

Abstract
The c-Jun N-terminal kinase (JNK) pathway potentially links together the three major pathological hallmarks of Alzheimer’s disease (AD): development of amyloid plaques, neurofibrillary tangles, and brain atrophy. As activation of the JNK pathway has been observed in amyloid models of AD in association with peri-plaque regions and neuritic dystrophy, as we confirm here for Tg2576/PSM146L transgenic mice, we directly tested whether JNK inhibition could provide neuroprotection in a novel brain slice model for amyloid precursor protein (APP)-induced neurodegeneration. We found that APP/amyloid β (Aβ)-induced neurodegeneration is blocked by both small molecule and peptide inhibitors of JNK, and provide evidence that this neuroprotection occurs downstream of APP/Aβ production and processing. Our findings demonstrate that Aβ can induce neurodegeneration, at least in part, through the JNK pathway and suggest that inhibition of JNK may be of therapeutic utility in the treatment of AD.
Keywords: JNK, APP, neurodegeneration, neuroprotection, biolistics, brain slice, drug discovery
Introduction
Multiple and distinct pathologies characterize Alzheimer’s disease (AD), including deposition of amyloid plaques, formation of neurofibrillary tangles and neurodegeneration (Huang and Jiang, 2009). Development of AD therapeutics has largely attempted to address these pathologies separately, but approaches that influence multiple pathologies may be advantageous. One pathway that potentially links all of the pathological hallmarks of AD is that through the c-jun N-terminal kinases (JNKs). The activation of JNKs leads to the phosphorylation of transcription factors controlling the apoptotic process, (Dhanasekaran and Reddy, 2008) thereby resulting in cell death in multiple neurodegenerative disorders (Bogoyevitch et al., 2004). Furthermore, the JNK pathway can be activated by amyloid β (Aβ) peptides (Morishima et al., 2001) and has also been reported to regulate the phosphorylation of amyloid precursor protein (APP) leading to modulation of Aβ levels (Colombo et al., 2009; Colombo et al., 2007). Finally, there is strong evidence that JNKs can phosphorylate tau in vitro (Yoshida et al., 2004).
The JNK pathway has been shown to be active in preclinical models of AD, including Tg2576 and Tg2576/PS1P264L transgenic mice by biochemical and immunohistochemical analyses (Flood et al., 2002; Puig et al., 2004). However, to date, there have been no reports on manipulation of the JNK pathway being directly tested in a model of AD to ask whether JNK activation may contribute to disease pathogenesis, and whether its inhibition may have therapeutic potential.
Thus, in the present study, we first used a transgenic animal model of AD, with mutations in presenilin and APP resulting in excessive Aβ generation and amyloid plaque formation, to examine the potential association of JNK activation with amyloid histopathogenesis. We then directly examined whether inhibition of JNK pathways could provide benefit in a novel AD model system in which particle-mediated gene transfer, or biolistics, is used to present an acute challenge of the amyloid cascade to ex vivo brain slice explants. This brain slice model has the ability to maintain the complex interplay among different resident cell types and their local connectivity, while retaining the ability to further investigate and manipulate the JNK pathways in the context of APP-induced neurodegeneration. Together, our findings from these in vivo and ex vivo models link amyloid, tau and neurodegenerative pathologies through the JNK pathway, and suggest that inhibition of JNK activity could provide therapeutic benefit in the context of AD.
Materials & Methods
Antibodies & Chemicals
Antibodies against amyloid β (6E10) and pan-axonal neurofilament marker (SMI-312) were purchased from Covance (Princeton, NJ), anti-phosphorylated JNK from Cell Signaling Technology (Danvers, MA), and anti-BACE1 (PA1-575) from Affinity Bioreagents (Rockford, IL). WYGSI-04 (Pu et al., 2009) was synthesized at Wyeth, SP600125 was purchased from Calbiochem (San Diego, CA), and JNK inhibitory peptides (L-JNKi1) and control peptides were purchased from EMD Chemicals (Gibbstown, NJ).
Immunofluorescence labeling
Animal protocols were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and approved by Wyeth’s Institutional Animal Care and Use Committee (IACUC). Mice were transcardially perfused with 4% paraformaldehyde, and brains extracted and then cryoprotected in 30% sucrose/TBS at 4° C for 2–3 days before sectioning. Coronal sections (30 μm thickness) were cut from frozen, fixed brains using a sliding microtome (Microm, Walldorf, Germany). Free floating sections were treated with 0.5% Triton X-100 in TBS at 4° C for 45 min, then blocked with normal goat serum (Vector Labs, Burlingame, CA) at 4° C for 45 min. For mouse primary monoclonal antibodies MOM reagent (Vector Labs, Burlingame, CA) was added to reduce binding to endogenous mouse immunoglobulins. Sections were incubated with the indicated primary antibodies at 4° C overnight. Following 5 extensive washes, sections were incubated with appropriate fluorescent-labeled secondary antibodies (Invitrogen, Carlsbad, CA) at 4° C for 48 hrs. After 5 additional washes, sections were mounted onto glass slides in Prolong Gold (Invitrogen).
Brain slice preparation and biolistic transfection
Brain slices were prepared from postnatal day 10 (P10) CD Sprague-Dawley rats (Charles River, Wilmington, MA). Rat pups were sacrificed in accordance with NIH guidelines and under Duke IACUC approval and oversight. 250 μm thick coronal sections from the middle third of the brain were cut using a Vibratome (Vibratome Company, St Louis, MO) and placed in long-term tissue culture medium as previously described (Wang et al., 2006; Yacoubian and Lo, 2000) and incubated under 5% CO2 in humidified chambers. A custom-modified biolistic device (Helios Gene Gun, BioRad, Hercules, CA) was used for particle-mediated transfection immediately after slicing. 1.6 μm elemental gold particles were used, upon which the desired DNA plasmids were precipitated as per manufacturer’s instructions and previously published detailed protocols (Lo, 1999; Wang et al., 2006). Compounds were added to the culture medium at the concentrations indicated and incubated for 2–3 days before biochemical and neurodegeneration outcome measures were made.
Expression constructs
APPWT and APPSw expression constructs were made in-house based on human neural APP sequences (695 aa form) and subcloned behind the CMV promoter/enhancer in the Gwiz expression vector (Genlantis, San Diego, CA). The YFP visual reporter was also subcloned into the Gwiz vector, which we have previously shown to yield sustained expression for >1 week in numerous brain regions and cell types including cortical pyramidal neurons (Southwell et al., 2008; Varma et al., 2007b; Wang et al., 2006).
Neuronal health and statistics
For the assessment of numbers of healthy cortical pyramidal neurons, brain slices were imaged on Leica MZIIIFL fluorescence stereomicroscopes using appropriate filter sets. Pyramidal neurons were readily discerned and unambiguously identified based on their characteristic positioning and orientation within the cortex, and on their striking morphological features, most notably the extension of a single, prominent apical dendrite radially towards the pial surface. These key features of pyramidal neuronal identity were used to determine the numbers of healthy, non-degenerating cortical pyramidal neurons at various times after slice preparation and APP transfection, namely 1) a robust and brightly labeled cell body positioned within the pyramidal neuronal layers of the cortex; 2) the retention of a clear apical dendrite extending radially towards the pial surface the slice; 3) the extension directly from the cell soma of >2 clear basal dendrites >2 cell body diameters long; and 4) clear and continuous cytoplasmic labeling with the YFP visual marker in the cell soma as well in all dendrites and the axon. Numbers of such non-degenerating cortical pyramidal neurons are expressed as raw average numbers per cortical region of each brain slice in Fig. 3, or as a percentage of the number of non-degenerating neurons scored for the positive control condition internal to the run, as is shown for the remainder of the figures. ANOVA followed by Dunnett’s post hoc comparison test at the 0.05 confidence level was used for assessing statistical significance with N=12 brain slices per condition. Each experiment was carried out at least 3 times.
Figure 3. Acute challenge with APP isoforms leads to its proteolytic processing into Aβ and the induction of neuronal degeneration.

(A) The co-transfected YFP fluorescent marker clearly delineates neuronal cell bodies and dendrites after 2–3 d in culture, shown in the left panel for a region in cortex (pia surface up). It can be seen that the majority of neurons transfected are cortical pyramidal neurons, easily distinguished by their extension of a single, prominent apical dendrite in the radial direction. Right panel shows overt neurodegeneration of such cortical pyramidal neurons 2–3 days after co-transfection with APP isoforms such as APPSw. (B) Expression of APP is readily detected in transfected brain slices via immunoprecipitation and immunoblotting with the human-specific APP antibody 6E10. Processing of APP into C99 and Aβ by native tissue proteases can also be seen, with higher levels of C99 and Aβ production as expected for APPSw relative to APPWT. (C) Induction of neurodegeneration by transfected APP is more severe with the Swedish mutation. Ordinate axis shows average, total numbers ± SEM of non-degenerating pyramidal neurons in the cortical regions of each explant, N=12 brain slices scored per condition. *, significant by ANOVA followed by Dunnett’s post hoc comparison test at the 0.05 confidence level.
Western blot analysis
APP-transfected cortical brain slices were washed 3 times with PBS on ice and homogenized by trituration through a 25 g needle in homogenization buffer (1% NP-40 in 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 2 mM EDTA) supplemented with Complete Protease Inhibitor Cocktail (Roche). Samples were cleared twice by centrifugation at 3000 g. For immunoprecipitation (IP), 6E10 antibodies were added to the samples and incubated overnight at 4° C followed by incubation with Protein G Sepharose (Sigma). The Protein G Sepharose immunocomplex was washed 5 times in homogenization buffer, and eluted in 0.15 mM glycine HCl, pH 2.5–3.0. For Western blot analysis, eluates were neutralized to physiological pH, and proteins separated on 10–20% Tricine gels (Invitrogen). Following transfer to PVDF membranes (0.2 μm pore size, Bio-Rad), Western blots were done using standard methods with 6E10 or BACE1 antibodies and an HRP-conjugated secondary IgG antibody with ECL visualization (Amersham) and documentation on a Bio-Rad VersaDoc MP5000.
Results
JNK activation in Tg2576/PS1M146L mice co-localizes with amyloid plaques and markers of neurodegeneration
To determine whether activation of the JNK pathway may be important for neurodegeneration in Alzheimer’s disease, and whether inhibition of the JNK pathway could be considered as a therapeutic approach, we sought to study a transgenic animal model with overt amyloid pathology. The Tg2576/PS1M146L mouse carries the APP Swedish mutation (KM670/671NL) and the presenilin mutation M146L, leading to highly elevated Aβ levels (Holcomb et al., 1998). In this mouse model, extensive amyloid plaque formation was observed at 4 months and progressively increased with age of the animals, whereas in wild-type animals no plaque deposition was detectable (Suppl. Fig. 1). We found that immunostaining for dual-phosphorylated JNK at Thr183 and Tyr185, an indicator of activated JNK, was associated with the periphery of amyloid plaques (Fig. 1A, B). Some of the smaller amyloid plaques were not associated with pJNK immunoreactivity, suggesting that activation of JNK may be subsequent to amyloid deposition. pJNK was also associated with hyperphosphorylated tau in these plaques as indicated by AT8 antibody staining, suggesting that JNK may contribute to tau phosphorylation in vivo (Fig. 1C). Importantly, we found pJNK colocalized with dystrophic neurons that surrounded plaques, which strongly suggests that JNK may play a role in Alzheimer-related neurodegeneration (Fig. 1D). These findings support and extend those previously reported in the similar Tg2576/PS1P264L double-transgenic line (Holcomb et al., 1998; Savage et al., 2002).
Figure 1. JNK is activated in dystrophic neurons in the peri-plaque region in Tg2576/PS1M146L mice.
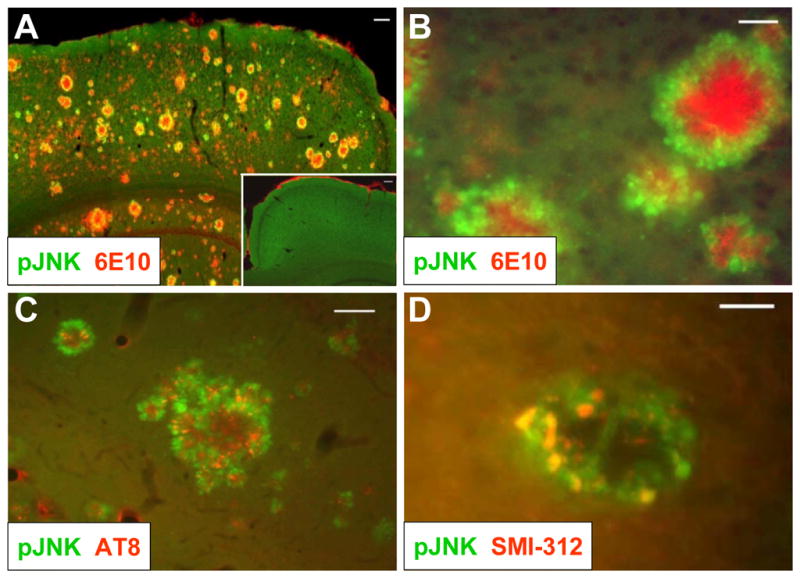
(A) 12-month old Tg2576/PS1M146L transgenic mice show strong 6E10 immunoreactivity indicative of Aβ deposition (red) and increased JNK phosphorylation (green) in the cortex compared to wild-type mice (inset). Scale bar, 100 μm. (B) At higher magnification, phosphorylated JNK (green) is seen to surround amyloid plaques (red) in cortex. Scale bar, 20 μm. (C) JNK phosphorylation (green) partially overlaps with immunoreactivity with the AT8 antibody, indicative of hyperphosphorylated tau. Scale bar, 50 μm. (D) JNK phosphorylation (green) also coincides with SMI-312 immunoreactivity, indicative of dystrophic neurons. Scale bar, 20 μm. All images are representative of observations from at least 3 animals.
Together, these findings indicate that significant levels of JNK activation are associated with amyloid plaques and dystrophic neurons in the Tg2576/PS1M146L AD mouse model. The critical question is thus whether inhibition of JNK would prevent or ameliorate AD symptoms in these model mice. However, currently available tool compounds, such as SP600125 and D-JNKi, are not sufficiently selective, brain penetrant, and/or stable for chronic in vivo studies. Thus, we developed an alternative brain tissue-based model system in which neurodegeneration is directly driven by APP/Aβ to ask directly whether such neurodegeneration can be ameliorated by JNK inhibition.
Development of a brain slice-based acute neurodegenerative AD model
Brain slice explants in organotypic culture have proven to be a valuable tool in drug discovery for identifying and evaluating potentially neuroprotective drugs and drug target candidates for a range of neurological disease including stroke, Huntington’s disease, and ALS (Cho et al., 2007). Such brain slice assays have offered an effective compromise between whole-animal experiments and cell line and primary culture-based assays and, importantly, retain the 3-dimensional milieu and local intercellular interactions of brain tissue that are intimately involved in disease pathogenesis in vivo.
In this brain slice AD model, we used biolistics to transfect human APP isoforms including wild-type (APPWT) and APP-Swedish (APPSw) into cortical pyramidal neurons, resulting in progressive neurodegeneration of these neurons over 3–5 days as visualized and quantified by co-transfection with a fluorescent protein reporter (Fig. 2A). As the co-transfection linkage rate of biolistics is >99%, this ensures that essentially all neurons expressing the fluorescent marker, such as yellow fluorescent protein (YFP), also express the APP construct of interest (Fig. 2B). Brain slices transfected with YFP alone after 2–3 days in culture exhibited cortical pyramidal neurons with a healthy morphology with long, intact, YFP-filled dendrites (Fig. 3A, left). In contrast, cortical neurons in brain slices transfected with APP isoforms appeared progressively less healthy over time, exhibiting discontinuous dendrites and collapsing cell soma (Fig. 3A, right).
Figure 2. Biolistic transfection is used to introduce APP isoforms into organotypic brain slice explants.

(A) 1.6 μm elemental gold particles coated with YFP and APP isoform DNA expression plasmids are accelerated with a gene gun to transfect P10 rat coronal brain slice explants, resulting in even transfection of resident neuronal cell types in the explants (lower right photomicrograph), including pyramidal neurons in the cortex (white boxed region). (B) Co-transfection linkage rate using biolistics is >99%. Co-coating gold particles with 3 different DNA plasmids each encoding a different fluorescent protein (YFP, CFP, and DsRed) leads to clear co-expression in essentially all transfected neurons shown here in a magnified portion of the cortex (merge at lower right).
Expression of these APP isoforms in transfected brain slices was readily detected by Western blot analysis using human-specific antibodies, and proteolytic processing of full-length APP into the C99 intermediate and Aβ peptides could be clearly observed (Fig. 3B). Such as acute challenge of APP isoforms to these wild-type rat brain slices was sufficient to generate significant neurodegeneration even in the case of APPWT (Fig. 3C). Transfection of APPSw induced increased levels of production of both C99 and Aβ as a proportion of total APP compared to APPWT, and was concomitant with increased levels/rates of neurodegeneration of transfected cortical neurons (Fig. 3B, C).
Inhibition of APP processing reduces Aβ levels and neurodegeneration
To demonstrate that APP processing into toxic Aβ species is necessary to induce neurodegeneration, we first tested the effects of inhibiting APP processing by genetic perturbation of its cleavage by γ-secretase using the APPK624S mutation which has been shown to completely prevent processing of APP into Aβ (Ren et al., 2007). As expected, the K624S mutation reduced Aβ levels to below detection limits in immunoblots but did not affect expression levels of full-length APP (Fig. 4A). Correspondingly, transfection of APPK624S did not induce significant levels of neurodegeneration compared to non-APP controls (Fig. 4B).
Figure 4. Inhibition of γ-secretase cleavage inhibits Aβ production and is neuroprotective.

(A) The APPK624S mutation inhibits γ-secretase cleavage of APP and decreases Aβ production to undetectable levels in 6E10 immunoblots of brain slice lysates, but does not significantly affect levels of expression of full-length APP. (B) Correspondingly, transfection with the APPK624S mutation does not result in measurable levels of neurodegeneration. (C) Treatment of transfected brain slices with the γ-secretase inhibitor WYGSI-04 (“GSI”) for 2 days resulted in selective, dose-dependent inhibition of Aβ production. Such immunoblot analysis showed that WYGSI-04 treatment had no effect on the expression of full-length APP, or on its initial cleavage by endogenous β-secretases to the intermediate fragment C99. Lanes were loaded with identical amounts of total protein from brain slice lysates, with equivalent transfection rates confirmed by direct immunoblotting against the co-transfected marker YFP (bottom band). “--” denotes treatment with the DMSO vehicle only. (D) Correspondingly, inhibition of Aβ production by WYGSI-04 provides dose-dependent neuroprotection in the same concentration range. Ordinate axes in (B) and (D) show average, total numbers ± SEM of non-degenerating pyramidal neurons in the cortical regions of each explant expressed as a percentage of control brain slices transfected with YFP only. *, significant by ANOVA followed by Dunnett’s post hoc comparison test at the 0.05 confidence level.
As this mutation may also cause other changes in APP processing, we confirmed these findings with direct inhibition of endogenous γ-secretases with a selective small molecule inhibitor. By Western blotting it is apparent that that the γ-secretase inhibitor WYGSI-04 selectively inhibits Aβ production in a dose-dependent manner with no effect on levels of either full-length APP or C99 (Fig. 4C). WYGSI-04 also provided inhibition of APP-induced cortical neurodegeneration in the same dose range (Fig. 4D). To confirm that the neuroprotection provided by WYGSI-04 was via its inhibition of Aβ production, we showed that this γ-secretase inhibitor had no general effects on the survival of control brain slices transfected with YFP only (Suppl. Fig. 2). Together, these genetic and pharmacological data show that expression of human APP in brain slices induces neurodegeneration requiring normal APP processing via γ–secretase to generate Aβ, supporting the use of this model system in investigating the potential benefits of intervening in signaling pathways leading to, as well as downstream of, Aβ production.
JNK inhibition reduces APP-induced neurodegeneration in brain slices
To determine the functional importance of the JNK pathway for APP-induced neurodegeneration in this AD model, cortical brain slices were first transfected with APP and incubated in the presence of varying concentrations of cell-permeable, biologically active peptides (JNKi) that specifically inhibit JNK activity by interfering with the interaction between JNKs and JNK interacting proteins (JIPs) and thus preventing JNK dual-phosphorylation (Barr et al., 2002; Borsello et al., 2003). The JNK inhibitor peptide fused to the N-terminal portion of HIV-TAT to confer cell permeability, but not a control peptide consisting of the HIV-TAT N-terminus alone, showed dose-dependent inhibition of APP-induced neurodegeneration (Fig. 5A).
Figure 5. Inhibition of JNK activity reduces neurodegeneration induced by APP/Aβ.

(A) Treatment of APP-transfected brain slices with the cell-permeable JNK inhibitory peptide JNKi (“JNKi pep”) protected against APP/Aβ-induced neurodegeneration in a dose-dependent manner, while a control peptide containing only the HIV-TAT N-terminal domain (“Cont pep”) had no effect. (B) The small molecule JNK inhibitor SP600125 also provided dose-dependent rescue of neurodegeneration induced by APP transfection. Ordinate axes show average, total numbers ± SEM of non-degenerating pyramidal neurons in the cortical regions of each explant expressed as a percentage of control brain slices transfected with YFP only and treated as indicated. *, significant differences by ANOVA followed by Dunnett’s post hoc comparison test at the 0.05 confidence level.
As independent confirmation of these findings, we next tested the small molecule JNK inhibitor SP600125 (Bennett et al., 2001). We found that SP600125 also provided dose-dependent rescue of the neurodegeneration induced by APP transfection (Fig. 5B). This neuroprotection appeared to be specific for the neurodegeneration induced by APP transfection and production of Aβ, as SP600125 did not provide any neuroprotection to control brain slices transfected with YFP only (Suppl. Fig. 3) as is observed with generally protective agents such as the pan-caspase inhibitor BOC-D-FMK.
The neuroprotective effect provided by JNK inhibition could either be through affecting APP processing, and hence levels of Aβ (Colombo et al., 2009; Colombo et al., 2007; Quiroz-Baez et al., 2009; Shen et al., 2008; Tamagno et al., 2009), or downstream of Aβ in apoptotic and/or other neurodegenerative pathways (Morishima et al., 2001). To address this, we investigated the processing of APP in brain slices treated with JNK inhibitors using Western blot analysis. We found no significant differences in the relative or absolute levels of Aβ generated in brain slices treated with the same dose ranges of SP600125 or with the JNKi inhibitory peptide (Fig. 6A, B). These findings suggest that BACE activity in the brain slice explants was not affected, and in fact Western blot analysis showed no changes in levels of BACE1 expression as a result of JNK inhibition (Suppl. Fig. 4). Furthermore, that JNK inhibition did not affect steady-state levels of C99 (Fig 6A) implies that γ-secretase activity was also unaffected. Together, these results indicate that the neuroprotective effects of JNK inhibition in this model likely interceded at a signaling pathway node(s) downstream of Aβ production (Fig. 6C).
Figure 6. JNK inhibition does not affect the production of Aβ.

Incubation of transfected brain slices with concentrations of the JNK inhibitor SP600125 (A) or JNKi peptide (B) across the ranges that provided neuroprotection showed no changes in expression levels of full-length APP, C99, or Aβ. Lanes were loaded with identical amounts of total protein from brain slice lysates, with equivalent transfection rates confirmed by direct immunoblot against the co-transfected marker YFP (bottom band). “--” denotes treatment with the DMSO vehicle only. (C) Together, these findings suggest that the primary site of action of JNK inhibition in this brain slice model for APP/Aβ-induced neurodegeneration lies downstream of the production of Aβ. However, other studies have suggested that JNK may also play roles in the production of Aβ itself (Colombo et al., 2009).
Discussion
Neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and stroke are typically late-onset conditions with multiple potential causes, pathological hallmarks and dysregulation of multiple pathways. Therefore, across a spectrum of neurodegenerative disorders there is increasing appreciation that targeting a single mechanism may not be sufficient to halt the progression of neurodegeneration and provide disease modification. Most notably, development of therapeutics against stroke has produced great success in preclinical models, but remarkable levels of failure in the clinic, perhaps because the complexity of the human disorder has not been fully addressed (Fisher et al., 2009).
Therapeutics for AD currently provide symptomatic relief, with the next frontier being development of therapeutics that are disease modifying (Salloway et al., 2008). The majority of efforts to this end is focused on specific pathologies; most advanced are efforts around Aβ. Mechanisms to prevent the generation of Aβ peptides through inhibition of enzymes involved in Aβ synthesis, clearance or catabolism (Vardy et al., 2005) are being widely tested. Similarly targeted approaches around tau (Schneider and Mandelkow, 2008) are also being evaluated. Considering the multiple pathological features of Alzheimer’s disease, however, approaches with broader scope than only a single mechanism may also be considered. Such approaches include broader-based processes such as reducing neuroinflammation, neurovascular modulation, and promoting neurogenesis (Sagisaka et al., 2010; Walker and Lue, 2007; Zlokovic, 2008). Despite compelling data on such mechanisms they are often difficult to selectively target for development of inhibitors as they are mediated by multiple receptors, proteins and signaling pathways, and have effects outside of the pathological state being targeted. Therefore, a defined mechanism, through a single target protein, that impacts multiple pathologies relevant to AD is desirable. One such candidate protein is JNK, which has been implicated in apoptotic cell death (Dhanasekaran and Reddy, 2008), tau phosphorylation (Reynolds et al., 1997), as well as amyloid pathology (Colombo et al., 2009; Flood et al., 2002) in a variety of in vitro, cellular and in vivo models.
In this context, we used a novel, brain slice-based AD model, in which acute challenge with APP transfection is used to initiate neuronal degeneration, to evaluate directly whether inhibition of JNK pathways can provide neuroprotective benefit. This tissue-based model for APP/Aβ-induced neurodegeneration was critical for these studies because it has been well-established that no significant neurodegeneration can be found in mouse transgenic models of amyloidosis. It is unclear why the condition in humans exhibits overt neurodegeneration whilst in murine models of amyloidosis little is observed. Perhaps differences in murine genetics, for example the difference in tau isoforms present (Himmler et al., 1989; Lee et al., 1988) or other proteins only present in mouse may lead to differential vulnerabilities. Alternatively, the long progression of AD in human, not replicable in mouse, may underlie these differences and restrict our observations to only certain aspects of AD pathologies in murine models.
To overcome this limitation, the brain slice model we developed was designed to recapitulate acute neurodegenerative effects of APP/Aβ in an experimental setting as close to the in vivo situation as possible. The goal was to achieve a rapid, high level expression of APP and its processing to Aβ that we could employ to activate, as well as follow, downstream signaling events leading to over neurodegeneration. In vivo methodologies would include the use of viral transduction, or inducible transgenic models; however, both of these approaches are limited in the rapidity and levels of expression that can be achieved. We have previously used particle-mediated gene transfer to introduce mutant huntingtin constructs into corticostriatal brain slice explants, inducing a striatal neurodegenerative phenotype that could be used for both mechanistic as well as drug discovery studies (Crittenden et al., 2010; Khoshnan et al., 2004; Southwell et al., 2008; Thompson et al., 2009; Varma et al., 2007a). The advantage of this system is that it allows the rapid delivery and expression of a disease gene of interest and its processing by endogenous mechanisms in the complex, native cell- and tissue-context.
We have shown here that an AD-relevant brain slice model can also be constructed using this approach, in which exogenous human APP is processed by endogenous tissue mechanisms to generate Aβ in a manner dependent on its cleavage by β- and γ-secretases. Importantly, the Aβ generated in this model induces clear neurodegeneration in contrast to in vivo mouse APP transgenic models. This may be due to the rapid nature of biolistic transfection, or to the acute nature of such transfection in an otherwise WT genetic background, precluding compensatory pathways that may counteract the toxic effects of the Aβ in in vivo models. In addition, the triggering of cell-based inflammatory responses in the brain slice preparation may provide an additional “second hit” that may enhance neurodegeneration.
To evaluate the impact of JNK inhibition on APP-induced neurodegeneration in this brain slice model, we used pharmacological intervention with two different compound classes that inhibit JNK by distinct mechanisms: SP600125, a competitive inhibitor of JNK kinase activity (Bennett et al., 2001), and JNKi, a peptidic inhibitor of the interaction between JNK and its interacting protein partners necessary for activity (Barr et al., 2002; Borsello et al., 2003). Both mechanisms of JNK inhibition provided nearly complete rescue of APP-mediated neurodegeneration. These results are consistent with and extend previous demonstration of JNK-mediated, neuronal apoptosis (Morishima et al., 2001) induced by applying the Aβ25–35 fragment to cultured neurons. Other studies have demonstrated involvement of the JNK pathway in regulation of the enzymes involved in APP processing in response to oxidative stress (Quiroz-Baez et al., 2009; Shen et al., 2008; Tamagno et al., 2009). In the brain slice model used here, we found no evidence of effects of JNK inhibition on either γ-secretase or BACE levels or activities. These differences may reflect the existence of two independent JNK-mediated pathways regulating Aβ production and/or its phenotypic effects: one upstream of APP processing, prominent in some models, that mediates regulation of APP processing by oxidative stress; the other downstream of APP processing, not affecting the production of Aβ per se, but rather providing direct protection against Aβ-induced neurodegeneration as we find here in the brain slice model.
To confirm that JNK activation is also relevant in a chronic model of APP-induced amyloidogenesis, we assessed how JNK is activated in an animal model of AD as has previously been investigated in the Tg2576/PS1P264L mouse (Savage et al., 2002). Here we used the Tg2576/PS1M146L mouse, differing in that it expresses the more common M146L mutation as opposed to the knock-in of the P264L mutation. Strikingly, we found that every amyloid plaque was surrounded by phosphorylated JNK, indicative of a very strong inductive influence of Aβ on this pathway. Intriguingly, we found that there was significant co-localization of phosphorylated JNK with a marker of dystrophic neurites. Together, these data support a specific role of JNK activation in the context of processes induced by APP leading to neurodegeneration and cell death.
There are several possible mechanisms by which JNK activation could be involved in such neurodegenerative processes, and by which its inhibition could provide neuroprotection. Recently, there have been reports that JNK affects the phosphorylation of APP, thereby modulating Aβ levels, and notably of its oligomeric forms (Colombo et al., 2009; Colombo et al., 2007). In our studies, however, we did not observe changes in APP processing by JNK inhibition, leading to the conclusion that the neuroprotection we observed in the brain slice model was not due to an overall reduction in levels of Aβ, but rather to the inhibition of classical JNK-mediated apoptotic pathways (Dhanasekaran and Reddy, 2008). The disparities between these findings may reflect the difference in experimental systems used; the study of Colombo et al. (2009) used APPSw transformed into the H4 cell line whereas native rat cortical brain tissue was used in the present study.
Collectively, our studies, along with those demonstrating JNK’s role in tau phosphorylation and regulation of Aβ levels, indicate that JNK inhibitors may be of therapeutic utility for AD as they may affect all of the major hallmarks of the disorder. Interestingly, the JNK3 isoform is enriched in the CNS (Kumagae et al., 1999) and may therefore provide a more selective target for AD. Furthermore, the JNK3 isoform appears to be of particular importance as its knockout is highly protective in multiple models of neurodegeneration (Brecht et al., 2005; Yang et al., 1997) and it can undergo significant autophosphorylation and thereby potentiate its own activity (Vogel et al., 2009). To take advantage of this, inhibitors will need to be developed that are both CNS-penetrant and isoform-specific, properties that neither SP600125 nor JNK inhibitory peptides possess. With such compounds, examination of the chronic pharmacological inhibition of JNKs in transgenic animal models of AD will be feasible to determine whether inhibition of this pathway can provide disease modification.
Supplementary Material
(A) Immunohistochemical analysis of amyloid plaques in coronal sections of brains from Tg2576/PS1M146L mice using Anti-β-Amyloid 40 antibody (Biosource, Carlsbad, CA) demonstrates an increase in numbers of amyloid plaques with age. Images are representative of at least 3 animals per time point. (B) Wild-type littermate controls show no amyloid plaques at 12 months of age. (C) Automated quantification (InterFocus Imaging, Linton, UK) of plaque number in Tg2576/PS1M146L mice shows strong age-dependent increase in plaque burden with age.
In contrast to the dose-dependent neuroprotective effect of WYGSI-04 for brain slice explants transfected with APP, WYGSI-04 (“GSI”) had no measurable effects on control brain slice explants transfected with YFP only, as shown here for the highest concentration of WYGSI-04 tested. Ordinate axis shows average, total numbers ± SD of non-degenerating pyramidal neurons in the cortical regions of each explant expressed as a percentage of control brain slices treated with the DMSO carrier only.
In contrast to the dose-dependent neuroprotection provided by SP600125 to APP-transfected brain slices, JNK inhibition by SP600125 had no measurable effects on control brain slice explants transfected with YFP only, as shown here for the same concentration range tested. Ordinate axis shows average, total numbers ± SEM of non-degenerating pyramidal neurons in the cortical regions of each explant expressed as a percentage of control brain slices treated with the DMSO carrier only.
JNK inhibition by SP600125 did not significantly affect levels of BACE1 expression in brain slice explants in the same concentration range that provided protection against APP-induced neurodegeneration (Fig. 5B), consistent with such treatment not affecting levels of Aβ production as shown in Fig. 6A. Lanes were loaded with equivalent amounts of total protein from brain slice lysates; “--” denotes treatment with the DMSO vehicle only.
Acknowledgments
This work was in part funded by NIH grant NS048181.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barr RK, et al. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–97. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- Bennett BL, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–6. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Brecht S, et al. Specific pathophysiological functions of JNK isoforms in the brain. Eur J Neurosci. 2005;21:363–77. doi: 10.1111/j.1460-9568.2005.03857.x. [DOI] [PubMed] [Google Scholar]
- Cho S, et al. Brain Slices as Models for Neurodegenerative Disease and Screening Platforms to Identify Novel Therapeutics. Current Neuropharmacology. 2007;5:19–33. doi: 10.2174/157015907780077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo A, et al. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol Dis. 2009;33:518–25. doi: 10.1016/j.nbd.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Colombo A, et al. The TAT-JNK inhibitor peptide interferes with beta amyloid protein stability. Cell Death Differ. 2007;14:1845–8. doi: 10.1038/sj.cdd.4402202. [DOI] [PubMed] [Google Scholar]
- Crittenden JR, et al. CalDAG-GEFI down-regulation in the striatum as a neuroprotective change in Huntington’s disease. Human Molecular Genetics. 2010;19:1756–1765. doi: 10.1093/hmg/ddq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–51. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, et al. Update of the Stroke Therapy Academic Industry Roundtable Preclinical Recommendations. Stroke. 2009 doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood DG, et al. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23:335–48. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- Himmler A, et al. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–8. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Huang HC, Jiang ZF. Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: relationship and links in Alzheimer’s disease. J Alzheimers Dis. 2009;16:15–27. doi: 10.3233/JAD-2009-0960. [DOI] [PubMed] [Google Scholar]
- Khoshnan A, et al. Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity. Journal of Neuroscience. 2004;24:7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagae Y, et al. Human c-Jun N-terminal kinase expression and activation in the nervous system. Brain Res Mol Brain Res. 1999;67:10–7. doi: 10.1016/s0169-328x(99)00013-3. [DOI] [PubMed] [Google Scholar]
- Lee G, et al. The primary structure and heterogeneity of tau protein from mouse brain. Science. 1988;239:285–8. doi: 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- Lo DC. Neuronal transfection using particle-mediated gene transfer. Current Protocols in Neuroscience. 1999;3:1–12. doi: 10.1002/0471142301.ns0315s05. [DOI] [PubMed] [Google Scholar]
- Morishima Y, et al. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–60. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J, et al. Synthesis and structure-activity relationship of a novel series of heterocyclic sulfonamide gamma-secretase inhibitors. Bioorg Med Chem. 2009;17:4708–17. doi: 10.1016/j.bmc.2009.04.052. [DOI] [PubMed] [Google Scholar]
- Puig B, et al. Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice. Neuropathol Appl Neurobiol. 2004;30:491–502. doi: 10.1111/j.1365-2990.2004.00569.x. [DOI] [PubMed] [Google Scholar]
- Quiroz-Baez R, et al. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of [alpha]-, [beta]- and [gamma]-secretase expression. Neurochemistry International. 2009;55:662–670. doi: 10.1016/j.neuint.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Ren Z, et al. Amyloid beta-protein precursor juxtamembrane domain regulates specificity of gamma-secretase-dependent cleavages. J Biol Chem. 2007;282:35350–60. doi: 10.1074/jbc.M702739200. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, et al. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem. 1997;68:1736–44. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- Sagisaka T, et al. Directed neural lineage differentiation of adult hippocampal progenitor cells via modulation of hippocampal cholinergic neurostimulating peptide precursor expression. Brain Research. 2010;1327:107–117. doi: 10.1016/j.brainres.2010.02.071. [DOI] [PubMed] [Google Scholar]
- Salloway S, et al. Disease-modifying therapies in Alzheimer’s disease. Alzheimers Dement. 2008;4:65–79. doi: 10.1016/j.jalz.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Savage MJ, et al. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–85. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Mandelkow E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 2008;5:443–57. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, et al. Hydrogen Peroxide Promotes AÎ2 Production through JNK-dependent Activation of Î3-Secretase. 2008;283:17721–17730. doi: 10.1074/jbc.M800013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwell AL, et al. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J Neurosci. 2008;28:9013–20. doi: 10.1523/JNEUROSCI.2747-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagno E, et al. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiology of Aging. 2009;30:1563–1573. doi: 10.1016/j.neurobiolaging.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Thompson LM, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. Journal of Cell Biology. 2009;187:1083–99. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardy ER, et al. Proteolytic mechanisms in amyloid-beta metabolism: therapeutic implications for Alzheimer’s disease. Trends Mol Med. 2005;11:464–72. doi: 10.1016/j.molmed.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Varma H, et al. Selective inhibitors of death in mutant huntingtin cells. Nature Chemical Biology. 2007a;3:99–100. doi: 10.1038/nchembio852. [DOI] [PubMed] [Google Scholar]
- Varma H, et al. Selective inhibitors of death in mutant huntingtin cells. Nat Chem Biol. 2007b;3:99–100. doi: 10.1038/nchembio852. [DOI] [PubMed] [Google Scholar]
- Vogel J, et al. The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology. 2009;57:539–50. doi: 10.1016/j.neuropharm.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Walker D, Lue LF. Anti-inflammatory and Immune Therapy for Alzheimer’s Disease: Current Status and Future Directions. Curr Neuropharmacol. 2007;5:232–43. doi: 10.2174/157015907782793667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JK, et al. Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proc Natl Acad Sci U S A. 2006;103:10461–6. doi: 10.1073/pnas.0600930103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoubian TA, Lo DC. Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci. 2000;3:342–9. doi: 10.1038/73911. [DOI] [PubMed] [Google Scholar]
- Yang DD, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–70. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yoshida H, et al. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–8. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics. 2008;5:409–14. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Immunohistochemical analysis of amyloid plaques in coronal sections of brains from Tg2576/PS1M146L mice using Anti-β-Amyloid 40 antibody (Biosource, Carlsbad, CA) demonstrates an increase in numbers of amyloid plaques with age. Images are representative of at least 3 animals per time point. (B) Wild-type littermate controls show no amyloid plaques at 12 months of age. (C) Automated quantification (InterFocus Imaging, Linton, UK) of plaque number in Tg2576/PS1M146L mice shows strong age-dependent increase in plaque burden with age.
In contrast to the dose-dependent neuroprotective effect of WYGSI-04 for brain slice explants transfected with APP, WYGSI-04 (“GSI”) had no measurable effects on control brain slice explants transfected with YFP only, as shown here for the highest concentration of WYGSI-04 tested. Ordinate axis shows average, total numbers ± SD of non-degenerating pyramidal neurons in the cortical regions of each explant expressed as a percentage of control brain slices treated with the DMSO carrier only.
In contrast to the dose-dependent neuroprotection provided by SP600125 to APP-transfected brain slices, JNK inhibition by SP600125 had no measurable effects on control brain slice explants transfected with YFP only, as shown here for the same concentration range tested. Ordinate axis shows average, total numbers ± SEM of non-degenerating pyramidal neurons in the cortical regions of each explant expressed as a percentage of control brain slices treated with the DMSO carrier only.
JNK inhibition by SP600125 did not significantly affect levels of BACE1 expression in brain slice explants in the same concentration range that provided protection against APP-induced neurodegeneration (Fig. 5B), consistent with such treatment not affecting levels of Aβ production as shown in Fig. 6A. Lanes were loaded with equivalent amounts of total protein from brain slice lysates; “--” denotes treatment with the DMSO vehicle only.