhoff K, Conzelmann E, Nehrkorn H. Hoppe-Seyler Z Physiol Chem. 1977;358:779–787. doi: 10.1515/bchm2.1977.358.2.779. [DOI] [PubMed] [Google Scholar]
- 21.Mahuran DJ, Lowden JA. Can J Biochem. 1980;58:287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 22.Brown CA, Mahuran DJ. Am J Hum Genet. 1993;53:497–508. [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkening G, Linke T, Sandhoff K. J Biol Chem. 1998;273:30271–30278. doi: 10.1074/jbc.273.46.30271. [DOI] [PubMed] [Google Scholar]
- 24.Klima H, Klein A, Van Echten G, Schwarzmann G, Suzuki K, Sandhoff K. Biochem J. 1993;292:571–576. doi: 10.1042/bj2920571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tommasini R, Endrenyi L, Taylor PA, Mahuran DJ, Lowden JA. Can J Biochem Cell Biol. 1985;63:225–230. doi: 10.1139/o85-032. [DOI] [PubMed] [Google Scholar]
- 26.Conzelmann E, Sandhoff K, Nehrkorn H, Geiger B, Arnon R. Eur J Biochem. 1978;84:27–33. doi: 10.1111/j.1432-1033.1978.tb12137.x. [DOI] [PubMed] [Google Scholar]
- 27.Xie B, Wang W, Mahuran DJ. Am J Hum Genet. 1992;50:1046–1052. [PMC free article] [PubMed] [Google Scholar]