

Abstract
Tay–Sachs or Sandhoff disease results from a deficiency of either the α- or the β-subunits of β-hexosaminidase A, respectively. These evolutionarily related subunits have been grouped with the “Family 20” glycosidases. Molecular modeling of human hexosaminidase has been carried out on the basis of the three-dimensional structure of a bacterial member of Family 20, Serratia marcescens chitobiase. The primary sequence identity between the two enzymes is only 26% and restricted to their active site regions; therefore, the validity of this model must be determined experimentally. Because human hexosaminidase cannot be functionally expressed in bacteria, characterization of mutagenized hexosaminidase must be carried out using eukaryotic cell expression systems that all produce endogenous hexosaminidase activity. Even small amounts of endogenous enzyme can interfere with accurate Km or Vmax determinations. We report the expression, purification, and characterization of a C-terminal His6-tag precursor form of hexosaminidase B that is 99.99% free of endogenous enzyme from the host cells. Control experiments are reported confirming that the kinetic parameters of the His6-tag precursor are the same as the untagged precursor, which in turn are identical to the mature isoenzyme. Using highly purified wild-type and Arg211-Lys-substituted hexosaminidase B, we reexamine the role of Arg211 in the active site. As we previously reported, this very conservative substitution nevertheless reduces kcat by 500-fold. However, the removal of all endogenous activity has now allowed us to detect a 10-fold increase in Km that was not apparent in our previous study. That this increase in Km reflects a decrease in the strength of substrate binding was confirmed by the inability of the mutant isozyme to efficiently bind an immobilized substrate analogue, i.e., a hexosaminidase affinity column. Thus, Arg211 is involved in substrate binding, as predicted by the chitobiase model, as well as catalysis.
The human lysosomal β-Hex1 isozymes are dimeric enzymes composed of α-, encoded by the HEXA gene 15q23-q24 (1) and/or β-, encoded by HEXB gene 5q13 (2), subunits. Since the primary structures of the α- and β-subunits share 60% sequence identity, both are evolutionarily and therefore structurally related (3). Thus, any of the three possible dimeric combination of these subunits generates an active isozyme. The three isozymes vary both in their pI and their heat stability, with Hex B (ββ) being both the most stable and the most basic [Hex B (ββ) > Hex A (αβ) ≫ Hex S (αα)]. Only Hex A and B can be readily detectable in normal human tissue, whereas these isozymes can hydrolyze many of the same neutral substrates, both natural (e.g., β-GlcNAc terminal Asn-linked oligosaccharides) and artificial (e.g., MUG); only Hex S and Hex A can efficiently hydrolyze negatively charged natural (e.g., β-GlcNAc-6-sulfate terminal keratan sulfate) and artificial (e.g., MUGS) substrates. Interestingly, only Hex A is able to hydrolyze negatively charged GM2 ganglioside in vivo; but to do so, it requires a substrate-specific cofactor, the GM2 activator protein (Activator) encoded by the GM2A gene [5q 31.3–33.1 (4)]. This small, heat stable protein forms a water-soluble complex with the ganglioside and likely blocks the normal H-bonding interaction between terminal residues of sialic acid and GalNAc (reviewed in ref 5). Thus, both Hex A and the Activator are required in vivo to prevent the lysosomal accumulation of GM2 (mainly in neuronal tissues where gangliosides are most actively synthesized) and any of the three forms of the severe neurodegenerative disease known as GM2 gangliosidosis. Whereas defects in either the α- or β-subunits comprising Hex A result in two of these three forms, Tay–Sachs disease (the most common) or Sandhoff disease, respectively, defects in the Activator protein result in the third, rare AB-variant form (reviewed in ref 6).
Understanding how a given mutation in any one of the above three genes produces its associated clinical phenotype is a goal of many clinical biochemists. To achieve this goal, a complete understanding of the structure–function relationships of the affected protein is necessary. Two major components in establishing such relationships are the development of an accurate three-dimensional structure and the identification residues involved in protein–substrate and/or protein–protein interactions. Although human Hex has been crystallized, diffraction was only to 3.2 Å, insufficient to produce an atomic model (7). However, all the members of a given hydrolase family (8) are believed to be evolutionarily related and have similar three-dimensional structures (e.g., ref 9). Thus, molecular modeling of human Hex has been carried out on the basis of the three-dimensional structure of bacterial chitobiase, as both are part of the glycosyl hydrolase “Family 20” (10). However, the accuracy of such modeling is variable and dependent on the degree of primary sequence similarity between the protein with the known structure and the protein being modeled (11). A sequence alignment of the active site regions of human α-subunit and the monomeric bacterial enzyme shows that there is only a 26% identity of amino acid residues. Furthermore, this degree of identity is based on an alignment that is only achieved by generating many large gaps in the human sequences, although these gaps are predicted to be at loop structures (10). Outside of this region, there is little sequence similarity. Thus, while the modeling of bacterial chitobiase may be helpful for characterizing the active site of human Hex, the validity of the model and the accuracy of the alignment on which it is based must be determined experimentally.
Recently, comparative molecular modeling of Hex from Streptomyces plicatus, Sp-Hex, also a member of Family 20, has been made from the chitobiase structure (12). The overall three-dimensional structure that was generated for Sp-Hex is similar to the predicted model of human Hex (10). However, Sp-Hex shares only 25% and 30% sequence identity to human Hex and chitobiase, respectively.
Analysis of missense mutations associated with GM2 gangliosidoses in hopes of identifying active site residues in human Hex has not been as informative as initially predicted, because most of these point mutations result in the retention of the mutant protein in the endoplasmic reticulum (ER), due to the ER’s tight “quality control system” (reviewed in ref 13 and 14). The most well-studied exception to this common scenario is the αArg178His (15, 16) substitution, which is associated with the B1 variant form of Tay–Sachs disease. B1 patient samples have a unique biochemical phenotype (15, 17, 18); they express near-normal levels of Hex A activity when assayed with neutral substrate (e.g., MUG) but have little or no activity toward α-specific substrates (e.g., MUGS). These observations lead to the hypothesis that Arg178 was at or near the active site of the α-subunit (15).
We analyzed the biochemical consequences of the αArg178-His mutation by mutating the aligned codons in the β-subunit to produce a stable mutant Hex B homodimer for analysis (19, 20). Kinetic analysis of our most conservative substitution, βArg211Lys, indicated that the mutant Hex B underwent normal intracellular transport, was stable in the lysosome, and had a nearly normal Km. However, its “specific activity at Vmax”, that is, a value proportional to kcat, was only 0.2–0.3% of the normal, indicating that βArg211 and by extension αArg178 take part in catalysis (19).
The above observations differ somewhat from the role of βArg211 predicted by the chitobiase molecular modeling (10, 12). Human βArg211/αArg178 aligns with c-Arg379 in chitobiase and sp-Arg162 in Sp-Hex. From the chitobiase model, it is known that Arg379 sits at the base of a binding pocket for NAG-A, the β-1,4-linked 2-acetamido-2-deoxy-glucopyranosyl residue at the nonreducing end of chitin and plays the most critical role of any residue in substrate binding and orientation (Figure 1). No role in catalysis was predicted from the model. However, while mutational analysis of purified Arg162His Sp-Hex expressed in bacteria did reveal that its Km had increased by 40-fold, its specific activity at Vmax (kcat) was also found to be reduced 5-fold, relative to wild type. Thus, while the sp-Arg162 appears to be principally involved in substrate binding, the reduction in the kcat of mutant Sp-Hex indicates that sp-Arg162 also has a role in catalysis.
Figure 1.
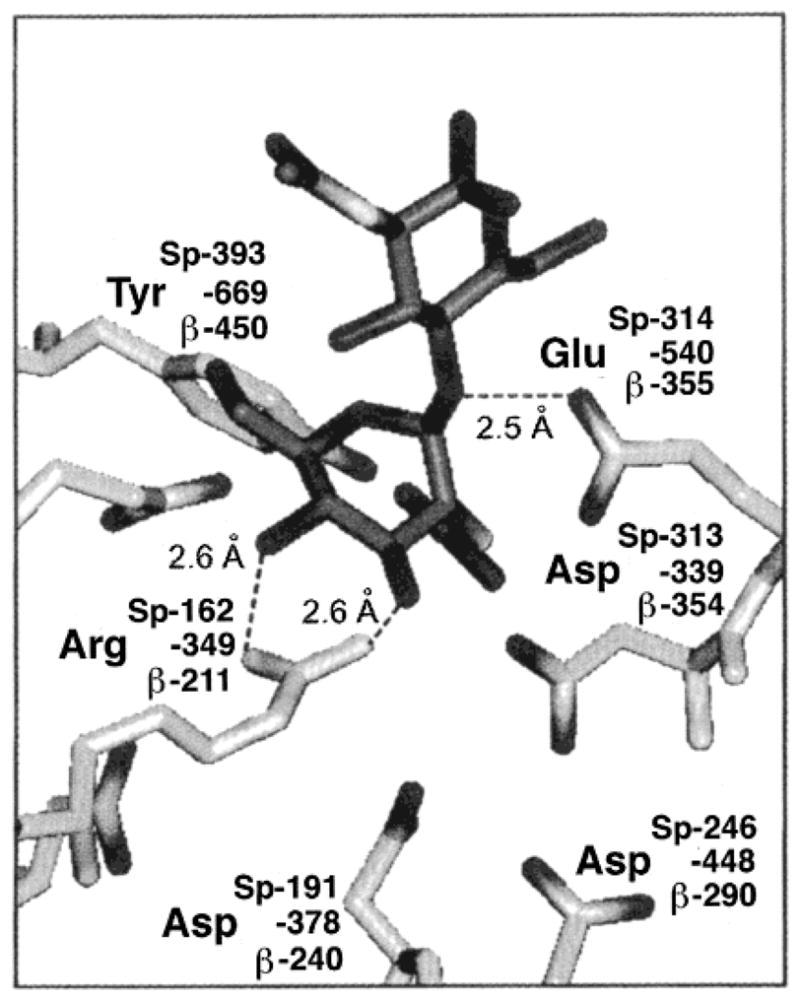
Residues of the active site of chitobiase (10) (residue numbering as -#), with the proposed aligning residues from S. plicatus Hex (residue numbering as Sp-number) and the human Hex β-subunit (residue numbering as, β-number), are shown binding to NAG-A of chitobiose (N,N′-diacetylchitobiose); darker structure shows both the NAG-A and NAG-B components. Figure was adapted from Mark et al. (12).
Because human Hex B cannot be functionally expressed in bacteria (data not shown), and all eukaryotic cells contain lysosomal Hex activity, it is difficult to determine an accurate Km for a mutant enzyme with a Vmax of only ~0.2%, that is, a signal-to-noise problem. As well, the limited solubility of MUG (~8 mM, normal Km ~0.7 mM) compounds this problem when trying to analyze a mutant isozyme with a greatly increased Km. Previously reported methods to increase the “signal (from translated human cDNA) to noise (endogenous Hex)” ratios have resulted in values of 30:1 (21), 75:1 (22), and 50–100:1 (20, 23). For purposes of determining if a naturally occurring mutation is neutral or disease-causing, this signal-to-noise level is more than sufficient (24–27). However, this ratio is too low to unequivocally identify a mutation that severely increases Km or one that neutralizes the catalytic acid or base residue present in the active sites of glycosidases. In the latter case, a decrease of several orders of magnitude would be expected (e.g., refs 28 and 29).
In the present study, we report a method for the generation of a C-terminal His6-tagged pro-Hex B from permanently transfected CHO cells. We demonstrate that this novel form of Hex B is secreted and is easily purified away from the endogenous CHO-Hex. We further demonstrate that the kinetic properties and thermostabilities of both pro-Hex B and pro-Hex B-His6 are the same as those of the mature form. As an initial attempt to test the validity of the bacterial chitobiase model using this approach, we reevaluated the effects of the conservative βArg211Lys substitution on the purified isozyme’s Km and kcat for MUG. As an additional control, we analyzed the ability of the mutant to bind immobilized CNAG, a Hex-substrate analogue (30). From these new data, we are able to definitely assign to βArg211, and by extension αArg178, a role in substrate binding as well as confirming its important role in catalysis.
MATERIALS AND METHODS
DNA Construction
Cloning procedures were as described by Sambrook et al. (31). To construct DNA encoding pro-β-Fxa-His6-COOH, a 1.8-kb fragment, containing β-chain cDNA and Factor Xa-His6 sequences, was amplified using the sense primer (5′-AGTAAGCTTGCGGCCGCAGAAGTC-GGGTCCCGAGGCT-3′) and the antisense primer (5′-CGG-GTCTAGAGCGGCCGCTTCAATGATGATGATGATGAT-GTCTACCCTCGATCATGTTCTCATGGTTACA-3′). The reactions were performed in a 100-μL mix containing 10 ng of plasmid DNA, 20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, 0.2 mM each of dNTPs, 0.4 μM each primer, and 1 unit of Vent DNA polymerase (BioLabs Inc., Beverly, MA). The cycling steps used were as follows: 1 cycle of heat denaturation at 95 °C for 5 min; 28 cycles of each consisting of denaturation at 95 °C for 30 s, annealing at 56 °C for 1 min, and extension at 73 °C for 1 min; and 1 cycle of 73 °C for 7 min in a Perkin-Elmer-Cetus thermal cycler 2400. The PCR product was digested with XbaI, purified with Geneclean kit (Bio 101 Inc., Vista, CA), and subcloned into expression vector pcDNA3.1HisA-1, which contains neomycin gene as a selective marker. pcDNA3.1HisA-1 is modified from pcDNA3.1HisA (Invitrogen Inc., Carlsbad, CA), in which the HindIII–XbaI polylinker sequence was replaced with HindIII–XbaI–NotI–XbaI linker to facilitate the subcloning procedure. The resulting fusion expression plasmid is designated as pcDNA-β-His6. For construction of pcDNA-β*-His6 encoding a Arg211Lys substitution, a 1.5-kb BstXI fragment was excised from pHexB43 (Arg Lys) (19) and used to replace the corresponding DNA fragment in pcDNA-β-His6. The mutation and the orientation of the insert were verified by DNA sequencing using T7 DNA polymerase sequencing kit (Pharmacia) before transfection of the DNA into CHO cells.
Cell Culture
CHO cells were maintained in α-MEM supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin, and 100 μg/mL penicillin, at 37 °C in 5% CO2. Transfected cells were grown in the same media containing 400 μg/mL G418. For large-scale purification of Hex, cells were plated and grown in the 850 cm2 roller bottle (Becton Dickinson Labware, Lincoln Park, NJ) at 37 °C in 5% CO2 incubator. When cells reached confluence, they were then washed twice with PBS and replaced with serum free media (GIBCO-BRL). The media were collected for subsequent protein purification (see below).
Transfection
Transfections were performed using Superfect Reagent (Qiagen Inc., Valencia, CA) by essentially following the manufacturer’s instructions. CHO cells were grown overnight until they were about 40% confluent. Ten micrograms of DNA was mixed with 40 μg of Superfect reagent (Qiagen) in 800 μL of serum-free MEM. The mixture was incubated for 10 min at room temperature to form DNA–Superfect complex. The complex was then added dropwise to the culture dishes and incubated for 2 hr at 37 °C. Next, the cells were washed and refed with α-MEM plus 10% FCS for 2 days. Following this, the cells were trypsinized and replated at a 1:8 dilution in α-MEM with 10% FCS and 400 μg/mL neomycin. Two weeks later, drug-resistant colonies were picked and grown in 24-well plates. The medium and the lysate from the surviving cells were assayed for Hex activity, and those producing the highest levels were selected for further growth and analyses. For cells transfected with pcDNA-β-His6* encoding a Arg211-Lys substitution, G418-resistant colonies were screened by Western blot analysis using human anti-Hex B antibody.
Enzyme and Heat Stability Assays
Cells were harvested and lysed in a buffer of 10 mM Tris-HCl (pH 7.5) and 5% glycerol through five sets of freeze–thaw cycles. For protein purification purposes, cells were directly lysed in a native binding buffer (10 mM sodium phosphate, pH 7.8, and 0.5 M NaCl) for Ni-NTA chromatography or 20 mM citrate–phosphate buffer (pH 4.5) containing 0.5 M NaCl for CNAG affinity chromatography (see below). Protein concentration was determined by the Lowry method (32). Human Hex activity from cell lysates and media was measured using a MUG substrate based on a MU fluorescent assay (20). Heat stability of purified human placental Hex B (mature form), Hex B isolated from the media of Tay–Sachs fibroblasts (pro-Hex B), or the His6-C-terminally tagged Hex B from the media of transfected CHO cells (pro-Hex B-His6) was determined at 60 °C, and the t1/2 was calculated from the best-fit line generated by plotting log (percent remaining activity) versus minutes of incubation (25).
Western Blot Analysis
Equal amounts of total protein from each sample of cell lysate or purified protein were resolved by SDS–PAGE using the Laemmli gel–buffer system (12.5% gel) and a Bio-Rad minigel system (33). Proteins were transferred to nitrocellulose, and the filter was blocked with 5% skim milk, as described previously (24, 34). The primary antibody was a rabbit antihuman Hex B. A horse-radish peroxidase-conjugated goat antirabbit IgG (1:10 000 dilution, Immux) was used as the secondary antibody. The nitrocellulose was developed using the Amersham ECL system and exposed to Hyperfilm.
Ni-NTA Chromatography
The Pro-Bond beads (Invitrogen Inc., Carlsbad, CA) were prepared by washing twice with sterile water and three times with the native binding buffer. One milliliter of gel was packed in a small column and equilibrated in the same buffer. Cell lysates or media from control CHO or transfected cells were supplemented with NaCl to a final concentration of 0.5 M to reduce the nonspecific binding before they were directly loaded onto the Pro-Bond column. Nonspecifically bound proteins were removed by washing the column twice with 3 mL of native binding buffer and three times with 3 mL of native wash buffer (10 mM sodium phosphate, pH 6.0, and 0.5 M NaCl). His6-containing proteins were eluted with increased concentration of imidazole (30 mM, 80 mM, and 500 mM) in the native wash buffer. Samples were concentrated to a point where 2–5 μL was needed per Hex assay using Centricon-10 (Amicon) membranes pretreated with 1% human serum albumin overnight and rinsed with large amounts of water. The 2 mL, 500 mM imidazole, human Hex-containing fraction was first concentrated to 50–100 μL and then diluted with 5 mL of citrate–phosphate buffer, pH 4.2, and reconcentrated to reduce the level of imidazole and correct the pH for Hex assays. Generally, it was found that ≤4 μL of the 500 mM imidazole-containing native wash buffer could be added to the 200-μL assay mix without affecting Hex activity measurements. The purity of the proteins was examined through SDS–PAGE followed by Coomassie Blue staining.
Circular Dichroism (CD) Spectra
CD spectra were recorded on a Jasco J-720 spectropolarimeter, from the mature placental Hex B, wild-type Hex B-His6, and mutant Hex B-His6, at concentration of 0.5 mg/mL in 10 mM phosphate (pH 6.0) buffer. Each curve was the average of four scans, recorded between 190 and 250 nm in a quartz cell (Jasco) with a path length of 1 mm.
Determination of Kinetic Parameters
The apparent Km and Vmax values were determined for the MUG substrate using concentrations ranging from 0.1 to 4 mM (34). These constants were calculated using a computerized nonlinear least-squares curve-fitting program for the Macintosh, KaleidaGraph 3.0. Thus, the individual substrate concentrations and their corresponding initial velocity measurements were directly fitted to the Michaelis–Menten equation, Vi = Vmax[S]/(Km + [S]), making possible the calculation of an accurate standard error (35). Values of kcat [mol of MU released h−1 (mol of each purified enzyme)−1] were calculated based on each enzyme’s calculated specific activity at Vmax and a Mr of 130 000 g/mol for Hex B. For the mutant Arg211Lys form of Hex B, accurate individual kcat and Km values could not be ensured because of the limited solubility of MUG; however, an accurate kcat/Km ratio could be determined from the slope of the best-fit straight line generated from a [S] (MUG from 0 to 1.25 mM) versus Vi plot using a known, constant amount of purified protein (1.6 μg).
Hex Affinity Chromatography
The affinity ligand, 2-acet-amido-N-(ε-aminocaproyl)-2-deoxy-β-D-glucopyranosylamine (CNAG), was coupled to Sephacryl S-200 according to Mahuran and Lowden (30). For purification of pro- and mature Hex B from Tay–Sachs fibroblasts, the media or lysate samples were dialyzed in 10 mM sodium phosphate buffer (pH 6.0) containing 0.2 M NaCl before being loaded on a CNAG column. After washing to remove nonspecific proteins with three gel vol of 10 mM sodium phosphate buffer and 0.5 M NaCl, the Hex protein was eluted with 10 mM Tris-HCl (pH 8.5).
The degree of binding to the affinity ligand beads was also used as a qualitative means of comparing the apparent Km’s of the wild-type and mutant forms of Hex B. The beads were first pretreated with 0.1% RNase A overnight to block any nonspecific binding sites. After washing the beads three times to remove the RNase A, 10 μg of purified Hex protein was loaded on a 0.3-mL minicolumn. The column was then washed twice with 2 mL of 10 mM sodium phosphate buffer, pH 6.0, and 0.2 M NaCl (wash buffer). Finally, the specifically bound Hex protein was eluted with the same wash buffer containing a competitive inhibitor, 150 μM δ-lactone (2-acetamido-2-deoxy-glucono-1,5-lactone, Toronto Research Chemicals Inc.). The binding affinity of each form of Hex B was assessed by the percent of Hex protein eluted with δ-lactone, as determined by the Lowry method (32).
RESULTS
Fusion constructs encoding the human prepro-β-polypeptide with either an N-terminal (placed after the signal peptide, data not shown) or C-terminal Factor-X-His6 tag (pro-β-Fxa-His6-COOH) were permanently transfected into CHO cells. No Hex activity was detected in the media of cells transfected with the N-terminal tagged β-cDNA. When lysates from these clones were analyzed by Western blotting, only bands corresponding to the precursor (ER) form were seen (data not shown). These data indicate that the N-terminal tag prevents the transport of the protein out of the ER. Media from clones transfected with the cDNA encoding the C-terminal tagged Hex B (pcDNA-β-His6) contained up to 5-fold higher levels of Hex activity than did medium from a nontransfected control (Table 1). The level of Hex activity in transfected cell lysates was similarly increased (Table 1).
Table 1.
Hex MUG Activity Levels in the Medium and Cell Lysates of Mock-Transfected and pcDNA-β-His6-transfected CHO Cells, Before and After Chromatography on a Ni2+-NTA Column
| CHO cells |
medium |
lysate |
||||
|---|---|---|---|---|---|---|
| transfected with | totala | freea,b | bounda,c | totala | freea,b | bounda,c |
| mock | 9.2 | 8.4 | 0.0008 | 1.03 | 0.9 | 0.0001 |
| pcDNA-β-His6 | 54 | 10.9 | 38.3 | 3.75 | 3.21 | 0.08 |
Hex activity [(nmol of MU) hr−1 (total mg of lysate protein)−1].
Hex activity that did not bind to the Ni2+-NTA beads or was eluted with 30 mM imidazole.
Hex activity eluted with 500 mM imidazole from the Ni2+-NTA beads.
Western blot analysis of lysate protein from pcDNA-β-His6-transfected cells, and cells transfected with pEFNEO-β [encoding the unmodified prepro-β-chain (23)] produced similar levels of immunoreactive bands with Mr corresponding to the mature, lysosomal βa-chain (26 and 28 kDa ± an N-linked oligosaccharide) (Figure 2, lanes β and β-His), indicating that the C-terminal His6-fusion enzyme is efficiently transported out of the ER and targeted to the lysosome (34).
Figure 2.
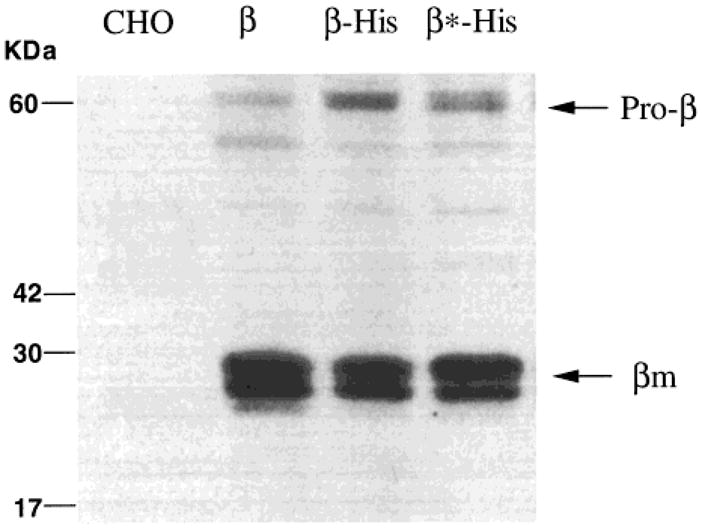
Western blot analysis using an antihuman Hex B IgG of lysates from mock-transfected CHO cells (CHO) or CHO cells transfected with pEFNEO-β, encoding wild-type prepro-β-chains (β); pcDNA-β-His6, encoding a C-terminal His-tagged form of prepro-β-chains (β-His); or pcDNA-His-β*, encoding a βArg211-Lys substitution C-terminal His-tagged form of prepro-β-chains (β*-His). Each lysate sample analyzed contains an equal amount of total protein (20 μg). The positions of bands corresponding to the pro-β- (65 kDa) and mature β-chains are indicated on the right. Protein standards are shown on the left.
We next attempted to purify the His-tagged wild-type Hex B from cell lysates by Ni2+-NTA chromatography. The level of Hex activity recovered from cell lysates by this method was very low, that is, only 2% of Hex activity initially added to the Ni2+ column (Table 1). This result indicated that the His-tag is cleaved when Hex reaches the lysosome. Since high levels of Hex activity are also secreted from the transfected cells, we attempted to purify the pro-Hex B-His6 from the expression medium; 71% of the Hex activity was recovered after imidazole elution from the Ni2+ column (Table 1). SDS–PAGE followed by Coomassie blue staining detected a single band corresponding to pro-β (65 kDa) polypeptide (Figure 3A). Western blotting using antihuman Hex B antibody further confirmed the identity of the 65-kDa polypeptide as the β-subunit of Hex (Figure 3B). In contrast, <0.01% of endogenous Hex from nontransfected CHO cell lysate or medium was bound and eluted from the Ni2+ column (Table 1).
Figure 3.

Coomassie blue staining (A) and Western blot (B) detection of purified proteins isolated from the serum-free medium of untransfected CHO cells (CHO) or CHO cells transfected with pcDNA-His-β, encoding the C-terminal His-tagged prepro-β-chain (β-His); pcDNA-His-β*, encoding a Arg211Lys substitution C-terminal His-tagged prepro-β-chain (β*-His); or cells transfected with pEFNEO-β, encoding the wild-type prepro-β-chain (β). The purification of Hex for the former three (lanes CHO, β-His, and β*-His) was carried out using Ni-NTA column under native conditions, while the untagged pro-Hex B form (lane β) was purified by CNAG affinity chromatography. The location of Mr standards and the pro-β are indicated.
Biochemical Properties of pro-Hex B and pro-Hex B-His6
Since Hex isolated from cell medium is in its precursor form (36), the biochemical properties of pro-Hex B, as well as pro-Hex B-His6 needed to be determined and compared with those of mature Hex B before the effects of any substitution mutation could be assessed. Pro- and mature forms were purified from the medium and the lysate, respectively, of Tay–Sachs fibroblasts by CNAG affinity chromatography (30). The specific activity for MUG of these purified enzymes and that previously reported for the purified human placental isozyme (30) were all similar (Table 2), as were their apparent Km values (Table 2, KFigure 4), i.e., m ~0.7 mM. Taken together, these data demonstrate that the pro-Hex B has the same kinetic parameters as the mature isozyme and that the C-terminal His-tags in pro-Hex B have no effect on these parameters. As well, the heat stabilities of the purified fibroblast pro-Hex B, mature placental Hex B, and the C-terminal His-tagged form of pro-Hex B were very similar (Table 2). Thus, the His-tag on each subunit does not appear to affect the stability of the folded dimer.
Table 2.
Analyses of Various Forms of Hex B: Heat Stability, Kinetic parameters (MUG), and Ability to Specifically Bind a CNAG Hex Affinity Column
| isozyme | t1/2 (min) 60 °C | SAa | Kmb (mM) | Hex B (ng/assay) | Vmaxb (nmol/h) | kcat × 10−3c (mol/mol h) | kcat × 10−3/Km | % binding to CNAGd |
|---|---|---|---|---|---|---|---|---|
| Hex B (mature) | 13 ± 1e | 10.1 ± 0.5 | 0.69 ± 0.09 | 0.78 | 11.3 ± 0.5 | 1900 ± 100 | 2700 ± 600 | 75 ± 5 |
| pro-Hex B | 14 ± 1 | 10.5 ± 0.6 | 0.71 ± 0.07 | 0.70 | 10.6 ± 0.3 | 2000 ± 100 | 2700 ± 500 | 72 ± 6 |
| pro-Hex B-His6 | 17 ± 1 | 13.8 ± 0.8 | 0.66 ± 0.07 | 0.51 | 10.0 ± 0.3 | 2500 ± 100 | 3800 ± 600 | 71 ± 4 |
| pro-Hex B- (Arg211Lys)-His6 |
NDh | 0.007 ± 0.0004 | ~8f | 1600 | ~64f | ~5f | 0.62 ± 0.02g | 7 ± 1 |
Specific activity, nmoles (MU) h−1 ng−1 (Hex B), at 1.6 mM MUG.
Km values are given in mM (MUG), and Vmax values are given as nmol of MU h−1, with the standard error reported as “±”. Note that Vmax values are those derived from Figure 4 and have not been normalized for the amount of Hex B protein used in the assay (given in column 5), i.e., they are not “specific activities” at Vmax.
kcat values are the maximum moles of MU released h−1 (mol of various forms of purified Hex B protein)−1, which are proportional to the specific activities at Vmax. They all assume an Mr for Hex B of 130 000.
Percent of Hex protein binding from three independent experiments: 100 × μg of protein-eluted (with δ-lactone) × (10 μg of protein loaded)−1 (on a CNAG minicolumn).
Standard error of at least three determinations.
Accuracy of the Vmax, Km, and kcat values could not be ensured due to the limited substrate solubility.
The kcat/Km ratio was accurately calculated from 130 000× the slope of the best-fit straight line of [MUG] (0–1.25 mM) versus (nmol of MU) h−1 [ng of pro-Hex B-(Agr211Lys)-His6 used in each assay]−1.
ND, not determined.
Figure 4.
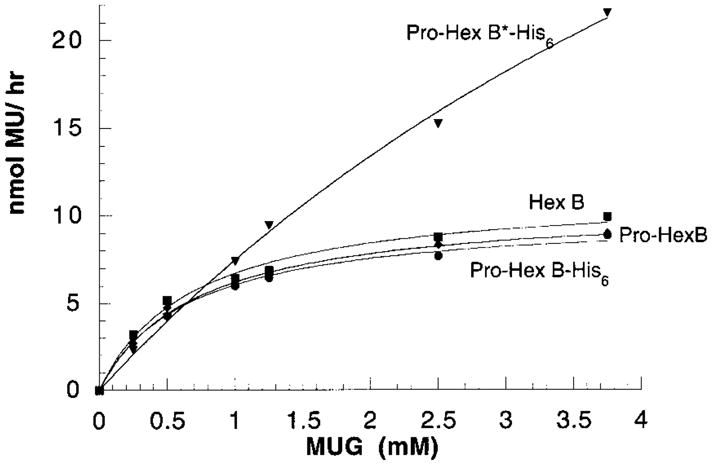
Kinetic analyses of purified pro-Arg211Lys-Hex B-His6, (pro-Hex B*-His6) solid triangles (1600 ng/assay); mature placental Hex B, solid squares (0.8 ng/assay); pro-Hex B, solid diamonds (0.7 ng/assay); and pro-Hex B-His6, solid circles (0.5 ng/assay). Actual experimental data points are plotted with the computer-generated best-fit curves overlaying them. R values were all ≥ 0.996. The Km and Vmax (not normalized for the amount of β protein used) values determined from each plot are given in Table 2.
Examination of the Role of βArg211 in the Active Site of Hex B
Whereas previous studies indicated that Arg211 is an active site residue in Hex B and that it is probably involved in catalysis (19), molecular modeling from chitobiase predicts that this residue is only involved in substrate binding. To determine the exact role of βArg211, pcDNA-β*-His6 encoding a Arg211Lys substitution was transfected into CHO cells. Western blot verified that lysates from cells transfected with pcDNA-β*-His6, like those from cells transfected with the wild-type β-cDNA, contained the mature (lysosomal) form of mutant Hex B (Figure 2, lane β*-His). Additionally, like the His-tagged wild-type enzyme (Table 1), the mature form of the mutant Hex B appears also to have lost the C-terminal His6-tag during its normal proteolytic processing in the lysosome (data not shown). Therefore, mutant Hex B*-His6 was purified from the expression medium. The elution from the Ni2+ column was carried out with increasing imidazole concentration. Kinetic parameters for the MUG substrate were determined (Table 3). Interestingly, the apparent Km value increased with increasing imidazole concentrations, peaking at >80 mM. The pooled fraction between 80 and 500 mM produced a Km ~8 mM (Figure 4); no further Hex was eluted at higher concentration. The specific activity of the enzyme in this fraction, using 1.6 mM MUG was reduced to 0.05% of the wild-type Hex B (Table 2). Accurate Km and Vmax values for the Hex B* eluted at 500 mM could not be assured because of the limited solubility of the MUG substrate. However, the Vmax/Km and thus the kcat/Km ratios were calculated from the initial velocity slope. Since this ratio represents the rate for the first irreversible step in substrate hydrolysis, the formation of the internal oxazoline intermediate (C-1 joined to the former acetamido-oxygen) (37), these data indicate that the wild-type enzyme accomplishes this step at a rate ~6000-fold greater than that of the mutant (Table 2). These results are consistent with the idea that (a) the 80 and 30 mM fractions of enzyme contained increasing amounts of endogenous as compared to mutant human Hex B; (b) the ~10-fold increase of Km in mutant Hex B as compared to wild-type enzyme indicated that Arg211 is indeed involved in substrate binding; and (c) additionally, the greatly reduced kcat/Km ratio indicates that Arg211 must also play an important role in catalysis.
Table 3.
Km Values for the Pro-Hex B-His6 and Pro-Hex B*-His6 (Arg211Lys) Forms of Hex B Elution at the Various Imidazole Concentrations from a Ni2+-NTA Columna
| imidazoleb (mM) |
Kmc |
|
|---|---|---|
| pro-Hex B-His6 | pro-Hex B*-His6 | |
| 0 | 0.65 ± 0.08 | 0.65 ± 0.10 |
| 30 | 0.70 ± 0.10 | 0.68 ± 0.07 |
| 80 | 0.68 ± 0.09 | 1.52 ± 0.12 |
| 500 | 0.66 ± 0.07 | ~8d |
The enzymes were expressed in transfected CHO cells and isolated from the media.
Eluting imidazole concentrations.
Km values are expressed in mM MUG, ± the standard error as calculated from the best-fit curve based on the Michaelis–Menten equation (R values were all 0.998).
The accuracy of the Km value could not be ensured due to low solubility of MUG substrate.
To verify that the Arg211Lys substitution in Hex B affects substrate binding as well as catalysis, the ability of the mutant protein to specifically bind a CNAG affinity minicolumn was assessed (30). While over 70% of wild-type pro-Hex B-His6 binds to and can be eluted from the CNAG column with δ-lactone (a strong competitive inhibitor), only 7.4% of mutant protein bound, consistent with its higher Km for MUG substrate.
Physical Properties of the Wild Type and Mutant Proteins
As a further control the CD spectrum of the nearly inactive mutant, His-tagged pro-Hex B was compared with that from the wild-type His-tagged pro-Hex B and the mature Hex B purified from human placenta (Figure 5). No significant differences were detected between the two His-tagged proteins, indicating that the secondary structures within the protein were unaffected by the mutation. Small variations in the spectrum were seen in comparing the tagged, precursor forms with the mature placental Hex B. However, such changes would be expected from the posttranslational removal of ~18 residues [in the lysosome (36)], converting the monomeric pro-β-subunit into its mature form of three polypeptide chains held together by disulfide bonds (38) and the addition of 12 residues comprising the tag.
Figure 5.
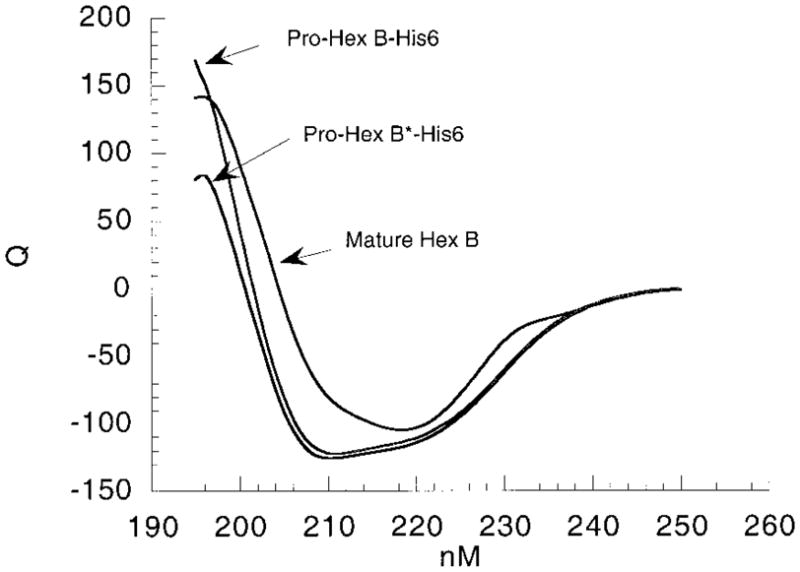
CD spectra (nM vs Q, molar ellipticity ×10−3) of two forms of pro-Hex B-His6-tagged, wild type and mutant (B*), as compared with mature Hex B purified from human placenta.
DISCUSSION
In vitro mutagenesis and mammalian cell expression is now a routine approach to demonstrating the links between mutations and biochemical causes of human disease. For Tay–Sachs and Sandhoff disease, all such studies to date have used eukaryotic cells to express human Hex. This is because in using prokaryotic expression systems, neither the α- or β-subunits are properly folded in vivo, nor can they be re-folded in vitro into functional dimers, possibly because of their need of N-linked oligosaccharides. Thus, all previously reported kinetic data include some contribution from the residual endogenous Hex of the host cells. Using various enrichment methods, the best results have still been ≤100/1, signal/noise ratio (19, 21–23). This ratio is not sufficient to characterize the role of active site residues where a signal/noise ratio of >6000/1 is often needed (28, 29). To circumvent the interference from endogenous Hex, we engineered cDNAs encoding a His6-tag at either the N- or C-terminus of Hex B. The C-terminal (Figure 2), but not the N-terminal, His6-tagged Hex B was transported to the lysosome; thus, it passed the quality control system in the ER indicating that no abnormal folding patterns were introduced into the protein (39, 40). Consistent with this idea, the heat stability of the pro-Hex B was not affected by the addition of the His-tag (Table 2). However, the C-terminal His-tag itself is apparently removed in the lysosome along with other pro-peptides during the normal formation of the mature Hex B form (36, 38, 41). The rapid loss of the tag is consistent with its being in an exposed position on the folded, dimeric isozyme. Fortunately, large amounts of human Hex are secreted into media of transfected cells. Therefore, we were able to purify the human pro-Hex B-His6 from the expression medium by Ni2+-NTA chromatography. A comparison of Hex isolated from the media of CHO cells transfected with the wild-type pro-Hex B-His6 and mock transfected cells indicated a signal-to-noise ratio of ~50,000/1 (Table 1). The thermostabilities, specific activities (using 1.6 mM MUG), Km, and kcat for MUG of the purified pro-Hex B, pro-Hex B-His6, and the mature Hex B forms were found to be nearly identical (Table 2, Figure 4). Consequently, this novel Hex construct makes mutational analyses of the isozymes’ active site possible.
Our previous data revealed that βArg211 is an active site residue in human Hex B. The very conservative Arg211Lys substitution as well as (a) Arg211His, (b) Arg211Thr, or (c) a double mutation (Arg211His and His212Arg) reversing the wild-type Arg211His212 sequence producing His211Arg212 resulted in similar, nearly inactive dimeric Hex B dimers, i.e., ~0.5% of the specific activity of wild type when expressed in COS cells (19). The conservative Lys substitution was the only one found to produce no detectable changes in the intracellular transport and/or stability of the Hex B protein (19). Furthermore, when the Lys211 mutant β-subunit was expressed with the human wild-type α in CHO cells, heterodimeric Hex A was formed, which was fully functional when assayed with labeled GM2 ganglioside and purified human GM2 activator protein (42). These data demonstrate that the Lys211 substitution mutation has no effect on protein folding and is therefore the best mutant construct to use in the characterization of the role of Arg211 in the enzyme’s active site.
In our early studies, we determined that the βArg211Lys mutant Hex B* dimer produced an apparent Km of 1.3 ± 0.4, and a specific activity (based on total lysate protein) at Vmax decreased 400-fold (19). It is possible that the kinetic parameters measured previously for Hex B* were distorted by the presence of a minor contaminant of endogenous COS or CHO cell Hex and/or a small amount of interspecies dimers, i.e., a Hex B dimer of COSβ–humanβ, co-immunoprecipitated with the mutant human Hex B. This would have the effect of lowering apparent Km values if the true Km value for the mutant were higher than that of the wild-type enzyme. This effect would be particularly pronounced if the mutant enzyme was also severely catalytically impaired. The presence of the His-tag on the mutant, but not on the contaminating endogenous enzyme, should allow better separation of these activities. Any untagged CHO Hex B or single-tagged interspecies Hex B would be bound less tightly to the Ni2+ column than would the double-tagged human isozyme. It is therefore significant that the samples of Hex that eluted at lower (30 and 80 mM) imidazole concentrations exhibited substantially lower Km values (0.7 and 1.5 mM, respectively) than that eluted at high (500 mM) imidazole (Km = 8 mM). These earlier fractions presumably therefore contained endogenous CHO β-subunits, and at least in the 80 mM fraction, a mixture containing significant amounts of the human βArg211Lys mutated protein (Table 3).
The apparent Km of 8 mM for MUG obtained with the pure βArg211Lys Hex B (Tables 2 and 3) is approximately 10-fold higher than that of the wild type. Thus Arg211 plays an important role in substrate binding, as predicted by the chitobiase model (Figure 1). However, the ~500-fold reduction in kcat also demonstrates that Arg211 must play an even more important role in catalysis (Table 2, Figure 4). Replacement of the equivalent residue in Sp-Hex, Arg162, by His yielded a mutant in which the Km was increased 40-fold and the specific activity at Vmax value was decreased by 5-fold relative to wild type (12). Thus in both cases, significant changes occurred to both kcat and Km; however, the relative magnitude of these changes differed between the two enzymes.
To further verify the above-described effect of the βArg211-Lys mutation on the Km of human Hex B, a substrate affinity column was used. We found that only ~7% of the mutant as compared to ~70% of the wild-type Hex B could be bound and eluted from the column with a strong competitive inhibitor, δ-lactone (10 μg of each were loaded, and the percent “bound and eluted” was calculated from Lowry protein assays, Table 2).
Three observations concerning the etiology of the GM2 gangliosidoses also indicate that it would be highly unlikely that a mutation whose major effect is on the binding of the terminal β-GalNAc residue of GM2 ganglioside would be the cause of human disease, e.g., the classic B1 variant (αArg178-His) (43). The first is the generally accepted “critical threshold” hypothesis that indicates that only 5–10% of normal Hex A levels are needed to prevent ganglioside storage (44). The second observation is that GM2 ganglioside levels are increased by ~500-fold in the brains of patients with GM2 gangliosidosis (45). Thus, even a major increase in the Km of Hex A can be compensated for in vivo by the 90–95% of “spare” Hex A. If this is still not sufficient to prevent storage, for a Hex A with a much higher Km the continual increase in substrate concentration in the lysosome would result in increased hydrolysis rates. Finally, the binding of the β-GalNAc residue makes up only a small portion of the binding strength for Hex A’s true in vivo substrate, the GM2 ganglioside/GM2 activator protein complex, which includes protein–protein interactions occurring outside the enzyme’s active site (34). The Km for this complex has been estimated at 0.2 μM (46).
The crystal structure of the Serratia marcescens chitobiase complexed with its substrate showed that the residue equivalent to βArg211, c-Arg349, is directly involved in substrate binding, interacting with both 3C-OH and 4C-OH of the β-GlcNAc residue in the –1 subsite (the binding site for NAG-A) and thereby apparently docking the substrate in its proper orientation in the active site (Figure 1). In an apparent contradiction to our experimental data demonstrating a 500-fold decrease in kcat, these authors suggested no role for c-Arg349 in catalysis (10). However, it is difficult to assign a distinct role based solely upon a single three-dimensional structure since, although the residue may appear to make good hydrogen bonds to the substrate in the complex, it is quite possible, in fact quite probable, that the interactions would be even stronger at the transition state, thereby assisting catalysis. Indeed, although not directly commented on in the paper, the chitobiase model does support such a role for c-Arg349 in catalysis. The structure of the chitobiase–substrate complex indicates that the nonreducing NAG-A (GlcNAc) residue is distorted when bound by c-Arg349, taking on an apparently energetically unfavorable 4-sofa confirmation, which results in the glycosidic oxygen being placed closer to the catalytic acid, c-Glu540 (10). This distortion of the sugar ring is likely critical to efficient catalysis by c-Glu540. Distortion of the sugar is also likely necessary to stabilize the oxazoline intermediate. Additionally, several recent crystallographic analyses of glycosidases trapped at various stages along the reaction coordinate diagram have provided evidence for such strengthening of interactions in the intermediate complex. Of particular interest in this regard are the structures of the Family 13 glycosyl transferase, cyclodextrin glycosyl transferase in its free enzyme form and trapped as the Michaelis complex and the covalent glycosyl–enzyme complex (47). Several interactions with the sugar, including that of a highly conserved Asp bridging the substrate 2- and 3-hydroxyl groups, are seen to tighten along the coordinate. Mutation of this residue results in severe effects on both kcat and Km. Other examples include complexes with the Family 5 cellulase Cel5A from Bacillus agaradhaerens (48) and with the Family 11 xylanase from Bacillus circulans (49). Indeed, in this latter case, a highly conserved arginine residue, Arg112, is found bridging the 2- and 3-hydroxyl group, very reminiscent of the case with the Family 20 hexosaminidases. Surprisingly, however, mutations at this center only affect catalytic parameters in a very modest way.
In conclusion, our data demonstrate that the conserved Arg residues in the subunits of human Hex, αArg178 and βArg211, like their aligned counterparts in Streptomyces Hex, spArg162, play important roles in the enzymes’ active site(s). Furthermore, these functions are consistent with those predicted for the corresponding c-Arg379 in the chitobiase model. They include (a) an involvement in the initial, ground-state binding of the substrate, as suggested by higher mutant Km values, and (b) the stabilization of the transition state(s) assisting catalysis, as suggested by lowered mutant kcat values. However, the latter function appears to be more important for catalysis by the human isozymes than by Streptomyces Hex. It will be interesting to see crystallographic data on complexes of enzymes from this family with both analogues of the oxazoline intermediate and of the transition state to find out how these tighter interactions are affected.
Acknowledgments
We thank Ms. A. Leung and Ms. M. Glibowicka for their excellent technical assistance.
Footnotes
This work was supported by a medical Research Council of Canada grant to D.M.
Abbreviations: Hex, β-hexosaminidase; GM2, GM2 ganglioside, GalNAcβ(1,4)-[NANAα(2,3)]-Galβ(1,4)-Glc-ceramide; GM2 activator protein, Activator; MU, 4-methylumbelliferone; MUG, 4-methylum-belliferyl-β-N-acetylglucosamine; MUGS, 4-methylumbelliferyl-β-N-acetylglucosamine-6-sulfate; CNAG, 2-acetamido-N-(ε-aminocaproyl)-2-deoxy-β-D-glucopyranosylamine; ER, endoplasmic reticulum; CRM, cross-reacting material; δ-lactone, 2-acetamido-2-deoxy-D-glucono-1,5-lactone; NTA, nitrilotriacetic acid; CD, circular dichroism; Q, molar ellipticity (deg cm2 dmol−1).
References
- 1.Nakai H, Byers MG, Nowak NJ, Shows TB. Cytogenet Cell Genet. 1991;56:164. doi: 10.1159/000133077. [DOI] [PubMed] [Google Scholar]
- 2.Bikker H, Meyer MF, Merk AC, deVijlder JJ, Bolhuis PA. Nucleic Acids Res. 1988;16:8198. doi: 10.1093/nar/16.16.8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korneluk RG, Mahuran DJ, Neote K, Klavins MH, O’Dowd BF, Tropak M, Willard HF, Anderson MJ, Lowden JA, Gravel RA. J Biol Chem. 1986;261:8407–8413. [PubMed] [Google Scholar]
- 4.Heng HHQ, Xie B, Shi XM, Tsui LC, Mahuran DJ. Genomics. 1993;18:429–431. doi: 10.1006/geno.1993.1491. [DOI] [PubMed] [Google Scholar]
- 5.Mahuran DJ. Biochim Biophys Acta. 1998;1393:1–18. doi: 10.1016/s0005-2760(98)00057-5. [DOI] [PubMed] [Google Scholar]
- 6.Mahuran DJ. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 7.Church WB, Swenson L, James MNG, Mahuran D. J Mol Biol. 1992;227:577–580. doi: 10.1016/0022-2836(92)90911-3. [DOI] [PubMed] [Google Scholar]
- 8.Henrissat B, Bairoch A. Biochem J. 1993;293:781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pons T, Olmea O, Chinea G, Beldarraín A, Márquez G, Acosta N, Rodríguez L, Valencia A. Proteins. 1998;33:383–395. doi: 10.1002/(sici)1097-0134(19981115)33:3<383::aid-prot7>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 10.Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. Nat Struct Biol. 1996;3:638–648. doi: 10.1038/nsb0796-638. [DOI] [PubMed] [Google Scholar]
- 11.Pennisi E. Science. 1996;273:426–428. doi: 10.1126/science.273.5274.426. [DOI] [PubMed] [Google Scholar]
- 12.Mark BL, Wasney GA, Salo TJS, Khan AR, Cao ZM, Robbins PW, James MNG, Triggs-Raine BL. J Biol Chem. 1998;273:19618–19624. doi: 10.1074/jbc.273.31.19618. [DOI] [PubMed] [Google Scholar]
- 13.Mahuran DJ. Biochim Biophys Acta. 1991;1096:87–94. doi: 10.1016/0925-4439(91)90044-a. [DOI] [PubMed] [Google Scholar]
- 14.Mahuran DJ. In: Protein Disfunction in Human Genetic Disease. Swallow D, Edwards Y, editors. Bios Scientific; Oxford, U.K: 1997. pp. 99–117. [Google Scholar]
- 15.Kytzia HJ, Hinrichs U, Maire I, Suzuki K, Sandhoff K. EMBO J. 1983;2:1201–1205. doi: 10.1002/j.1460-2075.1983.tb01567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohno K, Suzuki K. J Neurochem. 1988;50:316–318. doi: 10.1111/j.1471-4159.1988.tb13266.x. [DOI] [PubMed] [Google Scholar]
- 17.Li SC, Hirabayashi Y, Li YT. Biochem Biophys Res Commun. 1981;101:479–485. doi: 10.1016/0006-291x(81)91285-7. [DOI] [PubMed] [Google Scholar]
- 18.Hirabayashi Y, Li YT, Li SC. J Neurochem. 1983;40:168–175. doi: 10.1111/j.1471-4159.1983.tb12667.x. [DOI] [PubMed] [Google Scholar]
- 19.Brown CA, Mahuran DJ. J Biol Chem. 1991;266:15855–15862. [PubMed] [Google Scholar]
- 20.Brown CA, Neote K, Leung A, Gravel RA, Mahuran DJ. J Biol Chem. 1989;264:21705–21710. [PubMed] [Google Scholar]
- 21.Pennybacker M, Schuette CG, Liessem B, Hepbildikler ST, Kopetka JA, Ellis MR, Myerowitz R, Sandhoff K, Proia RL. J Biol Chem. 1997;272:8002–8006. doi: 10.1074/jbc.272.12.8002. [DOI] [PubMed] [Google Scholar]
- 22.Fernandes MJG, Yew S, Leclerc D, Henrissat B, Vorgias CE, Gravel RA, Hechtman P, Kaplan F. J Biol Chem. 1997;272:814–820. doi: 10.1074/jbc.272.2.814. [DOI] [PubMed] [Google Scholar]
- 23.Tse R, Vavougios G, Hou Y, Mahuran DJ. Biochemistry. 1996;35:7599–7607. doi: 10.1021/bi960246+. [DOI] [PubMed] [Google Scholar]
- 24.Hou Y, Vavougios G, Hinek A, Wu KK, Hechtman P, Kaplan F, Mahuran DJ. Am J Hum Genet. 1996;59:52–58. [PMC free article] [PubMed] [Google Scholar]
- 25.Brown CA, Mahuran DJ. Am J Hum Genet. 1993;53:497–508. [PMC free article] [PubMed] [Google Scholar]
- 26.Akalin N, Shi HP, Vavougios G, Hechtman P, Lo W, Scriver CR, Mahuran D, Kaplan F. Hum Mutat. 1992;1:40–46. doi: 10.1002/humu.1380010107. [DOI] [PubMed] [Google Scholar]
- 27.Trop I, Kaplan F, Brown C, Mahuran D, Hechtman P. Hum Mutat. 1992;1:35–39. doi: 10.1002/humu.1380010106. [DOI] [PubMed] [Google Scholar]
- 28.Malcolm BA, Rosenberg S, Corey MJ, Allen JS, de Baetselier A, Kirsch JF. Proc Natl Acad Sci USA. 1989;86:133–137. doi: 10.1073/pnas.86.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacLeod AM, Lindhorst T, Withers SG, Warren RA. Biochemistry. 1994;33:6371–6376. doi: 10.1021/bi00186a042. [DOI] [PubMed] [Google Scholar]
- 30.Mahuran DJ, Lowden JA. Can J Biochem. 1980;58:287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. 1–3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 32.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 33.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. J Biol Chem. 1998;273:21386–21392. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
- 35.Tommasini R, Endrenyi L, Taylor PA, Mahuran DJ, Lowden JA. Can J Biochem Cell Biol. 1985;63:225–230. doi: 10.1139/o85-032. [DOI] [PubMed] [Google Scholar]
- 36.Hasilik A, Neufeld EF. J Biol Chem. 1980;255:4937–4945. [PubMed] [Google Scholar]
- 37.Legler G, Bollhagen R. Carbohydr Res. 1992;233:113–123. [Google Scholar]
- 38.Mahuran DJ. J Biol Chem. 1990;265:6794–6799. [PubMed] [Google Scholar]
- 39.Ashkenas J, Byers PH. Am J Hum Genet. 1997;61:267–272. doi: 10.1086/514865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks DA. FEBS Lett. 1997;409:115–120. doi: 10.1016/s0014-5793(97)00423-7. [DOI] [PubMed] [Google Scholar]
- 41.Mahuran DJ, Neote K, Klavins MH, Leung A, Gravel RA. J Biol Chem. 1988;263:4612–4618. [PubMed] [Google Scholar]
- 42.Hou Y, Tse R, Mahuran DJ. Biochemistry. 1996;35:3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- 43.Dos Santos MR, Tanaka A, Sa Miranda MC, Ribeiro MG, Maia M, Suzuki K. Am J Hum Genet. 1991;29:287–293. [PMC free article] [PubMed] [Google Scholar]
- 44.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 45.Sandhoff K, Conzelmann E, Neufeld EF, Kaback MM, Suzuki K. In: The Metabolic Basis of Inherited Disease. Scriver CV, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill; New York: 1989. pp. 1807–1839. [Google Scholar]
- 46.Xie B, Rigat B, Smiljanic-Georgijev N, Deng H, Mahuran DJ. Biochemistry. 1998;37:814–821. doi: 10.1021/bi971211s. [DOI] [PubMed] [Google Scholar]
- 47.Uitdehaag JC, Mosi R, Kalk KH, van der Veen A, Dijkhuisen L, Withers SG, Dijkstra B. Nat Struct Biol. 1999;6:432–436. doi: 10.1038/8235. [DOI] [PubMed] [Google Scholar]
- 48.Davies GJ, Mackenzie L, Varrot A, Dauter M, Brzozowski AM, Schülein M, Withers SG. Biochemistry. 1998;37:11707–11713. doi: 10.1021/bi981315i. [DOI] [PubMed] [Google Scholar]
- 49.Sidhu G, Withers SG, Nguyen NT, McIntosh LP, Ziser L, Brayer GD. Biochemistry. 1999;38:5346–5354. doi: 10.1021/bi982946f. [DOI] [PubMed] [Google Scholar]