

Abstract
Cisplatin is one of the commonly-used chemotherapeutic drugs to efficiently treat malignant tumors in clinic, however, the adverse effects of cisplatin such as nephrotoxicity, neurotoxcity, and hemolytic uremic syndrome are often observed at its clinical doses (~60 mg/m2), which limit its broader application. In earlier studies, little attention was paid to the subtle changes in the architecture of lymphatic organs after low doses of cisplatin treatment. This paper reviews current understanding of cisplatin-induced erythrocyte injury, and presents our latest finding that a low dose of cisplatin (3.6 mg/m2/day, 14 days) could induce specific hemosiderin deposition in spleen of both normal and hepatoma-22 (H22) inoculated Balb/C mice. This dose of cisplatin significantly inhibited H22-induced acute ascites development. No significant toxicity was induced by this dose of cisplatin to tissues except for hemosiderin accumulation in the spleen of both normal and H22 tumor-bearing mice. Increased splenic iron content and erythrocyte injury were observed after treatment with the low dose of cisplatin. The mRNA levels of ferroportin (FPN1) and ferritin were upregulated by 25 and 5-fold in spleen, respectively. Overexpression of FPN1 and ferritin protein were also been observed at protein levels by Western blotting analysis. In addition, the mRNA expression of hepcidin was also increased, suggesting blockage of iron recycling through FPN1 in spleen with cisplatin treatment. In conclusion, cisplatin treatment damages the erythrocytes which accumulate in the red pulp of spleen with defective recycling of FPN1 and ferritin protein. Hepcidin inhibits the function of FPN1 as iron-exporter leading to iron overloaded inside ferritins of splenic cells, which are stained with abnormal hemosiderin accumulation. These results demonstrate that cisplatin-caused hemosiderin deposition in spleen provides a valuable clue for understanding the molecular basis of toxicity of cisplatin and hemosiderin accumulation and iron metabolism in vivo.
Keywords: CP, cis-diamminedichloroplatinum II, hemosiderin, ferroportin, ferritin, hepcidin
1. INTRODUCTION
Cisplatin (cis-diamminedichloroplatinum II, CP) is one of major chemotherapeutic agents at clinics to treat cancer patients [1]. It remains a cornerstone of present chemotherapy regimens, not only against epithelial malignancies (lung, ovarian, bladder, testicular, head, and neck), but also against a number of metastatic or advanced malignancies [2, 3, 4]. However, the significant adverse effect of cisplatin on normal cells and tissues remains as a significant obstacle to its broader clinical application. Intensive efforts have been made through cellular and molecular approaches to identifying the molecular mechanism of toxicity of cisplatin to different organs in vivo [5, 6].
The erythrocyte injury induced by cisplatin may be the main reason for cisplatin-induced anemia. Mahmud et al found that erythrocyte suicidal death was triggered by cisplatin, which can stimulate cell membrane scrambling and lead to cell shrinkage, and the resulting exposure of phosphatidylserine (PS) on the cell surface [7]. PS-exposing cells were rapidly cleared from circulation by reticuloendothelial system (RES), especially in spleen. However, there was little attention in previous studies paid to the subtle changes in the architecture of spleen as a lymphatic organ after cisplatin treatment [8]. Macrophages in the spleen play a major role in iron metabolism by recycling irons from erythrocytes. Iron recycling is achieved through the phagocytosis of damaged or senescent erythrocytes by macrophages. The iron is recycled to the circulation and ultimately to the erythron. The relationship between the injured erythrocytes by cisplatin treatment and iron metabolism in spleen remains poorly understood. Delineation of the mechanisms responsible for the cisplatin toxicity on spleen is important for improving its therapeutic index, and developing new agents that can prevent, overcome, or reverse its adverse effects.
2. DEVELOPMENT OF ASCITES IN TUMOR-BEARING MICE WAS PREVENTED BY A LOW DOSE OF CISPLATIN WITH SPECIFIC HEMOSIDERIN ACCUMULATION IN SPLEEN
In clinic, the routine therapeutic dose of cisplatin is 60 mg/m2 [9], which can kill cancer cells at least partially attributed to its DNA damage [10]. Side effects of cisplatin include nephrotoxicity [11, 12], neurotoxcity [13], and hemolytic uremic syndrome [14]. Cisplatin-based chemotherapy has been attributed to one of the causes for cumulative anemia in patients [15]. Oxidative stress and inflammation have been suggested to be the major mechanisms in the pathogenesis of cisplatin-induced toxicity [16]. Strategies have been taken to reduce the oxidative stress induced by cisplatin with administration of other compounds. Sung et al found that genistein significantly reduced the reactive oxygen species (ROS) produced in cisplatin-treated normal human kidney HK-2 cells [17]. Yin et al also demonstrated that different types of fullerene derivatives can scavenge various of physiologically relevant ROS, and may be valuable as cytoprotective agents in vivo [18]. Nephrotoxicity is also a frequent observed adverse effect of cisplatin-based chemotherapy and is one of the major limitations of its broader use in clinic.
The chemotherapeutic damage to body may be reduced by using a lower dose of cisplatin without affecting its efficacy of inhibiting tumor growth. To test this hypothesis, we designed the following experiments. For in vivo tumor treatment, 2×106 mouse hepatoma 22 cells (H22) (in 200μl saline) per mouse were injected into 5-week-old Balb/C mice in the peritoneal cavity. Each mouse was administered intraperitoneally (i.p.) with the saline or cisplatin on the second day of inoculation and continued to the day of sacrifice. Sixty mice (20±2 g) were randomly allotted into 2 groups (n=30 per group) and were treated according to the following protocol. Tumor-CP group mice were i.p. administered with cisplatin at the dose of 3.6mg/m2 once a day consecutively for a period of 14 days. Control animals were injected with saline solution only (Tumor-Saline group). Body weight of each mouse was measured in every 24h. Five mice in each group were sacrificed on Days 3, 4, 6, 8, 10, and 16 after peritoneal inoculation of H22 for further experiments. Blood was collected from the retro-orbital plexus of each mouse for blood smear assay, and the spleen, heart, liver, lung, and kidney of the mice were taken for hematoxylin-eosin (H&E) staining and Perls’s staining to analyze hemosiderin formation. The tissue slides were dehydrated through graded alcohols and embedded in paraffin wax. Histological changes were recorded for individual organs. In addition, eight normal mice (non H22 tumor bearing mice) were used as control compared with H22 tumor bearing mice. Eight mice (20±2g) were allotted into 2 groups (n=4 per group), one group was Normal-CP group and the other was Normal-Saline group. The treatment was same as mentioned above.
It is clearly shown in Fig. (1a) that body weight of H22 tumor-bearing mice increased up to 30 g without cisplatin treatment for two weeks in Tumor-Saline group. Cisplatin at dose of 3.6 mg/m2 could effectively inhibit the ascites development of H22 cells (Tumor-CP group) compared with the control groups (Tumor-Saline group). Reduced mice weights were observed with cisplatin administration in normal mice (Normal-CP group) compared with the Normal-Saline group (P<0.01) Fig. (1a). The tumor bearing mice treated with cisplatin appeared lethargy with decreased food appetite and tangle hair Fig. (1b). These results indicated that the low dose cisplatin can inhibit the development of ascites tumor, but still showed adverse effects to the body. The histopathological analysis of tissues revealed that there were no clear pathological changes of the heart, liver, lung, and kidney in the cisplatin treated groups (Tumor-CP group) compared to that of the saline-treated groups (Tumor-Saline group), while numerous brown granules occurred in the red pulp of spleen in tumor-bearing mice with low dose of cis-platin treatment (Tumor-CP group) Fig. (2a). Histological images in Fig. (2a) showed the localization and levels of hemosiderin formation in red pulp of spleen. Furthermore, histochemical analysis in Fig. (2b) demonstrated specific splenic hemosiderin deposition with H&E staining and Perls’s staining, the golden protocol used for identification and evaluation of ion accumulation in tissues.
Fig. 1.

a) Body weight measurement of the normal mice (non tumor-bearing mice) and H22 ascites tumor-bearing mice treated with cisplatin or saline. The mice in the Tumor-Saline group (-■-) were treated with 0.2 ml/day of saline; the mice in the Tumor-CP group (-●-) were treated with cisplatin at 3.6 mg/m2/day. The mice in the Normal-Saline group (-▲-) and Normal-CP group (-▼-) were treated with same condition as tumor-bearing mice. The error bars represent the standard deviation. b). Characters of H22 ascites tumor-bearing Balb/C mice measured at different days. Balb/C mice (five mice per group) were implanted with 2×106 mouse H22 cells per mouse into peritoneal cavity. Balb/C mice with developed ascites were identified on Day 2. The H22 ascites tumor-bearing mice were treated with cisplatin (CP) and saline. Cisplatin was given via intraperitoneal (i.p.) injection at 3.6 mg/m2/day, and saline was also administered via i.p. with the dose of 0.2 ml/day on the second day of inoculation and continued to the 16th day. Different measurements were taken on Day 3, 4, 6, 8,10, and 16. Cisplatin obviously inhibited the growth of ascites based on the reduced mice weight at enlonged administration points. Normal Balb/C mice were used as control. Images showed the normal mice treated with saline or cis-platin at 16th day. The behavior of mice treated by cisplatin is lethargy with decreased food appetite and tangle hair.
Fig. 2.

a) Hematoxylin-eosin (H&E) staining of different tissue sections in the Saline-treated (A–E) and cisplatin-treated (F–J) groups. The formation of brown hemosiderin granules were only observed in the red pulp of spleen in cisplatin-treated mice, but not in other tissues such as heart, liver, lung, and kidney, indicating the specific toxicity of low dose cisplatin in vivo. b) Representative histochemical measurements of spleen tissue sections of tumor-bearing mice in saline-treated (A–C) and cisplatin-treated (D–F) groups. The brown granules represent hemosiderin accumulation, which was mainly in the red pulp of spleen by the H&E staining (A and D). To clearly see the morphology of hemosiderin indicated by arrows, (B) and (E) are the enlarged portion of image (A) and (D), respectively. To confirm the conclusion of H&E staining, the same tissue sections were measured by the Perls’s staining (C) and (F). Scale bar =50μm.
To elucidate the mechanism of splenic hemosiderin deposition, we investigated the spleen of normal mice after cisplatin treatment. The results showed that spleen was shrunk due to the cisplatin treatment in normal mice. The size of spleen was clearly smaller in the Normal-CP group compared to the Normal-Saline group Fig. (3 A–B). The average of spleen specimens is 18.57±0.51mm of Normal-saline group and 12.63±0.15mm of Normal-CP group. The splenic index is defined as the [splenic weight (g)/body weight (g)]*100%, which is 0.54 of Normal-saline group and 0.36 of Normal-saline group. Histo-morphological analysis revealed that cis-platin treatment induced significant size reduction of splenic corpuscle Fig. (3 C–D). There was more hemosiderin deposition in the red-pulp of spleen than white-pulp demonstrated with Perls’s staining Fig. (3 E–F). Scrambling of cell membrane and bean-like erythrocytes aggregation were observed in normal mice after cisplatin treatment Fig. (3 G–H). It was reported that hemosiderin deposition in tissues was usually due to presence of the excessive amount of irons, caused by phagocytosed extravasated erythrocytes [19, 20]. There was no obvious hemosiderin deposition and abnormal erythrocyte measured in the Tumor-Saline group, which is consistent with the measurement in the Normal-Saline group Fig. (2a, 2b, 3). We speculated that the splenic injury and hemosiderin deposition might be mainly due to abnormal erythrocyte damage induced by cisplatin. These data also suggested that the hemosiderin deposition in the spleen were mainly initiated by cisplatin treatment and not associated with development of ascites tumor.
Fig. 3.

Effects of saline or cisplatin treatment on morphology of spleen and erythrocyte in normal mice for two weeks. (A–B) The size of spleen significantly reduced after treated with cisplatin; (C–D) splenic corpuscle size decreased and the brown granules deposited in the red pulp of spleen in the Normal-CP group; (E–F) Representative Perls’s staining of spleens tissue sections showed clear locations of iron accumulation in spleen of Normal-saline and Normal-CP groups; (G–H) Cell membrane scrambling and bean-like aggregation of erythrocytes were induced by cisplatin treatment in Normal-CP group.
3. HEMOSIDERIN AS THE DIAGNOSIS STANDARD AT CLINICS
Irons released from hemoglobin of phagocytosed erythrocytes were stored in ferritin and eventually converted to hemosiderins after ferritin partially degraded and stored in spleen. The hemorrhage of red pulp and hemosiderin deposition in spleen may increase due to the stimulated sequestration and destruction of erythrocytes [21]. Quantification of hemosiderin-laden macrophages in the bronchoalveolar lavage fluid (BALF) has been used to diagnose diffuse alveolar hemorrhage (DAH) in patients with diffuse alveolar damage (DAD). Clinical studies showed that there was a positive correlation between the percentage of hemosiderin-laden macrophages in the BALF and parenchymal hemorrhages, which were assessed semi-quantitatively by the histopathologic analysis. In brain disease research, prior subarachnoid hemorrhage (SAH) has been diagnosed by investigating subarachnoid hemosiderin deposition using T2*-weighted MRI method [22]. In venous disease research, impaired venous drainage in severe chronic venous insufficiency (CVI) often leads to microcirculatory overload, characterized by erythrocyte diapedesis and subsequent extravascular hemolysis, and typical dermal hemosiderin deposition. Urine hemosiderin is another novel marker to assess the severity of chronic venous disease with the sensitive, cost-effective, noninvasive, and repeatable results [23]. The disturbance of iron homeostasis in vivo caused by anemia and long-term repeated blood transfusion is closely related to the appearance of hemosiderin [24]. In anemia research, the degree of hemosiderin deposition in spleen helped distinguish patients with autoimmune hemolytic anemia from those with congenital hemolytic anemia. The major pathologic findings in autoimmune hemolytic anemia include increased polymorphonuclear neutrophil reaction, increased deposition of hemosiderin, and extramedullary hematopoiesis. In contrast, with the severity and the frequency of polymorphonuclear neutrophil reactions, deposition of hemosiderin, and extramedullary hematopoiesis were less in patients with congenital hemolytic anemia [25].
Previously, the amount of ferritin and hemosiderin in the liver has been used to diagnose patients with iron-loading diseases. Among patients with low iron overload, the bulk of iron deposition was present in ferritin. However, for individuals with heavy iron overload, iron deposition consisted of approximately equal amounts in hemosiderin (non-ferritin iron) and ferritin [26]. In pulmonary disease research, the hemosiderin content of BAL macrophages was once thought to have limited ability in giving diagnostic conclusions on a variety of lung diseases [27]. However, the presence of hemosiderin-laden macrophages (HLMs) in the BAL fluid or lung tissue has now become an important diagnostic biomarker for alveolar hemorrhage in clinical settings. As a result, the time course for clearance of HLMs within the BAL and lung tissue after blood aspiration could also be determined [28].
4. HEMOSIDERIN DEPOSITION IN THE SPLEEN OF PATIENTS WAS TIME-DEPENDENT
Hemosiderin is a granular brown degradation product of ferritin, which is derived from hemoglobin during hemolysis. In hemolytic anemia, when erythrocytes leave a ruptured blood vessel, the destroyed erythrocytes may release hemoglobin into the extracellular space. The damaged red cells or the released hemoglobin can be phagocytosized by macrophages. The iron released from heme can be stored as ferritin within cells. Intracellular ferritin is oxidized and degraded to form hemosiderin in cytoplasm. The iron-storing molecules ferritin and hemosiderin possess the unique ultrastructure feature, which can be identified by the electron microscopy because of the electron-density of iron concentrated within these particles. The homeostasis of iron is an important issue for most living cells. Hemosiderin may be formed in many diseases associated with iron overload. One of the defensive mechanisms of cells and tissues against iron overload is their capability for extra iron storage within ferritin and hemosiderin [29]. The accumulation of hemosiderin is observed in different organs such as liver, kidney, spleen, and brain, and can be used as a sign of disturbed iron metabolism in various diseases. In human, hemosiderin could only accumulate to a small amount in the spleen during a life span of 60–70 years [30]. Tissue iron overload can be caused by excessive absorption of irons from the diet as in hereditary hemochromatosis, or by excessive iron administration from multiple blood transfusions. Iron overload results in the excess deposition of iron in tissues of the body in the form of ultrafine particles of hydrated iron (III) oxyhydroxide (5Fe2O3·9H2O). The mineral deposits are associated with the iron storage compounds hemosiderin and ferritin. Organs which are particularly affected by iron overload are the liver, spleen, heart, pancreas, and adrenal gland [31].
To determine the effect of cisplatin administration on spleen and erythrocytes, H&E staining and Wright’s staining were employed to measure the morphology of spleen and erythrocytes in mice bearing with H22 cells at post-implant Day 3, 4, 6, 8, 10, and 16. Two pathologists independently scanned the spleen sections by microscopy using a 10× objective lens (with a 10× ocular lens), and determined the hemosiderin score based on the brown granules for each section: 0, shown without staining; 1, occasional staining with most fields negative; 2, focally abundant staining with most fields having no staining; 3, focally abundant staining with most fields showing positive staining; and 4, prominent staining throughout the section score. The total iron score was calculated by adding the scores based on individual pathologist’s observation. The results showed that hemosiderin deposition occurred on Day 8 after mice implanted with tumor cells, and reached the highest severity on Day 16 in the Tumor-CP group. However, no significant hemosiderin depositions were found in the Tumor-Saline group Fig. (4a, 4b). It showed that splenic hemosiderin deposition increased in a time-dependent manner with cisplatin administration in the Tumor-CP group Fig. (4b). On Day 6, hematological abnormality could be detected in the tumor-bearing mice with cisplatin treatment, and the erythrocyte aggregation into bean or necklace-like structures could be clearly observed under light microscopy. Most of the erythrocytes formed bean or necklace-like structure on Day 16 Fig. (4c), which demonstrated that cisplatin treatment could cause significant oxidative damage to the plasma membrane of the erythrocytes. Splenic iron concentration in the cisplatin treated group (Tumor-CP and Normal-CP groups) was significantly higher than that in the saline treated group (Tumor-Saline and Normal-Saline groups) detected by ICP-OES Fig. (4d). These findings about the erythrocyte changes helped to better understand the development of anemia following cisplatin treatment. Iron is released and deposited in the spleen through the phagocytosis of the injured erythrocytes by macrophages. Cisplatin primarily impairs erythrocytes and stimulates the destruction of injured erythrocytes in spleen. It may lead to the persistent hemosiderosis in spleen. Excessive hemosiderins phagocytosed in macrophage led to the destruction of macrophage and the release of the deposited contents such as irons, toxic compounds, and/or its metabolites into the spleen [32].
Fig. 4.

a). Hemosiderin accumulation in spleen tissues sections measured by H&E staining. The time-dependent progression of splenic hemosiderin accumulation was measured in tumor-bearing groups treated with saline (Tumor-Saline group) (A–F) or cisplatin (Tumor-CP groups) (G–L). Numerous brown granules accumulations of stained hemosiderin particals were presented in the red pulp between these focal aggregates of lymphoid tissue in the Tumor-CP group. Scale bar =50μm. b) Evaluation of hemosiderin deposition in the spleen of H22 tumor bearing mice with time. Hemosiderin score in the Tumor-CP (—●—) group was significantly higher than that in the Tumor-Saline (—■—) group. Hemosiderin deposition occurred on Day 8 after mice implanted with tumor cells, and reached the highest severity on Day 16 in the Tumor-CP group. However, no significant hemosiderin depositions were found in the Tumor-Saline group. It showed that splenic hemosiderin deposition increased in a time-dependent manner with cisplatin therapeutic administration in the Tumor-CP group. Two pathologists independently microscopically scanned the spleen sections using a 10× objective lens (with a 10× ocular lens), and determined the hemosiderin score based on the brown granules for each section: 0, if it showed no staining; 1, occasional staining with most fields negative; 2, focally abundant staining with most fields having no staining; 3, focally abundant staining with most fields showing positive staining; or 4, prominent staining throughout the section score. A total iron score was calculated by adding the scores based on each pathologist’s observation. c) Blood smear assay in the Tumor-Saline and Tumor-CP groups by Wright’s staining. Mice were administered intraperitoneally (i.p.) with saline (0.2 ml/day) or cisplatin (3.6mg/m2) on the second day of inoculation and continued to Day 16. The formation of bean-like erythrocytes occurred on Day 6 in the Tumor-CP group, but not in the Tumor-Saline group. Scale bar =50μm. d) The splenic iron content measured by ICP-OES. The splenic iron content in the cisplatin treated group (Normal-CP and Tumor-CP groups) was significantly higher than saline treated group (Normal-saline and Tumor-Saline groups). No significant difference between the Normal-Saline group and Tumor-Saline group was detected. Mice were treated with cisplatin at 3.6mg/m2/day for 14 days. The saline treatment was used as control group. Each bar represents mean +/− standard deviation (n=5). **P<0.01
Under the normal circumstances, hemosiderin deposition in the spleen of rat was clearly observed in at least 4 months old rats, although only few hemosiderin-laden macrophages appeared in the first 3 months. Hemosiderin deposition peaked at 12 months in the red pulp of rat spleen [33]. In our experiments, cisplatin treatment can increase the hemosiderin depositions in a short period of 2 weeks. Rapid formation of splenic hemosiderin accumulation induced by cisplatin treatment in two weeks may provide a useful model to screen compounds which can attenuate the oxidative stress damage in spleen, and to investigate the molecular mechanism of iron homeostasis disruption in vivo.
5. CISPLATIN INDUCED HEMOSIDERIN DEPOSITION WAS CLOSELY RELATED WITH IRON METABOLISM IN THE SPLEEN
Most iron in human body is contained in the hemoglobin of erythrocytes, and iron deficiency induced anemia is one of the most common types of anemia. Iron is an essential metal for mammalian cellular and tissue viability. Splenic iron content of mice treated with cisplatin (Normal-CP and Tumor-CP group) was significantly higher than that of mice treated with saline (Normal-saline and Tumor-Saline group) Fig. (4d). The exact mechanisms of the formation of most adult hemochromatosis, which make up most of the iron disorders, remain unsolved. Ferroportin (FPN1) is the first cellular iron exporter identified [34]. It is present on the surface of absorptive intestinal enterocytes, macrophages, hepatocytes, and placental cells, all of which release iron into plasma [34, 35]. Although an iron-export protein would be expected to reside on plasma membrane, the available immunofluorescence data in Kupffer cells indicated an intracellular distribution [34]. It is possible that the intracellular localization of macrophage FPN1 is affected by phagocytosis, as has been observed for the other transmembrane iron transporter Nramp1 [36]. The fact, that FPN1 is particularly abundant in RE macrophages of the liver, spleen, and bone marrow, indicates that this protein may also function to export iron from senescent erythrocytes after phatocytosis [37]. Mitchell et al examined the effect of iron status and erythrophagocytosis on FPN1 expression in J774 macrophages. They found that the mRNA and protein levels of FPN1 increased after phagocytosis of red cells, when iron liberated from heme significantly expanded the cellular iron pool [38]. Ferritin as an iron-binding protein plays important role to maintain balance of irons in most living cells [39]. Intracellular ferritin has the dual-function of storing irons in a biologically available form and segregating iron, which may participate the formation of ROS [40]. Hemosiderin as denatured product of ferritin is derived from hemoglobin released during hemolysis [39]. The accumulation of hemosiderin is observed in different organs such as liver, kidney, lung, and spleen, and can be used as a sign of disturbed iron metabolism in various diseases [23, 41, 42]. Hepcidin synthesized in liver is increased by inflammation and iron stores, and is decreased by hypoxia and anemia. The decreased hepcidin leads to tissue iron overloads, whereas hepcidin overproduction leads to hypoferremia and the anemia of inflammation [43]. Liu et al found that hepcidin is also synthesized in the spleen of normal mice and induced by lipopolysaccharide [44]. Nemeth et al reported that hepcidin can bound to FPN in cultured cells. After binding, FPN is internalized and degraded, which lead to decreased export of cellular irons [45]. Irons regulate the secretion of hepcidin, which in turn controls the concentration of ferroportin on the cell surface. These three factors mentioned above have the most important influences on iron metabolism in macrophages.
To investigate the effect of cisplatin on the splenic iron metabolism, the expression levels of ferroportin 1, ferritin and hepcidin were measured in normal mice treated with cisplatin (3.6 mg/m2/day) for 14 days, and with saline as control. Total RNA (1 μg) was collected from spleen and was reversely transcribed in a 20 μl reaction using reversely transcription cycle (Promega Incorporation) for 15 min at 42°C, 5 min at 95°C and 5°C, min at 4°C, TaqMan real time polymerase chain reaction was performed with SYBR Green Supermix (Bio-Rad, USA) according to the manufacturer’s protocol. Amplification conditions were 2 min at 95°C, followed by 40 cycles for 15 s at 95°C, 30 s at 55°C and 20 s at 68°C. The following primers were used:
FPN1: Forward 5′-AGACTTAAAGTGGCCCAGACGT-3′ and Reverse 5′-CAGGATGTAGCAGACAGTAAGGAC-3′.
Ferritin: Forward 5′-CGTCTATCTGTCTATGTCTTG-3′ and Reverse 5′-CACGGTCTGGTTTCTTTATATCC-3′.
Hepcidin: Forward 5′-AAGCAGGGCAGACATTGCGATA-3′ and Reverse 5′-TGCAACAGATACCACACTGGGA-3′.
β-actin: Forward 5′-TGCGTGACATCAAAGAGAAG-3′ and Reverse 5′-GATGCCACAGGATTCCATA-3′.
As shown in Fig. (5a–A), expressions of FPN1’s and ferritin-H’s mRNA significantly increased in the Normal-CP group (P<0.01). When normalized to β-actin, FPN1 and ferritin-H mRNA levels after cisplatin treatment were 25- fold and 5- fold higher than that of saline treatment. It was consistent with the increase of FPN1 and ferritin-H protein measured by Western blotting assay Fig. (5a–B). As shown in Fig. (4d) there were higher splenic iron contents in cisplatin treated group (Normal-CP group) compared with saline treated normal mice group (Normal-saline group). This finding supported the hypothesis that high level of iron in spleen induced the increased expression of FPN1 and ferritin. Immunohistochemical analysis showed that expression of ferroportin 1 protein in the red pulp of spleen of the cisplatin treatment group (Normal-CP group) was higher than that of saline treatment group (Normal- Saline group). More FPN1 expression was found in the cytoplasm rather than on the plasma membrane of the cells Fig. (5b). Quantification of hepcidin mRNA expression in the spleen by Real-time PCR indicated that significantly more expression occurred in the cisplatin treated group (P<0.05) Fig. (5c). Hepcidin secreted by the spleen may be bound to the ferroportin on the membrane of macrophage presented in the red pulp of spleen. After binding, ferroportin was internalized, leading to decreased export of cellular irons. Ferritin appears to chelate intracellular free iron pool, competing with FPN1, a membrane iron exporter protein, and cause iron remaining in the cells.
Fig. 5.
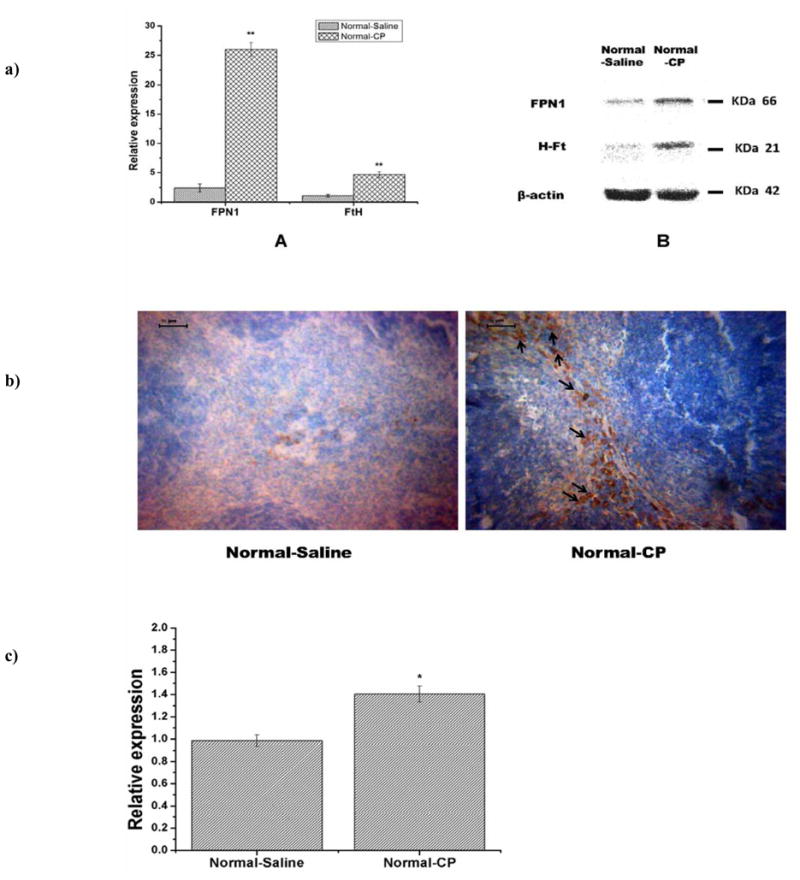
a). Expression of FPN1 and Ferritin-H were measured at transcriptional and translational levels. Expression of FPN1 and FtH increased significantly in spleen of normal mice with cisplatin treatment for 14 days. (A) Quantitative measurement of FPN1 and FtH mRNA expression. (B) Protein expression of FPN1 and H-Ft in spleen by Western blotting measurement. Expression values are normalized to β-actin and expressed as the mean ±standard deviation (n=3). **P<0.01. b) FPN1 expression in spleen tissue sections of normal mice measured by immunohistochemistry. There are more ferroportin1 expression in the red pulp of spleen in the Normal-CP group than that of Normal-Saline group. More FPN1 expression was found in the cytoplasm rather than on the plasma membrane of the cells (black arrow). Scale bar =50μm. c) Hepcidin mRNA expression in the spleen measured by Real-time PCR. There was more hepcidin mRNA expression in Normal-CP than that of Normal-Saline group by semi-quantification measurement. Data are means ±standard deviation (n=5), *P<0.05.
These results clearly demonstrate that hemosiderin deposition is modulated in response to erythrophagocytosis induced by cisplatin. High level of splenic iron content stimulate the FPN1 and ferritin up-regulations on both translation and transcription levels. The function of FPN1 is inhibited due to the direct competition from ferritin and its own internalization by hepcidin, leading to hemosiderin deposition as shown in the cartoon Fig. (6). These observations indicated the significant role of FPN1 in iron recycling by macrophages, and improved our understanding of how this process is regulated.
Fig. 6.

Hemosiderin accumulation in the splenic macrophage initiated by cisplatin. (Left), usually under normal condition, iron recycling is achieved through the phagocytosis of damaged or senescent erythrocytes. The irons released from erythrocyte was bound by ferritin and stored as ferric. Ferrous releasing from ferritin is exported by FPN1. (Right), under abnormal conditions with cisplatin treatment, cisplatin treatment may increase the amounts of damaged erythrocytes, which release more irons inside plasma. High concentrations of intracellular irons induce the overexpression of ferritin and FPN1 proteins. The function of FPN1 is inhibited due to its competition with ferritin. The deactivated FPN1 is then internalized with hepcidin and blocks the iron exporting out of cells. As a result, ferritin accumulates abnormally and aggregates into hemosiderin.
6. SUMMARY AND CONCLUSIONS
The main idea of this work was to investigate whether the low dose cisplatin can cure H22 ascites tumor and evaluate its toxicity effect on different organs. To explore the relationship between the iron metabolism in spleen and cisplatin treatment, a much lower intraperitoneal injection dose of cisplatin was chosen compared to the generally clinical dose in previous studies. The low dose of cisplatin can effectively inhibit tumor growth, which may provide a reference for clinical cancer therapeutic protocol. Interestingly, the specific hemosiderin deposition in the mice spleen was found by routine examination with H&E staining in short period (just 2 weeks) with cisplatin treatment. Other than spleen, there were no detectable damages observed in organs such as kidney, liver, heart, and lung treated with the low dose cisplatin, which suggested the high specificity of splenic injury with cisplatin treatment in vivo. This specific spleen toxicity may be due to the erythrocyte suicide triggered by cisplatin. The mice splenic iron content increased significantly after cisplatin treatment compared to the control group. It was found that ferroportin and ferritin in spleen were upregulated with cisplatin treatment. These results provide compelling evidence that cisplatin can induce the damage of erythrocytes, which leads to release irons in spleen. High levels of released irons up-regulated the ferroportin and ferritin expression. Increased hepcidin in spleen inhibited ferroportin to export iron. Several observations in this study could provide some guidance for studying iron metabolism in cisplatin treatment in vivo.
In conclusion, low dose of cisplatin administration can inhibit the development of ascites carcinoma and initiate hemosiderin specific deposition in spleen. Hemosiderin accumulation could be caused by the destruction of erythrocytes treated with cisplatin. Hepcidin overexpression might inhibit the function of FPN1 as an iron-exporter. Altered function of FPN1 and ferritin protein led to iron loading in the cells, which were shown with H&E and Perls’s measurements for iron staining. Cisplatin-caused hemosiderin deposition in spleen provides a valuable clue for our understanding the molecular basis of hemosiderin accumulation and iron metabolism in vivo.
Acknowledgments
The authors thank Chinese Natural Science Foundation project (No. 30970784 and 30870265), National Key Basic Research Program of China (2009CB930200), Chinese Academy of Sciences (CAS) “Hundred Talents Program” (07165111ZX), and China-Finland Bilateral Nanotechnology Collaboration (No. 2008DFA-01510) for financial supports.
Footnotes
Authors declare there are no any conflicts of interest that relate to this article.
References
- 1.Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 2.Shen D, Pastan I, Gottesman MM. Cross-resistance to methotrexate and metals in human cisplatin-resistant cell lines results from a pleiotropic defect in accumulation of these compounds associated with reduced plasma membrane binding proteins. Cancer Res. 1998;58(2):268–275. [PubMed] [Google Scholar]
- 3.Mavligit GM, Zukwiski AA, Ellis LM, Chuang VP, Wallace S. Gastrointestinal leiomyosarcoma metastatic to the liver. Durable tumor regression by hepatic chemoembolization infusion with cisplatin and vinblastine. Cancer. 1995;75(8):2083–2088. doi: 10.1002/1097-0142(19950415)75:8<2083::aid-cncr2820750809>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 4.Nogueira March JL, Ojea A, Jamardo D, Diez E, Figueiredo L. Evaluation of the effectiveness of cisplatin in the treatment of disseminated cancer of the prostate. Arch Esp Urol. 1985;38(1):28–34. [PubMed] [Google Scholar]
- 5.Liang XJ, Shen DW, Garfield S, Gottesman MM. Mislocalization of membrane proteins associated with multidrug resistance in cisplatin-resistant cancer cell lines. Cancer Res. 2003;63(18):5909–5916. [PubMed] [Google Scholar]
- 6.Liang XJ, Yin JJ, Taylor B, Winkovitch SM, Garfield SH, Shen DW, Gottesman MM, Aszalos A. Disruption of microfilaments by cytochalasin B decreases accumulation of cisplatin in human epidermal carcinoma and liver carcinoma cell lines. Cancer Chemother Pharmacol. 2008;62(6):977–984. doi: 10.1007/s00280-008-0687-9. [DOI] [PubMed] [Google Scholar]
- 7.Mahmud H, Foller M, Lang F. Suicidal erythrocyte death triggered by cisplatin. Toxicology. 2008;249(1):40–44. doi: 10.1016/j.tox.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Milicevic Z, Slepcevic V, Nikolic D, Zivanovic V, Milicevic NM. Effects of cis-diamminedichloroplatinum II (cisplatin) on the splenic tissue of rats: a histoquantitative study. Exp Mol Pathol. 1994;61(2):77–81. doi: 10.1006/exmp.1994.1027. [DOI] [PubMed] [Google Scholar]
- 9.Woo HY, Bae SH, Park JY, Han KH, Chun HJ, Choi BG, Im HU, Choi JY, Yoon SK, Cheong JY, Cho SW, Jang BK, Hwang JS, Kim SG, Kim YS, Seo YS, Yim HJ, Um SH Korean Liver Canc Study, G. A randomized comparative study of high-dose and low-dose hepatic arterial infusion chemotherapy for intractable, advanced hepatocellular carcinoma. Cancer Chemother Pharmacol. 2010;65(2):373–382. doi: 10.1007/s00280-009-1126-2. [DOI] [PubMed] [Google Scholar]
- 10.Havelka AM, Berndtsson M, Olofsson MH, Shoshan MC, Linder S. Mechanisms of action of DNA-damaging anticancer drugs in treatment of carcinomas: is acute apoptosis an “off-target” effect? Mini Rev Med Chem. 2007;7(10):1035–1039. doi: 10.2174/138955707782110196. [DOI] [PubMed] [Google Scholar]
- 11.Chirino YI, Pedraza-Chaverri J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp Toxicol Pathol. 2009;61(3):223–242. doi: 10.1016/j.etp.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Chirino YI, Sanchez-Gonzalez DJ, Martinez-Martinez CM, Cruz C, Pedraza-Chaverri J. Protective effects of apocynin against cisplatin-induced oxidative stress and nephrotoxicity. Toxicology. 2008;245(1–2):18–23. doi: 10.1016/j.tox.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Scott RH, Woods AJ, Lacey MJ, Fernando D, Crawford JH, Andrews PL. An electrophysiological investigation of the effects of cisplatin and the protective actions of dexamethasone on cultured dorsal root ganglion neurones from neonatal rats. Naunyn Schmiedebergs Arch Pharmacol. 1995;352(3):247–255. doi: 10.1007/BF00168554. [DOI] [PubMed] [Google Scholar]
- 14.Vandendries ER, Drews RE. Drug-associated disease: hematologic dysfunction. Crit Care Clin. 2006;22(2):347–355. viii. doi: 10.1016/j.ccc.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Wood PA, Hrushesky WJ. Cisplatin-associated anemia: an erythropoietin deficiency syndrome. J Clin Invest. 1995;95(4):1650–1659. doi: 10.1172/JCI117840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahman I, Biswas SK, Kirkham PA. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem Pharmacol. 2006;72(11):1439–1452. doi: 10.1016/j.bcp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Sung MJ, Kim DH, Jung YJ, Kang KP, Lee AS, Lee S, Kim W, Davaatseren M, Hwang JT, Kim HJ, Kim MS, Kwon DY, Park SK. Genistein protects the kidney from cisplatin-induced injury. Kidney Int. 2008;74(12):1538–1547. doi: 10.1038/ki.2008.409. [DOI] [PubMed] [Google Scholar]
- 18.Yin JJ, Lao F, Meng J, Fu PP, Zhao YL, Xing GM, Gao XY, Sun BY, Wang PC, Chen CY, Liang XJ. Inhibition of tumor growth by endohedral metallofullerenol nanoparticles optimized as reactive oxygen species scavenger. Mol Pharmacol. 2008;74(4):1132–1140. doi: 10.1124/mol.108.048348. [DOI] [PubMed] [Google Scholar]
- 19.Honma T, Hamasaki T. Association between hemosiderin deposition and blood vessel regression during involution of foreign-body granuloma. Histochemical and ultrastructural study. J Submicrosc Cytol Pathol. 2001;33(1–2):173–186. [PubMed] [Google Scholar]
- 20.Ishikawa-Sekigami T, Kaneko Y, Okazawa H, Tomizawa T, Okajo J, Saito Y, Okuzawa C, Sugawara-Yokoo M, Nishiyama U, Ohnishi H, Matozaki T, Nojima Y. SHPS-1 promotes the survival of circulating erythrocytes through inhibition of phagocytosis by splenic macrophages. Blood. 2006;107(1):341–348. doi: 10.1182/blood-2005-05-1896. [DOI] [PubMed] [Google Scholar]
- 21.Barlas N, Aydogan M. Histopathologic effects of maternal 4-tert-octylphenol exposure on liver, kidney and spleen of rats at adulthood. Arch Toxicol. 2009;83(4):341–349. doi: 10.1007/s00204-008-0354-2. [DOI] [PubMed] [Google Scholar]
- 22.Imaizumi T, Chiba M, Honma T, Niwa J. Detection of hemosiderin deposition by T2*-weighted MRI after subarachnoid hemorrhage. Stroke. 2003;34(7):1693–1698. doi: 10.1161/01.STR.0000075771.88719.CE. [DOI] [PubMed] [Google Scholar]
- 23.Zamboni P, Izzo M, Fogato L, Carandina S, Lanzara V. Urine hemosiderin: a novel marker to assess the severity of chronic venous disease. J Vasc Surg. 2003;37(1):132–136. doi: 10.1067/mva.2003.64. [DOI] [PubMed] [Google Scholar]
- 24.Gaunt SO, Baker DC. Hemosiderin in leukocytes of dogs with immune-mediated hemolytic anemia. Vet Clin Pathol. 1986;15(3):8–10. doi: 10.1111/j.1939-165x.1986.tb00643.x. [DOI] [PubMed] [Google Scholar]
- 25.Chang CS, Li CY, Liang YH, Cha SS. Clinical features and splenic pathologic changes in patients with autoimmune hemolytic anemia and congenital hemolytic anemia. Mayo Clin Proc. 1993;68(8):757–762. doi: 10.1016/s0025-6196(12)60633-8. [DOI] [PubMed] [Google Scholar]
- 26.Zuyderhoudt FM, Sindram JW, Marx JJ, Jorning GG, van Gool J. The amount of ferritin and hemosiderin in the livers of patients with iron-loading diseases. Hepatology. 1983;3(2):232–235. doi: 10.1002/hep.1840030216. [DOI] [PubMed] [Google Scholar]
- 27.Grebski E, Hess T, Hold G, Speich R, Russi E. Diagnostic value of hemosiderin-containing macrophages in bronchoalveolar lavage. Chest. 1992;102(6):1794–1799. doi: 10.1378/chest.102.6.1794. [DOI] [PubMed] [Google Scholar]
- 28.Epstein CE, Elidemir O, Colasurdo GN, Fan LL. Time course of hemosiderin production by alveolar macrophages in a murine model. Chest. 2001;120(6):2013–2020. doi: 10.1378/chest.120.6.2013. [DOI] [PubMed] [Google Scholar]
- 29.Iancu TC. Ferritin and hemosiderin in pathological tissues. Electron Microsc Rev. 1992;5(2):209–229. doi: 10.1016/0892-0354(92)90011-e. [DOI] [PubMed] [Google Scholar]
- 30.Ward RJ, Legssyer R, Henry C, Crichton RR. Does the haemosiderin iron core determine its potential for chelation and the development of iron-induced tissue damage? J Inorg Biochem. 2000;79(1–4):311–317. doi: 10.1016/s0162-0134(99)00237-8. [DOI] [PubMed] [Google Scholar]
- 31.Bovell E, Buckley CE, Chua-Anusorn W, Cookson D, Kirby N, Saunders M, St Pierre TG. Dietary iron-loaded rat liver haemosiderin and ferritin: in situ measurement of iron core nanoparticle size and cluster structure using anomalous small-angle x-ray scattering. Phys Med Biol. 2009;54(5):1209–1221. doi: 10.1088/0031-9155/54/5/007. [DOI] [PubMed] [Google Scholar]
- 32.Fujitani T, Tada Y, Yoneyama M. Chlorpropham-induced splenotoxicity and its recovery in rats. Food Chem Toxicol. 2004;42(9):1469–1477. doi: 10.1016/j.fct.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Masuda T, Satodate R, Tsuruga K, Kasai T. Quantitative assessment of a change of hemosiderin deposition with age in splenic compartments of rats. Tohoku J Exp Med. 1993;170(3):169–179. doi: 10.1620/tjem.170.169. [DOI] [PubMed] [Google Scholar]
- 34.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275(26):19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 35.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403(6771):776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 36.Gruenheid S, Pinner E, Desjardins M, Gros P. Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med. 1997;185(4):717–730. doi: 10.1084/jem.185.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277(42):39786–39791. doi: 10.1074/jbc.M201485200. [DOI] [PubMed] [Google Scholar]
- 38.Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102(12):4191–4197. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- 39.Theil EC. Ferritin: structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu Rev Biochem. 1987;56:289–315. doi: 10.1146/annurev.bi.56.070187.001445. [DOI] [PubMed] [Google Scholar]
- 40.Theil EC. Mining ferritin iron: 2 pathways. Blood. 2009;114(20):4325–4326. doi: 10.1182/blood-2009-08-239913. [DOI] [PubMed] [Google Scholar]
- 41.Turkmen N, Eren B, Fedakar R, Akgoz S. The significance of hemosiderin deposition in the lungs and organs of the mononucleated macrophage resorption system in infants and children. J Korean Med Sci. 2008;23(6):1020–1026. doi: 10.3346/jkms.2008.23.6.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D’Anna MC, Veuthey TV, Roque ME. Immunolocalization of ferroportin in healthy and anemic mice. J Histochem Cytochem. 2009;57(1):9–16. doi: 10.1369/jhc.2008.951616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102(3):783–788. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- 44.Liu XB, Nguyen NB, Marquess KD, Yang F, Haile DJ. Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis. 2005;35(1):47–56. doi: 10.1016/j.bcmd.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 45.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]