

Abstract
In amyotrophic lateral sclerosis (ALS), the exogenous temporal triggers that result in initial motor neuron death are not understood. Overactivation and consequent accelerated loss of vulnerable motor neurons is one theory of disease initiation. The vulnerability of motor neurons in response to chronic peripheral nerve hyperstimulation was tested in the SOD1G93A rat model of ALS. A novel in vivo technique for peripheral phrenic nerve stimulation was developed via intra-diaphragm muscle electrode implantation at the phrenic motor endpoint. Chronic bilateral phrenic nerve hyperstimulation in SOD1G93A rats accelerated disease progression, including shortened lifespan, hastened motor neuron loss and increased denervation at diaphragm neuromuscular junctions. Hyperstimulation also resulted in focal decline in adjacent forelimb function. These results show that peripheral phrenic nerve hyperstimulation accelerates cell death of vulnerable spinal motor neurons, modifies both temporal and anatomical onset of disease, and leads to involvement of disease in adjacent anatomical regions in this ALS model.
Keywords: motor neuron, neurodegeneration, ALS, amyotrophic lateral sclerosis, SOD1, phrenic nerve, diaphragm, diaphragm pacing, diaphragm stimulation, respiratory, disease onset, environment
Introduction
Potential exogenous triggers or risk factors for motor neuron degeneration and the development of amyotrophic lateral sclerosis (ALS) have been hypothesized, including infectious etiologies such as viruses (Mattson, 2004), toxic or heavy metal exposures (Sutedja et al., 2008b), central nervous system or peripheral nerve trauma (Mitsumoto et al., 1998), occupation (Sutedja et al., 2008a), exercise and body habitus (Chen et al., 2008), amongst others. More recently, the hunt for disease modifying genes has also been undertaken (Ravits and Traynor, 2008). However, little is known about the factors that influence the anatomical site of disease onset in ALS. Why does disease start with limb onset in one patient, while it begins with speech, swallowing or breathing problems in another? It is also not appreciated which factors play a role in the anatomical progression of disease to adjacent regions of the neuraxis. For example, patients with weakness in one leg generally develop weakness in the other leg prior to the development of speech or swallowing abnormalities. This pattern is also manifested in the caudal-to-rostral disease progression observed in mutant human SOD1-expressing transgenic rodent models.
The vast majority of ALS cases are sporadic, while approximately 10% are familial. Twenty percent of familial cases are linked to various point mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene on chromosome 21 (Rosen et al., 1993). Transgenic mice (Bruijn et al., 1997; Gurney et al., 1994; Wong et al., 1995) and rats (Howland et al., 2002; Matsumoto et al., 2006; Nagai et al., 2001) carrying mutant human SOD1 genes (G93A, G37R, G86R, G85R) have been generated, and, despite the existence of other animal models of motor neuron loss, are currently the most highly used models of the disease.
ALS patients ultimately succumb to disease approximately 2-5 years following diagnosis because of respiratory compromise due to loss of phrenic motor neuron innervation of the diaphragm. Furthermore, the extent of respiratory involvement has been reported as a major prognostic factor (Haverkamp et al., 1995). Interventions targeting respiratory function have resulted in impressive, albeit incomplete, efficacy on ALS patient survival (Lo Coco et al., 2006). We have also shown cervical motor neuron loss, phrenic nerve axonal loss, diaphragm atrophy and progressive reduction of phrenic nerve compound muscle action potential amplitudes in SOD1G93A rats (Llado et al., 2006). These results demonstrate a vulnerability of phrenic motor neurons in SOD1G93A rats similar to human disease, and demonstrate that decline in respiratory function also plays a central role in disease progression in the rat model of ALS.
By targeting respiratory function in the SOD1G93A rat, we have developed a model that influences the site of disease onset, the temporal course of the disease, and results in the anatomical involvement of disease in anatomically adjacent regions. Specifically, we have developed a novel in vivo technique for chronic bilateral phrenic nerve stimulation in the SOD1G93A rat via intra-diaphragm muscle electrode implantation at phrenic nerve motor endpoints. By hyperstimulating this nerve-muscle connection, we can induce early onset of respiratory dysfunction, symptoms which normally are not observed in the SOD1G93A rat until very late in the disease course. Interestingly, these abnormalities also resulted in development of forelimb weakness prior to any hindlimb symptoms, a process that is different from the normal caudal to rostral disease course in this model.
This paradigm has several unique features: 1) It regionally activates a defined population of motor neurons (phrenic); 2) It maintains the integrity of the motor unit (no axotomy or traumatic injury is required); 3) It allows for the study of the most clinically relevant aspect of human ALS biology (diaphragm and respiratory function); 4) It is reproducible; 5) It allows for examination of the effects of peripheral nervous system activity/overactivity on central nervous system biology; 6) It is a novel approach for the study of a focally-restricted process in a well-studied ALS animal model with an identical mutation and thus a more homogenous neurodegenerative biology.
Materials and Methods
Animal models
SOD1G93A rats
Transgenic rats carrying the human SOD1 gene with the G93A mutation were used (Howland et al., 2002). Male and female rats were obtained from Taconic, and maintained as an in-house colony. Typically, untreated SOD1G93A mutants first develop hindlimb disease onset, followed by the development of forelimb onset with subsequent progression to endstage, as has been described previously (Howland et al., 2002; Lepore et al., 2008). For all studies, equal numbers of males and females were included in all groups, and animals from the same litter were distributed amongst groups. In addition, age-matched wild-type (WT) Sprague-Dawley rats from Taconic were also used.
Care and treatment of animals
The care and treatment of animals in all procedures was conducted in strict accordance with the guidelines set by the European Communities Counsel Directive (November 24th, 1986), the NIH Guide for the Care and Use of Laboratory Animals, the Guidelines for the Use of Animals in Neuroscience Research and the Johns Hopkins University IACUC, and measures were taken to minimize any potential pain or animal discomfort. Rats were housed at standard temperature (21°C) and in a light controlled environment with ad libitum access to the food and water, and were maintained in racks of ventilated cages located in the same room. In order to avoid dehydration, Aqua-Jel packs were provided when animals started to show disease symptoms.
Electrode stimulation
Electrode implantation
85-90 day old WT and SOD1G93A rats received intraperotineal injections of anaesthetic cocktail [acepromazine maleate (0.7mg/kg; Fermenta Animal Health, Kansas City, MO), ketamine (95mg/kg; Fort Dodge Animal Health; Fort Dodge, IA), and xylazine (10mg/kg; Bayer, Shawnee Mission, KS)]. A laparotomy was performed to access the posterior surface of the diaphragm, and stimulating electrodes were bilaterally implanted into the center of both hemi-diaphragms to achieve phrenic nerve motor endpoint activation upon external stimulation. By placing the electrodes in different parts of the diaphragm intraoperatively and examining contraction during stimulation, we identified that optimal full diaphragm muscle contractions can be obtained in the adult rat by placing electrodes centrally in the hemi-diaphragm (Fig 1A). Electrode wires were fed out of the abdomen, and were tunneled subcutaneously towards the back of the animal, just posterior to the base of the skull. The ends of wires were secured via 4-0 suture, and gold sockets were attached to the ends of the wires for daily connection to the external stimulator. In addition, anodes were placed subcutaneously in the back of the animal. All WT and SOD1G93A rats were implanted with the full array of electrodes, regardless of whether the animal was part of the stimulated or unstimulated group. Those animals referred to as “unstimulated” underwent the surgical implantation of electrodes and had electrodes in place for the duration of the study. All incisions were closed with 4-0 suture, and animals were placed on a circulating-H2O heating pad and were closely monitored for 24 hours post-surgery.
Figure 1. Phrenic nerve hyperstimulation: technique development in SOD1G93A rats.
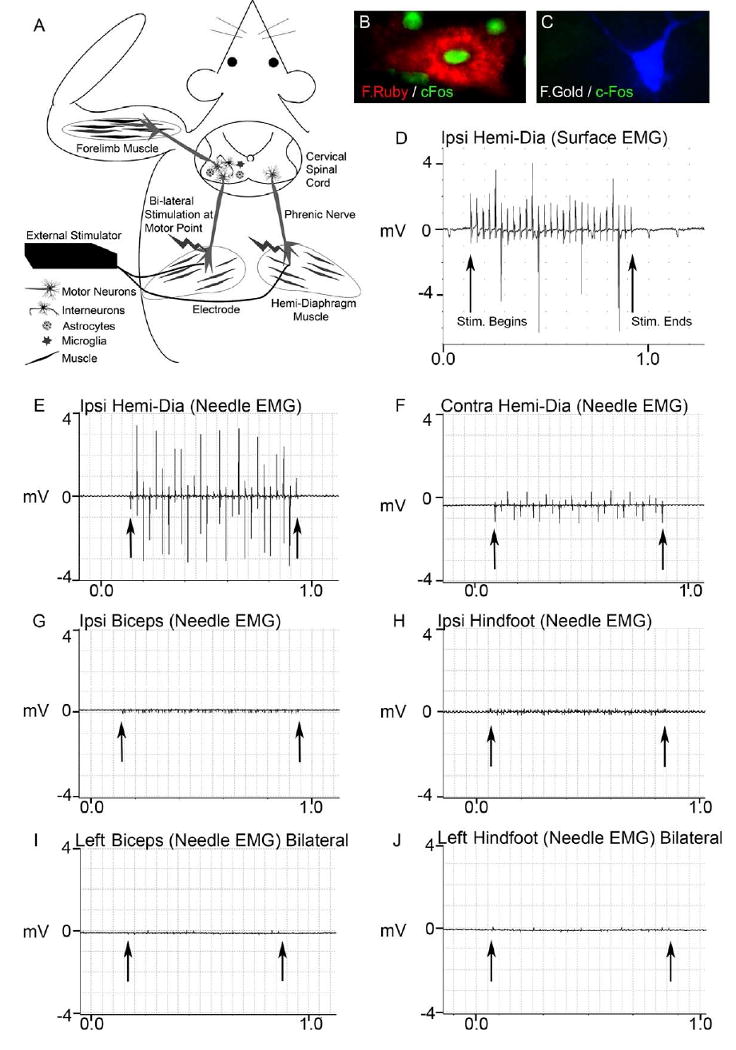
The diagram illustrates the phrenic nerve motor endpoint stimulation technique (A). Stimulating electrodes were bilaterally implanted into the center of both hemi-diaphragm muscles to achieve phrenic nerve activation upon external stimulation. Even though placed directly into muscle, the electrode ends were positioned near the phrenic nerve motor endpoints to allow for stimulation of the phrenic nerve (stimulation/contraction is unsuccessful if there is not phrenic nerve innervation of the diaphragm). The stimulator is found externally, and the ends of the 2 electrodes are connected to the stimulator each day during stimulation, while the animal is awake and freely-behaving. Motor neurons innervating the diaphragm originate in the cervical spinal cord, in regions where forelimb motor neurons also reside. This paradigm allows for the study of focal effects on the peripheral nervous system (muscle and nerve), as well as central nervous system effects on motor neurons, astrocytes, and microglia - all key cell types in mutant SOD1-mediated disease (A).
Following bilateral phrenic nerve stimulation, c-Fos expression was detected in fluororuby retrogradely-labeled phrenic motor neurons (Fig 1B), but not in fluorogold-labeled forelimb motor neurons (Fig 1C), suggesting that phrenic nerve stimulation activated phrenic motor neurons, but not forelimb motor neurons. To also examine whether external diaphragm stimulation resulted in ectopic stimulation of other motor neuron / muscle groups (besides phrenic nerve / diaphragm), a comprehensive series of intra-muscular needle EMG and muscle surface EMG recordings were obtained during unilateral phrenic nerve stimulation of SOD1G93A rats. Stimulation resulted in consistent surface electrical responses in the ipsilateral diaphragm lasting the duration of the programmed stimulus duration (D). Robust intramuscular electrical activity (∼3.5mV) was obtained in the ipsilateral hemi-diaphragm (E) with needle EMG recordings, while a smaller response (∼1.0mV) was also noted in the hemi-diaphragm contralateral to stimulation (F). On the contrary, significant electrical responses during phrenic nerve stimulation were not found with intra-muscular needle EMG recordings in the ipsilateral biceps (G) or ipsilateral hindfoot (H). Needle EMG recordings were also conducted in the leftt biceps (Fig 1I) and left hindfoot (Fig 1J) during bilateral phrenic nerve stimulation, and significant electrical activity was not observed. Arrows denote beginning and end of stimulation.
Stimulation paradigm
Three stimulation paradigms were conducted: 1) SOD1G93A rats with high stimulation parameters; 2) SOD1G93A rats with low stimulation parameters; 3) WT rats with high stimulation parameters. Except for preliminary experiments (see Results section), all rats received 2 hours of continuous stimulation 5 days per week. Stimulation began for all animals at 90 days of age, and stimulation was always conducted on awake, freely-behaving animals that were restricted only to their home cage. All SOD1G93A rats continued to receive stimulation until disease endstage, while WT rats received stimulation for 8 weeks, and were subsequently sacrificed. External stimulators were attached to the ends of the 2 stimulating electrodes and to the anode, and animals were constantly monitored during the 2 hour period of stimulation to prevent detachment of electrodes from the stimulator. Refer to Table 1 for an outline of the study design and numbers and types of animals used for each experiment.
| Rat Type | Sex | Number of Rats | Stimulation Duration | Surgery/Implant | Stimulation | Parameters | Analysis |
|---|---|---|---|---|---|---|---|
| Wild-Type | M/F | 4 | 8 weeks: start at 90 days | Yes | High stimulation | Varied | Development and validation of technique |
| SOD1G93A | M/F | 6 | 8 weeks: start at 90 days | Yes | High stimulation | Varied | “ “ |
| SOD1G93A | M/F | 3 | Acute stim at 90 days | Yes | High stimulation | Acute stimulation | Needle and Surface EMGs, c-fos staining |
| SOD1G93A | M/F | 7 | 90 days -endstage | Yes | High stimulation | 2 hrs/day 5 days/wk | Weight, Grip strength, Survival, CMAPs at 126 and 138 days of age, Motor neuron counts at endstage |
| SOD1G93A | M/F | 7 | Unstim | Yes | Unstim (High) | NA | “ “ |
| SOD1G93A | F | 3 | 90-146 days | Yes | High stimulation | 2 hrs/day 5 days/wk | Retrograde axonal tracing, Iba1, NMJ |
| SOD1G93A | F | 3 | 90-146 days | Yes | Unstim (High) | NA | “ “ |
| Wild-Type | M/F | 4 | 90-146 days | Yes | High stimulation | 2 hrs/day 5 days/wk | Weight, Grip strength, CMAPs at 146 days of age, Motor neuron counts at 146 days of age, Retrograde axonal tracing, Iba1, NMJ |
| Wild-Type | M/F | 4 | 90-146 days | Yes | Unstim (High) | NA | “ “ |
| SOD1G93A | M/F | 4 | 90 days -endstage | Yes | Low stimulation | 2 hrs/day 5 days/wk | Weight, Grip strength, Survival, CMAPs at 111 days of age, Motor neuron counts at endstage |
| SOD1G93A | M/F | 5 | Unstim | Yes | Unstim (Low) | NA | “ “ |
The stimulus was presented through single helix wound stainless steel electrodes with 4.0 mm of exposed length and 5.0 mm2 of surface area. High stimulation parameters consisted of stimulation delivered at the respiratory rate of 15.0 bursts / minute, with stimulus burst duration of 0.8 sec, 20.0 μs pulse duration, 5.0mAmp amplitude and an intra-burst frequency of 33.0 Hz. The delivered charge density on a per pulse basis was 0.02 μC / mm2. With these stimulation parameters, we intentionally hyperstimulated the phrenic nerve at the motor endpoint. Low stimulation parameters consisted of stimulation delivery at the respiratory rate of 15.0 bursts / minute, with a 1.0 mAmp amplitude, 10.0 μsec pulse width (with no ramping) and 12.0 Hz pulse frequency. All variables (including electrodes and anodes, surgical implantation) were the same between high and low stimulation experiments, except for the parameters delivered by the stimulator.
Behavioral analysis
Animal weighing and grip strength measurements were conducted starting 1 week post-electrode implantation. Weighing was conducted once per week until endstage, while grip strength measurements were only conducted on specific days (see Results section).
Hindlimb and forelimb grip strength
Hind- and forelimb muscle grip strengths were separately determined using a “Grip Strength Meter” (DFIS-2 Series Digital Force Gauge; Columbus Instruments, OH) (Lepore et al., 2008). Grip strength testing was performed by allowing the animals to grasp a thin bar attached to the force gauge. This was followed by pulling the animal away from the gauge until the hind- or forelimbs released the bar. This provides a value for the force of maximal grip strength. The force measurements were recorded in three separate trials, and the averages were used in analyses.
Survival/endstage analysis
To determine disease endstage in a reliable and ethical fashion, an artificial endpoint was used for all SOD1G93A mutant rats (Lepore et al., 2008). Endstage was defined by the inability of rats to right themselves within 30 seconds when placed on their sides. The moribund rats were scored as “dead”, and were subsequently euthanized.
Compound muscle action potential (CMAP) recordings
Under anesthesia, phrenic nerve conduction studies (Llado et al., 2006) were performed with stimulation (0.5 ms single stimulus; 1.0 Hz supramaximal pulses) at the neck via near nerve needle electrodes placed 0.5 cm apart along the phrenic nerve. Recording was obtained via a surface strip along the costal margin, and CMAP amplitude was measured baseline to peak. Recordings across the nerve segment were made using an ADI Powerlab 8SP stimulator and BioAMP amplifier (Powerlab), followed by computer assisted data analysis (Scope 3.5.6; ADI). Distal motor latency of evoked potentials includes duration of nerve conduction between stimulating and recording electrodes plus time of synaptic transmission.
Histological analysis
Tissue processing
SOD1G93A were sacrificed during the course of disease for immunohistochemistry and tracer analysis or at disease endstage for total cervical motor neuron counts. WT rats were sacrificed at either 146 days of age for immunohistochemistry and motor neuron counts. Animals were transcardially perfused with 0.3% saline, followed by ice-cold 4% paraformaldehyde (Fisher Scientific; Pittsburgh, PA). Spinal cords were removed from the animal, followed by preparation of C4-C6 and L4-L5 spinal cord segments by: 1) cryoprotection in 30% sucrose (Fisher) / 0.1 M phosphate buffer at 4°C for 3 days for immunohistochemistry; 2) washing in 0.1M phosphate buffer, followed by paraffin-embedding, for motor neuron counts. For immunohistochemistry, tissue was embedded in OCT (Fisher), fast frozen with dry ice, and stored at −80°C until processed. Spinal cord tissue blocks were cut in the sagittal or transverse planes at 8μm or 20μm thicknesses. Sections were collected on glass slides and stored at -20°C until analyzed. Subsets of spinal cord slices were collected in PBS for free-floating histochemistry.
Immunohistochemistry
GFAP (Chemicon) was used to identify astrocytes, and Iba1 (Wako) was used to detect microglia. c-Fos (AdSerotec) was used to detect activated motor neurons. Samples were incubated for 2 hours at room temperature with goat anti-mouse and goat anti-rabbit secondary antibodies (1:200; Jackson, West Grove, PA) conjugated to rhodamine or FITC. Samples were counterstained with DAPI (1:1000; Sigma) to identify nuclei, and cover-slipped with anti-fade mounting media (Fluorosave, CN Biosciences; La Jolla, CA). Slides were subsequently stored at 4°C. Images were acquired on either a Zeiss fluorescence microscope using a Photometric Sensys KAF-1400 CCD camera (Roper Scientific; Trenton, NJ) or on a Zeiss laser confocal microscope. Images were analyzed using either Metamorph or Zeiss confocal software. Adobe Photoshop 7.0 (Adobe, San Jose, CA) was used to prepare figures.
Motor neuron survival
The cervical (C4-C6) spinal cord from endstage animals was serially sectioned (14 μm), dehydrated in a graded series of alcohol solutions, embedded in paraffin, and stained with cresyl violet to quantify motor neuron numbers. Motor neurons were counted in every 7th section at 20× magnification in order to avoid repetitive counting. Only motor neurons with a clearly identifiable nucleus and nucleolus, a cell soma over 100 μm2 and located within the ventral horn were counted at a 200-fold magnification (Llado et al., 2006).
Neuromuscular Junction Analysis
Following electrode implantation, three groups of rats were sacrificed at the same age (following 9 weeks of stimulation) for neuromuscular junction analysis: stimulated wild-type; unstimulated SOD1G93A, stimulated SOD1G93A. A group of unstimulated SOD1G93A rats was also analyzed at disease endstage. 1 hemi-diaphragm muscle was dissected from each animal for whole-mount immunohistochemistry (Wright et al., 2007). Muscle was stretched, pinned down to Sylgard (Fisher) media, and extensively cleaned to remove any connective tissue to allow for antibody penetration. Motor axons and their terminals were labeled with SMI-312R (Covance) and SV2-s (DSHB), respectively, and both antibody labelings were detected with FITC anti-mouse IgG secondary (Invitrogen). Post-synaptic acetylcholine receptors were labeled with Alexa Fluor 647-conjugated alpha-bungarotoxin (Invitrogen). Labeled muscles were analyzed for total numbers of NMJs, fully innervated NMJs, partially denervated NMJs, completely denervated NMJs, NMJs with terminal sprouting, NMJs with axonal sprouting/partial reinnervation.
Fluorescent Tracer Injections and Analysis
Fluororuby (dextran tetramethylrhodamine; Invitrogen) and fluorogold (Fluorochrome, LLC; Denver, CO) retrograde axonal tracers were injected unilaterally into 1 hemi-diaphragm muscle and ipsilateral forelimb muscles (deltoid and triceps), respectively (Boulenguez et al., 2007). Injections were conducted during the same surgery as electrode implantation. Because tracers were injected into muscles prior to the commencement of stimulation, the tracing evaluated the degree of motor neuron loss (but not denervation at the neuromuscular junction) following stimulation. Following laparotomy, 30uL of fluororuby (10% solution in 2% DMSO/sterile saline vehicle) was injected into 1 hemidiaphragm (8-10 injection sites per muscle) using a 10uL Hamilton syringe with an attached 33-gauge needle (45° bevel). For delivery of fluorogold (2.5% solution in dH2O vehicle), the ipsilateral forelimb was shaved, and a midline incision in the skin was made on the lateral surface of the limb from the elbow to the shoulder. 75uL of solution was injected throughout the deltoid and triceps muscles (5uL per site) using a 10uL Hamilton syringe with an attached 33-gauge needle (30° bevel). Skin was stapled following forelimb injections.
Three groups of rats were injected with both tracers in identical fashion at the same age (stimulated wild-type; unstimulated SOD1G93A, stimulated SOD1G93A), and all rats were sacrificed at the same age (following 9 weeks of stimulation). Briefly, sagittal sections of the entire cervical spinal cord and the rostral part of thoracic spinal cord were cut free-floating at 30μm thickness. Slices were washed in 0.1M TBS, mounted on slides, allowed to dry for 2 hours, dehydrated through a series of alcohols, and cover-slipped with DPX. Total numbers of fluororuby+ and fluorogold+ motor neurons were counted in all sections. In order to rule out the possibility of lost motor neuron labeling (or substantially decreased fluorescent signal below detection levels) at long time points following tracer injection, unstimulated wild-type rats were injected with both fluororuby and fluorgold using the same protocol as described above and were sacrificed at an early time point of 14 days.
Statistical analysis
Kaplan-Meier analysis of the SOD1G93A rats was conducted using the statistical software Sigmastat (SAS Software) to analyze animal survival. Weight results were analyzed via ANOVA. In all other analyses, Student t-test was performed to compare data between groups of animals. All data are presented as mean ± S.E.M., and significance level was set at p ≤ 0.05.
Results
Development of phrenic nerve stimulation technique in wild-type rats and in the SOD1G93A rat model of ALS
We first sought to develop the technique of intra-diaphragm electrode implantation at phrenic motor endpoints for stimulation in wild-type (WT) and SOD1G93A rats (see diagram: Fig 1A). In these initial experiments, we bilaterally implanted both male and female WT and SOD1G93A rats with intra-diaphragm electrodes successfully in both hemi-diaphragms at phrenic motor endpoints via laparotomy. All WT (n = 4) and SOD1G93A (n = 6) rats survived surgery and implantation. Peripheral stimulation of phrenic nerves with resulting diaphragm muscle contraction bilaterally was successfully carried out on awake, freely-behaving WT and SOD1G93A rats, resulting in consistent bilateral contractions of both hemi-diaphragms upon external stimulation. Stimulation at high parameters (see Materials and Methods section for details) could be continued chronically for at least 8 weeks post-implantation in all WT and SOD1G93A rats. During the first week post-implantation, rats were subjected to only 30 minutes of continuous stimulation per day for 3 days a week to allow for accommodation to the prescribed intervention. During weeks 2 and 3, rats received 3 days of stimulation for 2 hours per day. Over the remainder of the study, we increased the stimulation regimen to 2 hours per day for 5 days a week. Rats were able to tolerate the stimulation without any apparent signs of pain or discomfort during daily stimulation. Furthermore, all electrodes remained in place, and bilateral stimulation consistently resulted in contractions of both hemi-diaphragms, even up to 8 weeks post-implantation. Stimulation never resulted in adverse events such as diaphragm tissue necrosis, animal infection or sudden death. These results demonstrate that intra-diaphragm electrode implantation and subsequent external phrenic nerve motor endpoint stimulation: 1) is feasible, 2) functions in successfully obtaining robust and consistent bilateral diaphragm muscle contraction, and 3) is well tolerated by both WT and SOD1G93A rats for extended durations of time.
To examine whether diaphragm stimulation via intramuscular electrode implantation resulted in ectopic stimulation of other motor neuron groups besides phrenic motor neurons, we performed c-Fos labeling of specific populations of motor neurons in the cervical spinal cord. Forelimb motor neurons and phrenic motor neurons were retrogradely labeled with the fluorescent tracers, fluorogold (FG) and fluororuby (FR), respectively, to allow for identification of specific motor neuron populations. Two weeks later, animals received 2 hours of bilateral phrenic nerve stimulation, and were sacrificed 1.5 hours following stimulation. Spinal cord sections were immunolabeled with c-Fos. c-Fos expression was detected in scattered FR-labeled phrenic motor neurons (Fig 1B), but not in FG-labeled forelimb motor neurons (Fig 1C), suggesting that phrenic nerve stimulation activated phrenic motor neurons, but not forelimb motor neurons.
To also rule out the possibility of ectopic stimulation of motor neurons / muscle groups (besides phrenic nerve / diaphragm), we performed a comprehensive series of intra-muscular needle EMG and muscle surface EMG recordings during unilateral phrenic nerve stimulation of SOD1G93A rats. Surface EMG recordings of the diaphragm at the costophrenic interface demonstrate electrical activity in ipsilateral diaphragm muscle during stimulation (Fig 1D). The recordings displayed in Figure 1 do not represent motor unit potentials from diaphragmatic contraction but rather demonstrate stimulation from the adjacent electrode. During recording from intramuscular diaphragmatic electrodes, robust intra-muscular activity (∼3.5mV) was obtained in the ipsilateral hemi-diaphragm (Fig 1E), while less activity (∼1.0mV) was also noted in the hemi-diaphragm contralateral to the site of stimulation (Fig 1F). On the contrary, significant electrical activity during phrenic nerve stimulation was not found with intra-muscular needle EMG recordings in the ipsilateral biceps (Fig 1G), contralateral biceps (not shown), ipsilateral hindfoot (Fig 1H) or base of tail (not shown). A similar pattern was also found with surface EMG recordings of these same muscles during phrenic stimulation (not shown). In addition, only diaphragmatic contraction (ipsilaterally more than contralaterally) was noted. We did not visually observe muscle contraction in either forelimb or hindlimb muscles. These results demonstrate that phrenic nerve stimulation resulted in a regionally (ipsilateral diaphragm) concentrated stimulus. We also conducted needle EMG recordings in the left biceps (Fig 1I) and left hindfoot (Fig 1J) during bilateral phrenic nerve stimulation, and did not observe significant electrical activity.
Phrenic nerve hyperstimulation shortened life span and accelerated weight loss and decline in forelimb motor performance in SOD1G93A rats
The effects of phrenic nerve motor endpoint stimulation were next examined in SOD1G93A rats. Ninety day-old SOD1G93A rats were implanted bilaterally, and caged individually to avoid removal of electrodes. Half of the SOD1G93A rats received high stimulation 5 days per week for 2 hours per day until disease endstage (n = 7), and the other half of the rats with electrode implants in place for the duration of the study did not receive stimulation and are referred to as “unstimulated: unstim” (n = 7). Stimulation at high parameter settings (see Methods for description), a stimulation paradigm previously noted to cause neuromuscular fatigue in canine models (Oda et al., 1981), accelerated death by 24 days (unstim: 167.30 ± 2.12 days; stim: 144.30 ± 2.91; p < 0.0001; Fig 2A) and hastened weight decline (p < 0.01 at a number of time points; Fig 2B) compared to unstimulated controls.
Figure 2. Phrenic nerve hyperstimulation accelerated disease in SOD1G93A rats.
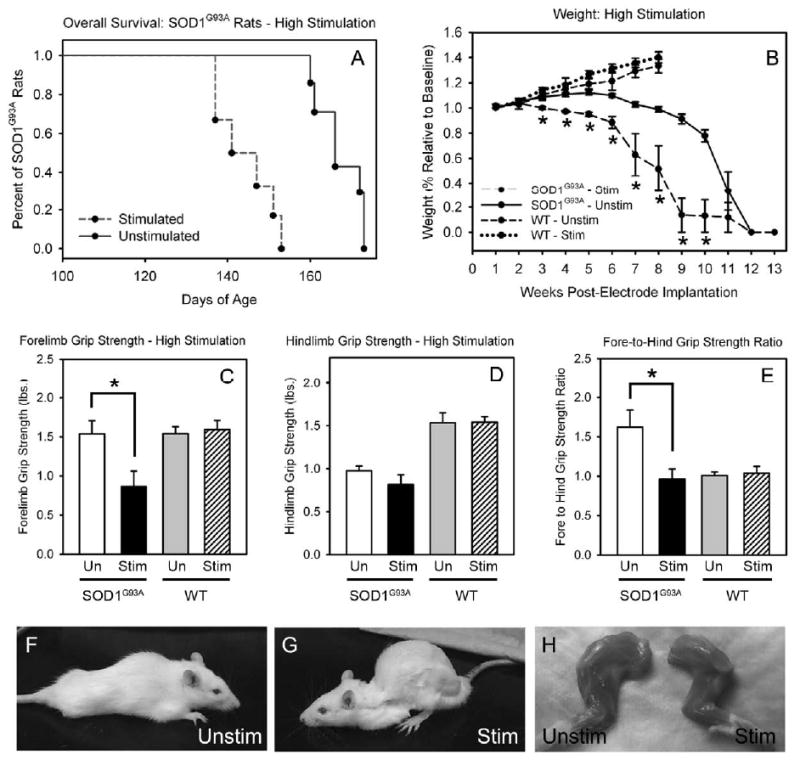
Bilateral phrenic nerve stimulation at high parameter settings accelerated death in SOD1G93A rats by 24 days (A) and weight decline (B) compared to unstimulated SOD1G93A rat controls. Compared to unstimulated SOD1G93A controls, stimulated SOD1G93A rats had significantly reduced forelimb grip strength at 141 days of age (C). No differences in hindlimb grip strength were noted between groups at this same age (D). The ratio of fore-to-hindlimb grip strength is significantly decreased in stimulated SOD1G93A rats (E). Therefore, selective phrenic nerve motor point stimulation selectively accelerated decline in forelimb, but not hindlimb, grip strength. This phenotype can be appreciated by comparing representative images of unstimulated (F) and stimulated (G) SOD1G93A litter mates. Forelimb muscle atrophy was also accelerated in hyperstimulated SOD1G93A rats (H). Unlike SOD1G93A rats, bilateral phrenic nerve stimulation in wild-type rats at high parameter settings had no effect on the rate of weight decline at all time points (B) or on forelimb grip strength (C), hindlimb grip strength (D) and fore-to-hindlimb grip strength ratio (E).
It is important to note that phrenic nerve stimulation did not result in immediate death of stimulated rats. On the contrary, the SOD1G93A rats were stimulated for at least 2-3 months before reaching endstage. Similar to sham unstimulated rats, the stimulated animals reached endstage over a range of ages, except that their survival curve was significantly shifted towards earlier ages. In addition, these animals did not quickly reach endstage once they developed disease onset in the forelimbs. Instead, the animals proceeded through a typical progression of increasing limb weakness before succumbing to disease. These findings show that stimulation accelerated the natural course of disease, but did not result in acute electrical injury to motor neurons. We did not appreciate any muscle tissue necrosis or inflammation at the site of stimulation in the diaphragm (electrode implantation site) to account for reduced respiratory parameters (not shown).
Interestingly, phrenic nerve hyperstimulation selectively accelerated loss of other behavioral functions associated with cervical spinal cord function. Compared to unstimulated sham SOD1G93A rats, stimulated SOD1G93A rats had significantly reduced forelimb grip strength at 141 days of age (unstim: 1.54 ± 0.17 lbs. of force; stim: 0.86 ± 0.20; p < 0.05; Fig 2C). However, no differences in hindlimb grip strength were noted between groups at this same age (unstim: 0.97 ± 0.06; stim: 0.81 ± 0.11; p > 0.05; Fig 2D). The typical phenotype in this model results in SOD1G93A rats developing weakness first in hindlimb function, followed by development of forelimb weakness, as we have described previously (Howland et al., 2002; Lepore et al., 2008). By expressing fore- and hindlimb grip strength as a ratio of fore-to-hind function, our data suggest that forelimbs retain greater grip strength than the hindlimbs at 141 days of age in unstimulated sham SOD1G93A rats. This ratio is significantly decreased in stimulated rats (unstim: 1.62 ± 0.22; stim: 0.96 ± 0.13; p < 0.05; Fig 2E), suggesting that selective phrenic nerve motor endpoint stimulation focally accelerated decline in forelimb, but not hindlimb, grip strength. This reproducible phenotype of forelimb weakness and kyphosis can be appreciated by comparing representative images of unstimulated (Fig 2F) and stimulated (Fig 2G) animals. Furthermore, forelimb muscle atrophy was accelerated in hyperstimulated SOD1G93A rats (Fig 2H). These results show that hyperstimulation of vulnerable SOD1G93A phrenic nerves in these animals resulted in acceleration of disease progression, including hastened animal death, and altered the phenotype from a predominantly hindlimb onset to a forelimb disease onset.
Phrenic nerve hyperstimulation accelerated decline in phrenic nerve compound muscle action potentials (CMAPs)
Compared to unstimulated SOD1G93A rats (Fig 3B), SOD1G93A rats that received high stimulation (Fig 3C) had reduced phrenic nerve compound muscle action potentials (CMAPs), a functional electrophysiological assessment of diaphragm function (Llado et al., 2006). CMAPs were recorded at 2 different points during disease progression, and a significant decrease in peak response amplitude was found in stimulated rats at both times (unstim - 126 days: 6.44 ± 0.49 mV; unstim - 138 days: 5.05 ± 0.58; stim - 126 days: 4.5 ± 0.54; stim - 138 days: 2.25 ± 0.95; p < 0.05 at both time points; Fig 3A). Stimulation had no effect on latency of response (unstim - 126 days of age: 2.70 ± 0.07 msec; unstim - 138 days: 3.61 ± 0.21; stim - 126 days: 2.85 ± 0.16; stim - 138 days: 4.08 ± 0.24; p > 0.05 at both time points; data not shown).
Figure 3. Phrenic nerve hyperstimulation accelerated decline in phrenic nerve compound muscle action potentials (CMAPs) in SOD1G93A rats.
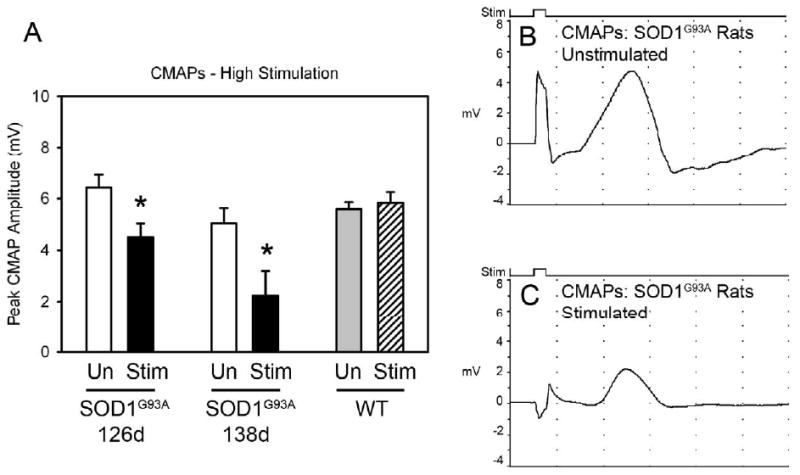
Compared to unstimulated SOD1G93A rats (B), SOD1G93A rats that received high stimulation (C) had reduced phrenic nerve compound muscle action potentials (CMAPs), a functional electrophysiological assay of diaphragm function. CMAPs were recorded at 2 different points during disease progression, and a significant decrease in peak response amplitude was found in stimulated rats at both times (A). No effects of high stimulation on CMAP amplitude was noted in wild-type rats (A).
Phrenic nerve hyperstimulation accelerated loss of cervical spinal cord motor neurons in SOD1G93A rats
Unstimulated SOD1G93A rats had significantly greater numbers of total cervical motor neurons at disease endstage than SOD1G93A rats that received high stimulation (unstimulated: 17.10 ± 1.02 motor neurons/section; stimulated: 10.15 ± 1.86; p < 0.05; Fig 4A).
Figure 4. Phrenic nerve hyperstimulation accelerated the loss of both phrenic motor neurons and forelimb motor neurons in SOD1G93A rats.
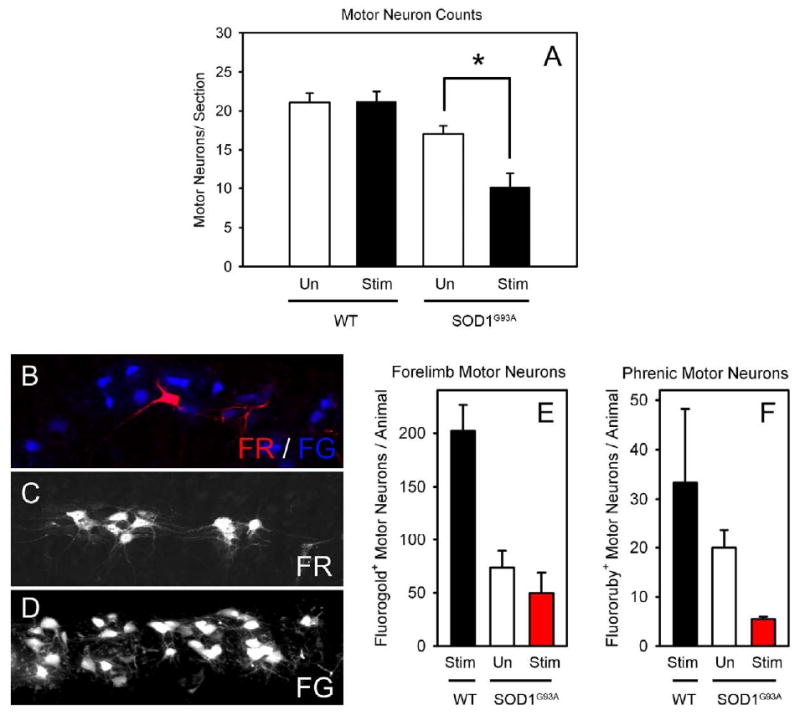
Unstimulated SOD1G93A rats had significantly greater numbers of cervical motor neurons at disease endstage than SOD1G93A rats that received hyperstimulation (A). On the contrary, hyperstimulation of wild-type rats had no effects on numbers of cervical motor neurons (A). Within cervical spinal cord ventral horn (∼C4-C5), phrenic motor neurons (fluororuby-labeled: red; C) and motor neurons innervating the ipsilateral forelimb (fluorogold-labeled: blue; D) were in close proximity (B). Compared to wild-type rats that received phrenic nerve hyperstimulation, both SOD1G93A stimulated and SOD1G93A unstimulated rats had significantly reduced numbers of forelimb (E) and phrenic (F) motor neurons. The numbers of phrenic and forelimb motor neurons in the stimulated SOD1G93A rats showed a reduced trend compared to age-matched unstimulated SOD1G93A rats.
To selectively examine the loss of specific motor neuron populations, phrenic motor neurons were retrogradely labeled with the fluorescent tracer, Fluororuby (FR), via intra-muscular injections into ipsilateral hemi-diaphragm (Fig 4C). Forelimb motor neurons in the same animals were retrogradely labeled with a second fluorescent tracer, Fluorogold (FG), via unilateral intra-muscular injections into triceps and deltoid muscles (Fig 4D). Within cervical spinal cord ventral horn (∼C4-C5), phrenic motor neurons and motor neurons innervating the ipsilateral forelimb were in close proximity (Fig 4B). Compared to wild-type rats that received phrenic nerve hyperstimulation (33.3 ± 14.9 FR+ motor neurons/animal; 210.5 ± 39.5 FG+ motor neurons/animal), both SOD1G93A high stimulated (5.6 ± 0.3 FR+; 49.3 ± 19.3 FG+) and SOD1G93A unstimulated (19.6 ± 16.4 FR+; 75.0 ± 27.0 FG+) rats had significantly reduced numbers of forelimb (Fig 4E) and phrenic (Fig 4F) motor neurons (p < 0.05). In addition, the numbers of forelimb and phrenic motor neurons in the stimulated SOD1G93A rats showed a trend towards a reduction when compared to age-matched unstimulated SOD1G93A rats (p = 0.06 for phrenic). This suggests that phrenic nerve hyperstimulation accelerated loss of motor neurons (particularly phrenic motor neurons) within the cervical spinal cord. It is important to note that tracer injections were conducted during the same surgery as electrode implantation. Because tracers were injected into muscles prior to the commencement of stimulation, the tracing evaluated the degree of motor neuron loss (but not denervation at the neuromuscular junction) following stimulation.
To rule out the possibility of lost motor neuron labeling (or substantial decrease in fluorescence signal below detection levels) at long time points, unstimulated wild-type rats (n = 3) were sacrificed at 14 days following fluororuby and fluorogold injections to compare the efficiency of motor neuron labeling shortly after injection with much longer time points. No significant differences were found between motor neuron counts in unstimulated wild-type rats at 14 days (33.0 ± 10.3 FR+ motor neurons/animal; 183.3 ± 23.3 FG+ motor neurons/animal) and 9 weeks (see data above) post-injection, demonstrating that both tracers persist within labeled motor neurons even at long time points.
Phrenic nerve hyperstimulation had no effect on wild-type rats
To test if the effects of bilateral phrenic nerve hyperstimulation were specific to vulnerable motor neurons in the neurodegenerative SOD1G93A model, WT rats (n = 4 unstim; n = 4 stim) were subjected to the same stimulation paradigm as SOD1G93A rats, including stimulation parameters, age of animals, and frequency and duration of daily stimulation. Unlike SOD1G93A rats, high stimulation had no effect in wild-type rats on the rate of weight decline at all time points (p > 0.05; Fig 2B) or on: forelimb grip strength (unstim: 1.53 ± 0.12 lbs. of force; stim: 1.54 ± 0.06; p > 0.05; Fig 2C), hindlimb grip strength (unstim: 1.54 ± 0.09; stim: 1.59 ± 0.12; p > 0.05; Fig 2D), fore-hind grip strength ratio (unstim: 1.01 ± 0.04; stim: 1.04 ± 0.08; p > 0.05; Fig 2E), cervical motor neuron loss (unstim: 21.1 ± 1.18 motor neurons/section; stim: 21.2 ± 1.32; p > 0.05; Fig 4A), CMAP peak amplitude (unstim: 5.59 ± 0.27 mV; stim: 5.83 ± 0.42; p > 0.05; Fig 3A) or latency (unstim: 3.94 ± 0.19 msec; stim: 3.76 ± 0.06; p > 0.05; data not shown) at 146 days of age.
Decreased phrenic nerve stimulation had no effect on SOD1G93A rats
To test if the effects of bilateral phrenic nerve hyperstimulation on vulnerable cervical motor neurons in SOD1G93A rats are specific to stimulation intensity, a separate cohort of SOD1G93A rats was subjected to the same stimulation paradigm that accelerated disease in earlier experiments with SOD1G93A rats, except that stimulation intensity (see Materials and Methods section for specifics of changes) was reduced greater than 10-fold (n = 5 unstim; n = 4 stim). Similar to WT rats that received high stimulation, no effects of low stimulation on SOD1G93A rats were found in any outcome measures (data not shown).
Phrenic nerve hyperstimulation accelerated pathological changes at the diaphragm neuromuscular junction in SOD1G93A rats
Previous studies have demonstrated that alterations at the neuromuscular junction are some of the earliest pathological signs occurring in SOD1 rodent models of ALS (Fischer et al., 2004). To examine whether phrenic nerve hyperstimulation induced pathological changes at diaphragm neuromuscular junctions, hemi-diaphragm muscle was examined from age-matched wild-type stimulated, SOD1G93A unstimulated and SOD1G93A stimulated rats at a single time point (following 9 weeks of stimulation) prior to symptomatic onset of forelimb and respiratory dysfunction. Specifically, motor axons and their terminals were labeled with SMI-312R and SV2-s, respectively, and post-synaptic acetylcholine receptors were labeled with Alexa Fluor 647-conjugated alpha-bungarotoxin. There were no differences in total numbers of NMJs, as assessed by total numbers of alpha-bungarotoxin+ junctions (WT-stim: 267.3 ± 37.0 junctions; SOD1G93A unstim: 304.7 ± 11.2; SOD1G93A stim: 271.7 ± 17.9; p > 0.05 for all comparisons; n = 3/group; Fig 5A). Nearly all wild-type NMJs were completely intact, characterized by: complete overlap of the pre-synaptic axon and pre-synaptic vesicles with post-synaptic acetylcholine receptors, no signs of multiple innervation, absence of pre-synaptic axon thinning (Fig 5D, F). While both groups of SOD1G93A rats showed signs of synaptic disruption, stimulated SOD1G93A rats had significantly fewer intact junctions, with greater than 60% of junctions being affected (WT-stim: 99.5 ± 0.2%; SOD1G93A unstim: 85.7 ± 0.8; SOD1G93A stim: 37.0 ± 5.6; p < 0.05 for all comparisons; Fig 5B, E). To specifically examine the types of changes occurring in stimulated diaphragm muscle, NMJ changes were broken down into a number of phenotypic categories. Compared to unstimulated SOD1G93A rats, a significantly greater percentage of junctions from stimulated SOD1G93A rats showed signs of partial denervation (WT-stim: 0.0 ± 0.0%; SOD1G93A unstim: 2.0 ± 0.4; SOD1G93A stim: 20.6 ± 1.4; p < 0.05 for all comparisons; Fig 5C, H), complete denervation (WT-stim: 0.0 ± 0.0%; SOD1G93A unstim: 1.7 ± 0.4; SOD1G93A stim: 11.7 ± 4.8; p < 0.05 for all comparisons; Fig 5C), thinning of the pre-synaptic axon (WT-stim: 0.0 ± 0.0%; SOD1G93A unstim: 2.8 ± 0.4; SOD1G93A stim: 6.8 ± 1.0; p < 0.05 for all comparisons; Fig 5C, I), terminal sprouting (WT-stim: 0.4 ± 0.2%; SOD1G93A unstim: 2.8 ± 0.4; SOD1G93A stim: 6.8 ± 1.0; p < 0.05 for all comparisons; Fig 5C, G) and reinnervation of denervated junctions by terminal sprouts from adjacent junctions (WT-stim: 0.0 ± 0.0%; SOD1G93A unstim: 1.3 ± 0.2; SOD1G93A stim: 2.7 ± 0.2; p < 0.05 for all comparisons; Fig 5C, G). In many instances, individual junctions showed signs of more than 1 change. Very few instances of any of these junctional pathologies were found in wild-type stimulated muscles, in accordance with previous work (Love et al., 2003). These results show that, even at an early point in disease prior to symptomatic onset, external phrenic nerve hyperstimulation induced pathological changes at diaphragm neuromuscular junctions, as well as motor neuron loss. The degree of neuromuscular junction loss was not more significant than motor neuron loss at this point, suggesting that a distal “dying back” process, a phenomenon observed in SOD1G93A mouse hindlimbs (Fischer et al., 2004), was not evident at least at this timepoint. However, pathological evaluations at even earlier time points could reveal whether early neuromuscular junction changes preceded motor neuron loss.
Figure 5. Phrenic nerve hyperstimulation stimulation accelerated pathological changes at diaphragm neuromuscular junctions in SOD1G93A rats.
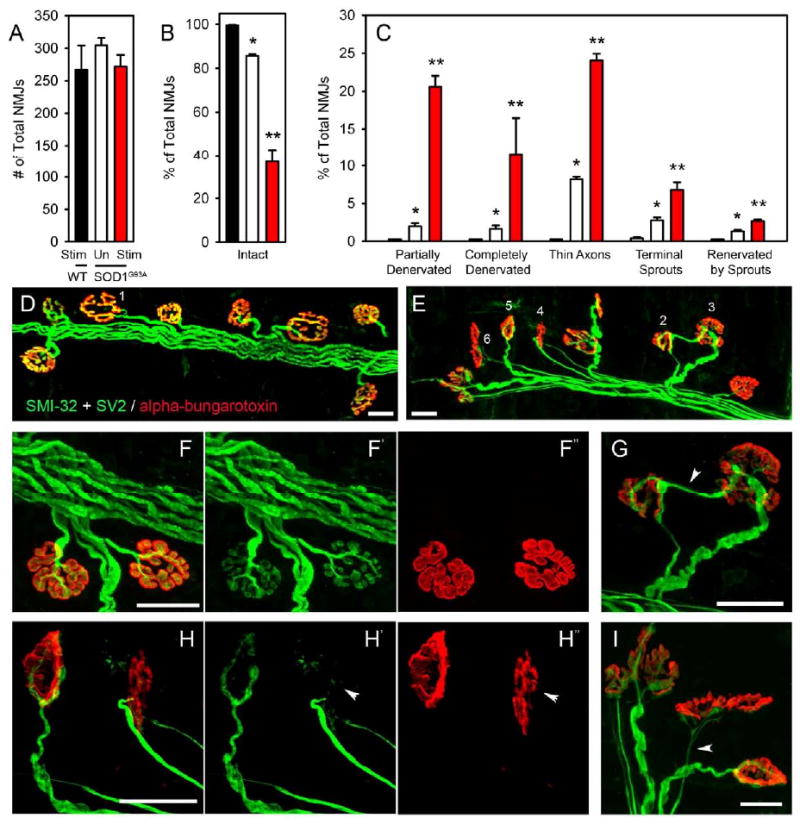
There were no differences in total numbers of NMJs among any groups, as assessed by total numbers of alpha-bungarotoxin+ junctions (A). Nearly all wild-type NMJs were completely intact, characterized by: complete overlap of the pre-synaptic axon and pre-synaptic vesicles with post-synaptic acetylcholine receptors, no signs of multiple innervation, absence of pre-synaptic axon thinning (D: NMJ #1, F). While both groups of SOD1G93A rats showed signs of synaptic disruption, stimulated SOD1G93A rats had significantly fewer intact junctions, with greater than 60% of junctions being affected (B, E). To specifically examine the types of changes occurring in stimulated diaphragm muscle, NMJ changes were broken down into a number of phenotypic categories. Compared to unstimulated SOD1G93A rats, a significantly greater percentage of junctions from stimulated SOD1G93A rats showed signs of partial denervation (C, E: NMJ #4 and #5, H - arrowhead), complete denervation (C), thinning of the pre-synaptic axon (C, E: NMJ #6, I -arrowhead), terminal sprouting (C, E: NMJ #3, G - arrowhead) and reinnervation of denervated junctions by terminal sprouts from adjacent junctions (C, E: NMJ #2, G). In many instances, individual junctions showed signs of more than 1 change. Only rare instances of any of these junctional pathologies were found in wild-type stimulated muscles. Scale bars: 25μm.
In order to address whether these morphological changes were consistent with the natural progression of changes at the NMJ observed in the SOD1G93A rat, we performed NMJ analysis of unstimulated endstage SOD1G93A rat diaphragm. At endstage, the total numbers of alpha-bungarotoxin+ junctions were unchanged compared to wild-type stimulated, SOD1G93A unstimulated and SOD1G93A stimulated rats (245.5 ± 2.5 total junctions; not shown). Compared to SOD1G93A unstimulated and SOD1G93A stimulated rats, we observed similar morphological changes in the diaphragm of endstage SOD1G93A rats, including signs of partial denervation (43.8 ± 0.6% of junctions; not shown), complete denervation (44.2 ± 3.0% of junctions; not shown), and thinning of the pre-synaptic axon (9.2 ± 2.7% of junctions; not shown). However, the degree of all of these NMJ pathologies was more pronounced in endstage animals (3.0 ± 0.8% of junctions were completely intact). Therefore, phrenic nerve stimulation accelerated pathological changes at the diaphragm NMJ normally associated with SOD1G93A rat disease progression. In addition, stimulation did not induce novel types of NMJ alterations not normally observed in SOD1G93A-based disease.
Phrenic nerve hyperstimulation is accompanied by cervical spinal cord microgliosis
In addition to motor neuron loss, we sought to examine whether other markers of SOD1-mediated disease were active. Microgliosis occurring focally in regions of SOD1G93A rat pathology has previously been reported (Lepore et al., 2008). Furthermore, microglial activation has been used in mutant SOD1 mouse models as both a sign of disease activity and a potential contributor to SOD1 disease progression (Boillee et al., 2006; Kriz et al., 2002; Van Den Bosch et al., 2002; Zhu et al., 2002). To examine whether peripheral nerve hyperstimulation resulted in an altered microglial response, Iba1 (a microglial marker) immunostaining was performed in age-matched cervical spinal cords. Immunohistochemistry revealed that compared to wild-type spinal cord (Fig 6A; n = 3), there was a significantly elevated microglial response specifically in the ventral gray matter of both SOD1G93A unstimulated (n = 3; Fig 6B) and SOD1G93A stimulated (n = 3; Fig. 6C) SOD1G93A rat groups. However, the response was significantly increased in SOD1G93A stimulated rats compared to unstimulated SOD1G93A animals (WT-stim: 57.9 ± 4.1 arbitrary units; SOD1G93A unstim: 83.4 ± 5.7; SOD1G93A stim: 123.9 ± 4.2; p < 0.05; n = 3/group; Fig 6D). However, significant differences in GFAP immunostaining between the SOD1G93A groups was not prominent (not shown) at the single time point evaluated.
Figure 6. Phrenic nerve hyperstimulation increased cervical spinal cord microgliosis in SOD1G93A rats.
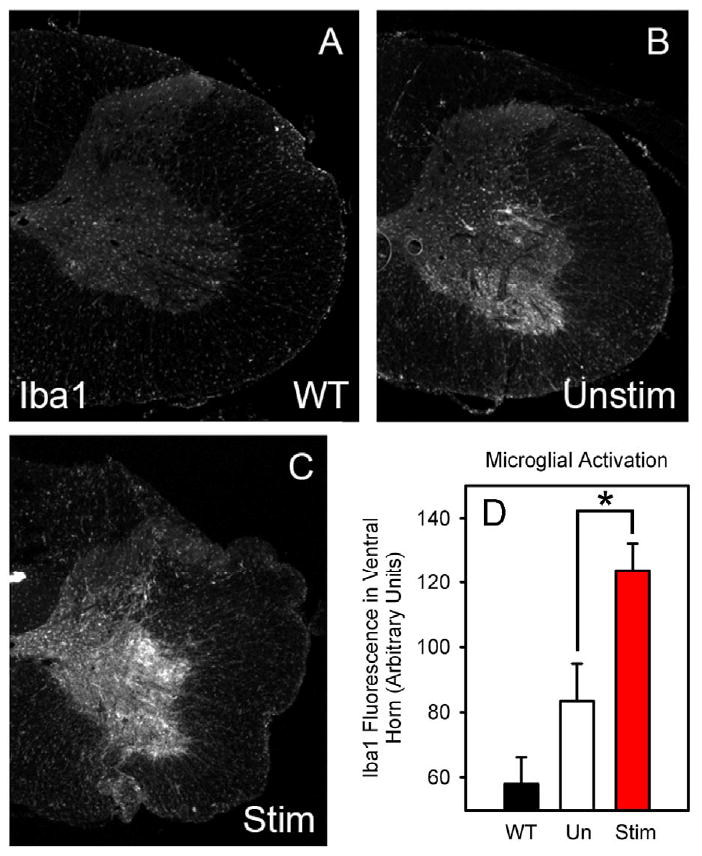
The inflammatory response was examined with the microglial marker, Iba1, in age-matched cervical spinal cords. Immunohistochemistry revealed that compared to wild-type spinal cord (A), there was a significantly elevated microglial response specifically in the ventral gray matter of both SOD1G93A unstimulated (B) and SOD1G93A stimulated (C) SOD1G93A rat groups. The response was significantly increased in SOD1G93A stimulated rats compared to unstimulated SOD1G93A animals (D).
Discussion
Phrenic motor neuron vulnerability
Previous electrophysiological and pathological studies have demonstrated that respiratory function in the SOD1G93A rat is temporally associated with disease progression, cervical motor neuron loss, and distal denervation (Llado et al., 2006). These data suggest that SOD1G93A respiratory pathobiology models human ALS respiratory dysfunction. Furthermore, using cell-transplantation studies, focal neuroprotective effects on motor neurons in this region have resulted in the maintenance of respiratory physiology, prolongation of forelimb strength, and prolonged survival in this model (Lepore et al., 2008). Taken together, our current study suggests that the focal decline in phrenic CMAPs, loss of phrenic motor neurons and distal phrenic denervation, coupled with the sparing of hindlimb grip strength, is likely responsible for the acceleration in stimulated SOD1G93A rat death, rather than non-specific systemic effects of this paradigm.
In addition to focal loss of cervical phrenic motor neurons, we also report a significant pathological effect of hyperstimulation at diaphragm neuromuscular junctions (NMJ), including accelerated denervation, axonal thinning and terminal sprouting. Previous work has demonstrated that distal changes at the NMJ can precede actual motor neuron loss in models of ALS (Fischer et al., 2004). It is intriguing to hypothesize that phrenic nerve-diaphragm hyperstimulation in SOD1G93A rats worsens disease by accelerating a distal motor neuropathy at the NMJ. However, we also find elevated central motor neuron death at the same time point examined for NMJ changes. Whether a distal motor neuropathy is the first, very early feature in this paradigm is not known since earlier time points following initiation of the experimental paradigm were not evaluated. Nevertheless, at the time point examined, both motor neuron loss and distal denervation are seen. What is evident in the assessment of distal axons and neuromuscular junctions is that hyperstimulated phrenic motor neurons and their axons in SOD1G93A rats are more vulnerable than wild-type motor neurons and that this vulnerability can be induced by activity at the nerve-muscle junction.
Mechanisms and temporal course of disease onset in ALS
Prevailing evidence suggests that, at least for sporadic ALS, there is no unifying hypothesis for the cause of disease development. However, it is clear that there are unique presentations, including most generally spinal- or bulbar-onset ALS (Mitsumoto et al., 1998). Interestingly, even within groups of patients with familial ALS and/or identical gene mutations, variability in site of onset and disease progression occurs (Andersen et al., 1997; Cudkowicz et al., 1997; Mitsumoto et al., 1998). Small reports of ALS following traumatic injury to the peripheral nervous system with onset in a particular associated limb have been published. However, the heterogeneity of the ALS population, as well as an appreciation for the severity of the injury and the time course between injury and development of clinical symptoms, has made such relationships difficult to interpret. Some epidemiological evidence also points to a relationship between developing ALS and environmental factors such as traumatic insult (Kurtzke, 1991), electrical shock (Jafari et al., 2001), military service (Haley, 2003; Horner et al., 2003), and physically-fit lifestyles, although methodologies make these small observations difficult to interpret (Armon, 2007). Though inconclusive, these small analyses point to a possible role of peripheral nervous system injury or overactivity in the focal onset of ALS and/or in hastening of disease progression. Given that most cases of ALS are not linked to known genetic causes and that familial-linked cases are heterogeneous even within families, the suggestion that environmental insults are modifiers of disease presentation and progression is an appealing concept.
Motor neurons are particularly vulnerable to cellular insult in ALS. While a number of studies in mouse models of the disease have demonstrated a beneficial effect (or at least a lack of deleterious effects) of moderate exercise on disease parameters (Chen et al., 2008; Kirkinezos et al., 2003; Liebetanz et al., 2004; McCrate and Kaspar, 2008; Veldink et al., 2003), other studies have also shown that intense exercise paradigms can actually accelerate disease (Mahoney et al., 2004). This threshold effect was observed in the present study. Hyperstimulation of SOD1G93A rats via a paradigm that bilaterally activates the phrenic nerves resulted in focal onset of respiratory dysfunction, involvement of adjacent forelimb function and overall accelerated disease progression, while stimulation at a reduced charge density in a separate cohort of SOD1G93A rats did not worsen disease. Also, hyperstimulation of wild-type rats did not have these effects, demonstrating a selective vulnerability of motor neurons with a genetic predisposition (SOD1G93A mutation) to a high intensity focal insult. These data would suggest that focal injury, in the context of a genetic predisposition to motor neuron disease, may alter both the timing and site of disease onset.
Several studies have demonstrated increased vulnerability of motor neurons in SOD1 rodents to peripheral nerve injury, including exacerbated motor neuron loss following facial nerve avulsion (Ikeda et al., 2005) transection (Mariotti et al., 2002) and sciatic nerve crush (Sharp et al., 2005). These findings support the idea that the SOD1 genotype increases motor neuron vulnerability to peripheral stimuli. Interestingly, previous work has also shown that peripheral axotomy (L5 spinal nerve, sciatic nerve) or nerve crush (tibial nerve) of lumbar motor neurons in SOD1G93A models slows loss of motor neurons (Franz et al., 2009; Kong and Xu, 1999). A possible explanation for this paradoxical finding, as well as a connection to the present results, is that axotomy prevented chronic activation of, and consequent focal injury to, these vulnerable motor neurons. Large motor neurons may be especially vulnerable because of their increased somal and axonal volumes, neurofilament content, excitatory input and metabolic requirements.
We report that hyperstimulation elevated microgliosis in the cervical spinal cord of SOD1G93A rats, suggesting that this cellular response may be a marker of this focal disease activity. A number of studies have documented elevated levels of microglial factors, including interleukins, TNF, TGF, COX2 and interferons in mouse models of ALS. These factors seem to be increased in abundance and variety as disease progresses (Sargsyan et al., 2005), consistent with the hypothesis that these cells and their factors are part of a cascade following initial injury. Conversely, administration of minocycline to transgenic mutant SOD1 mice resulted in reduction in microglial activation and prolonged survival (Kriz et al., 2002; Van Den Bosch et al., 2002; Zhu et al., 2002). While microgliosis and other factors may account for the loss of both phrenic motor neurons and adjacent forelimb motor neurons, it is also possible in our experimental paradigm that the decline in forelimb function may be the result of some spread of electrical stimulation to adjacent forelimb motor neurons, rather than only the targeted phrenic motor neuron populations. Therefore, while recent studies have implicated several cell types in both disease onset (Boillee et al., 2006) and progression (Boillee et al., 2006; Yamanaka et al., 2008), the specific mechanisms resulting in local disease pathology and disease involvement among adjacent motor neuron populations remains to be fully elucidated.
Conclusions
Even though heterogeneities in timing and site of disease onset are well recognized in ALS, few studies have been conducted to elucidate the mechanisms of these clinical phenomena. This study using the SOD1G93A rat focused on the chronic stimulation of the phrenic nerve at the motor endpoint. The results reported demonstrate that such peripheral nervous system stimulation can modify both the temporal and anatomical onset of disease in this ALS model and suggest that focal peripheral nervous system overactivity could influence clinical presentations on a genetically susceptible background. This system lays the framework for studying the mechanisms and specific factors that affect site of ALS disease onset, as well as the temporal course and regional involvement of disease. More detailed studies of focal peripheral injury in a more genetically homogeneous human ALS population (such as patients with identical SOD1 mutations) could reveal the importance of peripheral nervous system effects on anatomical sites and timing of disease onset.
Acknowledgments
We thank: all members of the Maragakis lab for discussion; Project ALS (NJM) and NIH (F32-NS059155: ACL) for funding.
Footnotes
Disclosure of financial interest: Drs. Ray Onders and Anthony Ignagni have a financial interest and are associated with Synapse Biomedical Inc., which provided the materials and equipment for diaphragm stimulation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Andersen PM, et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain. 1997;120(Pt 10):1723–37. doi: 10.1093/brain/120.10.1723. [DOI] [PubMed] [Google Scholar]
- Armon C. Sports and trauma in amyotrophic lateral sclerosis revisited. J Neurol Sci. 2007;262:45–53. doi: 10.1016/j.jns.2007.06.021. [DOI] [PubMed] [Google Scholar]
- Boillee S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, et al. Specific and artifactual labeling in the rat spinal cord and medulla after injection of monosynaptic retrograde tracers into the diaphragm. Neurosci Lett. 2007;417:206–11. doi: 10.1016/j.neulet.2007.02.047. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–38. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Chen A, et al. The role of exercise in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19:545–57. ix–x. doi: 10.1016/j.pmr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41:210–21. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- Fischer LR, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–40. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Franz CK, et al. A conditioning lesion provides selective protection in a rat model of Amyotrophic Lateral Sclerosis. PLoS One. 2009;4:e7357. doi: 10.1371/journal.pone.0007357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–5. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haley RW. Excess incidence of ALS in young Gulf War veterans. Neurology. 2003;61:750–6. doi: 10.1212/wnl.61.6.750. [DOI] [PubMed] [Google Scholar]
- Haverkamp LJ, et al. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain. 1995;118(Pt 3):707–19. doi: 10.1093/brain/118.3.707. [DOI] [PubMed] [Google Scholar]
- Horner RD, et al. Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology. 2003;61:742–9. doi: 10.1212/01.wnl.0000069922.32557.ca. [DOI] [PubMed] [Google Scholar]
- Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–9. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, et al. Motoneuron degeneration after facial nerve avulsion is exacerbated in presymptomatic transgenic rats expressing human mutant Cu/Zn superoxide dismutase. J Neurosci Res. 2005;82:63–70. doi: 10.1002/jnr.20621. [DOI] [PubMed] [Google Scholar]
- Jafari H, et al. Motor neuron disease after electric injury. J Neurol Neurosurg Psychiatry. 2001;71:265–7. doi: 10.1136/jnnp.71.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkinezos IG, et al. Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosis. Ann Neurol. 2003;53:804–7. doi: 10.1002/ana.10597. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Peripheral axotomy slows motoneuron degeneration in a transgenic mouse line expressing mutant SOD1 G93A. J Comp Neurol. 1999;412:373–80. doi: 10.1002/(sici)1096-9861(19990920)412:2<373::aid-cne13>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Kriz J, et al. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–78. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF. Risk factors in amyotrophic lateral sclerosis. Adv Neurol. 1991;56:245–70. [PubMed] [Google Scholar]
- Lepore AC, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11:1294–301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebetanz D, et al. Extensive exercise is not harmful in amyotrophic lateral sclerosis. Eur J Neurosci. 2004;20:3115–20. doi: 10.1111/j.1460-9568.2004.03769.x. [DOI] [PubMed] [Google Scholar]
- Llado J, et al. Degeneration of respiratory motor neurons in the SOD1 G93A transgenic rat model of ALS. Neurobiol Dis. 2006;21:110–8. doi: 10.1016/j.nbd.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Lo Coco D, et al. Noninvasive positive-pressure ventilation in ALS: predictors of tolerance and survival. Neurology. 2006;67:761–5. doi: 10.1212/01.wnl.0000227785.73714.64. [DOI] [PubMed] [Google Scholar]
- Love FM, et al. Activity alters muscle reinnervation and terminal sprouting by reducing the number of Schwann cell pathways that grow to link synaptic sites. J Neurobiol. 2003;54:566–76. doi: 10.1002/neu.10191. [DOI] [PubMed] [Google Scholar]
- Mahoney DJ, et al. Effects of high-intensity endurance exercise training in the G93A mouse model of amyotrophic lateral sclerosis. Muscle Nerve. 2004;29:656–62. doi: 10.1002/mus.20004. [DOI] [PubMed] [Google Scholar]
- Mariotti R, et al. Altered reaction of facial motoneurons to axonal damage in the presymptomatic phase of a murine model of familial amyotrophic lateral sclerosis. Neuroscience. 2002;115:331–5. doi: 10.1016/s0306-4522(02)00448-7. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, et al. Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res. 2006;83:119–33. doi: 10.1002/jnr.20708. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev. 2004;3:105–20. doi: 10.1016/j.arr.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrate ME, Kaspar BK. Physical activity and neuroprotection in amyotrophic lateral sclerosis. Neuromolecular Med. 2008;10:108–17. doi: 10.1007/s12017-008-8030-5. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, et al. Amyotrophic lateral sclerosis. F.A. Davis; Philadelphia: 1998. [Google Scholar]
- Nagai M, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–54. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, et al. Evaluation of electrical parameters for diaphragm pacing: an experimental study. J Surg Res. 1981;30:142–53. doi: 10.1016/0022-4804(81)90006-8. [DOI] [PubMed] [Google Scholar]
- Ravits J, Traynor BJ. Current and future directions in genomics of amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19:461–77. viii. doi: 10.1016/j.pmr.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sargsyan SA, et al. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia. 2005;51:241–53. doi: 10.1002/glia.20210. [DOI] [PubMed] [Google Scholar]
- Sharp PS, et al. The effect of peripheral nerve injury on disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neuroscience. 2005;130:897–910. doi: 10.1016/j.neuroscience.2004.09.069. [DOI] [PubMed] [Google Scholar]
- Sutedja NA, et al. What we truly know about occupation as a risk factor for ALS: A critical and systematic review. Amyotroph Lateral Scler. 2008a:1–19. doi: 10.3109/17482960802430799. [DOI] [PubMed] [Google Scholar]
- Sutedja NA, et al. Exposure to chemicals and metals and risk of amyotrophic lateral sclerosis: A systematic review. Amyotroph Lateral Scler. 2008b:1–20. doi: 10.3109/17482960802455416. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, et al. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13:1067–70. doi: 10.1097/00001756-200206120-00018. [DOI] [PubMed] [Google Scholar]
- Veldink JH, et al. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul Disord. 2003;13:737–43. doi: 10.1016/s0960-8966(03)00104-4. [DOI] [PubMed] [Google Scholar]
- Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–16. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Wright MC, et al. Distinct patterns of motor nerve terminal sprouting induced by ciliary neurotrophic factor vs. botulinum toxin. J Comp Neurol. 2007;504:1–16. doi: 10.1002/cne.21439. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–3. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–8. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]