

Abstract
Background
Mitogen activated protein (MAP) kinases and nuclear factor kappa-B (NF-κB) are implicated in early stages of acute pancreatitis pathogenesis. We investigated the relationship between the p38 MAP kinase and NF-κB in isolated acinar cells.
Methods
Isolated rodent acinar cells were stimulated with agonists after infection with an adenovector containing a luciferase promotor driven only by NF-κB and an adenovector containing the dominant negative (DN) form of p38 (empty vector in controls).
Results
Initial immunoblots confirmed that the agonist stimulated p38 activation in acinar cells was substantially attenuated by DN p38 over expression. Stimulation of native CCK-A receptors or TNF-α receptors promoted a significant increase in NF-κB-dependent gene transcription in cells infected with the empty vector, while over expression of DN p38 significantly abrogated NF-κB-dependent luciferase activity.
Conclusion
These findings support our hypothesis that p38 is involved in the activation of proinflammatory nuclear transcription factors such as NF-κB in pancreatic exocrine cells.
Keywords: MAP kinase, acinar cell, acute pancreatitis, rat, mouse, p38, NF-κB, CCK, TNF-α, adenoviral vector
Introduction
Cholecystokinin (CCK) and tumor necrosis factor-α (TNF-α) induce pancreatic acinar cells to express high levels of pro-inflammatory mediators (1;2). Nuclear factor kappa-B (NF-κB) is a transcription factor that induces various pro-inflammatory mediators (3–5). Although the p38 mitogen activated protein (MAP) kinase is known to regulate pro-inflammatory cytokine expression, its role in pancreatic acinar cells has not been elucidated. We have shown that NF-κB-dependent gene transcription in the AR42J exocrine pancreatic cancer cell line requires an active extracellular signal-regulated kinase (ERK) (6). In the current study, we ask if the p38 MAP kinase has a role in pro-inflammatory gene expression via regulating NF-κB-dependent transcription in pancreatic acini. The isolated acinar cell primary culture used in the present study is a well established model but the use of a luciferase reporter for measurement of NF-κB-dependent transcription has to the best of our knowledge not previously been validated in this model. Using adenoviral vector delivery of a specific dominant negative (DN) form of p38, our experiments show that the absence of p38 MAP kinase activity abrogates CCK- or TNF-α-stimulated NF-κB-dependent transcription in isolated acinar cells.
Materials and Methods
Materials
Dulbecco’s modified Eagle medium (DMEM), penicillin and streptomycin were purchased from Invitrogen (Grand Island, NY). Trypsin inhibitor was from Sigma (St. Louis, MO; Cat. No. T6522). Probumin Millipore Universal Grade bovine serum albumin (BSA) was obtained from Millipore (Kankakee, IL; Cat. No. 81-003-3), and purified collegenase was from Worthington Biochemical (Lakewood, NJ; Cat. No. LS005273). Replication-deficient adenoviruses expressing GFP (Ad.GFP), NF-κB-luciferase (Ad.NF-κB-luc), and empty vector (Ad.EV) were purchased from the University of Iowa Vector Core Facility (7). Adenovirus expressing a recombinant dominant negative form of murine p38-α was procured from Cellbiolabs (San Diego, CA; Cat. No. ADV-105). Sulphated CCK-8 was bought from Sigma (St Louis, MO; Cat. No. C2175) and rat recombinant TNF-α from R&D Systems, Inc., (Minneapolis, MN; Cat. No. 510-RT-010). Specific antibody against phospho-NF-κB p65 (serine 536; Cat. No. 3033) and rabbit polyclonal specific antibody recognizing the phosphorylation site Thr180/Tyr182 of phospho-p38 (Cat. No. 9211) were obtained from Cell Signaling (Danvers, MA). ECL Plus Western Blotting detection reagent was from Amersham Pharmacia Biotech (Piscataway, NJ; Cat. No. RPN2132). Sprague Dawley rats weighing 200–250 g were from Harlan Laboratories (Indianapolis, Indiana) and C57/BL6 mice were from Jackson Laboratories (Bar Harbor, ME). All studies were approved by the Institutional Animal Care and Use Committees.
Cell Culture
Primary cultures of pancreatic acinar cells were isolated from the freshly excised pancreata of healthy mice or rats by methods previously described by Dr. John A. Williams (University of Michigan, Ann Arbor, MI) (8), with additional modifications suggested by Dr. Williams. In brief, the pancreata of mice weighing between 30–50 g or rats between 200–250 g were excised under general anesthesia, digested with DMEM (containing collagenase, 0.25% BSA, penicillin and streptomycin, and trypsin inhibitor), titurated, filtered through 150 μm mesh, and purified using a linear 4% BSA gradient. After a 2 hr room temperature equilibration, acinar cells were seeded in 12-well non-treated tissue culture plates at approximately 350–400 μg protein/ml using 1 ml/well of DMEM containing BSA, trypsin inhibitor, and penicillin and streptomycin. Protein concentrations were determined using the BCA reagent (Pierce, Rockford, IL; Cat No. 23250) according to manufacturer’s instructions. All incubations were carried out at 37°C in 95% air and 5% CO2. Mouse pancreas was used in all studies except the luciferase studies where the rat pancreas was used to enable us to harvest more cells. Cell viability in culture was evaluated using an ATP assay (CellTiter-Glo® Luminescent Cell Viability assay, Promega, Madison, WI; Cat. No. G7573), and cell injury was assessed with an LDH assay (CytoTox96, Promega, Madison, WI; Cat. No. G1780). Quantitation of α-amylase activity was done using the Phadebas test (Magle Life Sciences, Lund, Sweden; Cat. No. 1301).
Time course and dose response studies
To determine the optimal response time for acinar cell p38 MAP kinase activation, cells were stimulated with 10 μM CCK or 1 ng/ml TNF-α for 0, 1, 5, or 20 min prior to harvest. Optimal CCK concentration was tested at 10 nM, 100 nM, 1 μM, or 10 μM while TNF-α was tested at 1, 10, 25, or 50 ng/ml. Immunoblotting was performed as previously described (9). Blots were probed with antibody against phosphorylated p38, visualized using ECL Plus Western Blotting detection reagent (Amersham Pharmacia Biotech, Piscataway, NJ, USA), stripped for 30 min at 50°C with a mild stripping buffer (69.3 mM SDS, 125 mM Tris pH 6.8, 243 mM B-mercaptoethanol), rinsed in TBS with 1% tween 20 (v:v) and reblocked. Blots were then re-probed for α-tubulin to evaluate sample loading.
Viral infections
To assess infection efficiency, freshly isolated acinar cells were incubated with either Ad.GFP or Ad.EV (1E7 pfu/ml for 24h) and expression of GFP was observed on an Olympus IX51 Inverted Florescent Microscope (Leeds Precision Instruments, Inc., Minneapolis, MN). This was followed by an ATP assay to assess cell viability. To determine the effect of the DN p38 MAP kinase on CCK- or TNF-α-stimulated acinar cells, we infected cultures with 1E7 pfu/ml Ad.DN p38 or Ad.EV for 48 hrs and evaluated p38 activation by immunoblotting. To evaluate of the role of p38 activation on NF-κB-dependent gene transcription, 1E7 pfu/ml of Ad.NF-κB-luc was co-infected with 1E7 pfu/ml Ad.DN p38 or Ad.EV for 48h. Cells were stimulated with 10 μM CCK for 24 hrs or 10 ng/ml TNF-α for 6 hrs prior to harvest. Cells were harvested and induction of NF-κB-mediated transcription was evaluated by measuring luminescence using the Promega luciferase assay system (Promega, Madison, WI). Measurements in relative light units (RLU) were normalized using protein concentration.
Statistical Analysis
One-way ANOVA or the paired t-test were used (SigmaStat software, SPSS Inc., Chicago, IL). Three wells were studied in each group and results expressed as mean±SEM. P values <0.05 were considered significant.
Results
Acinar cell morphology and cell viability
Photomicrographs of isolated mouse acinar cells show morphology suggestive of healthy looking acini when freshly isolated and also at 24 hrs, while at 48 hr we see some clumping of cells along with diminished zymogen granules (Fig. 1). Viability of cells in culture was assessed with ATP assay of cell lysates that showed mild to moderate decreases in ATP activity over time compared to 0-h control value taken as 100% [71±10% at 24 h, and 67±1%# at 48 h, in the absence of viral infection; 81±5% at 24 h, and 68±6%# at 48hr, in the presence of empty virus at 1E7 pfu/ml; paired t-test, pound sign (#) indicates p<0.05 vs 0 and 24 h]. Cell injury was assessed by measuring LDH release from cells into the medium over time, where the 0-h control value was considered as 0% LDH release: 42±3%* at 24 h and 48±3%* at 48hrs, in the absence of virus; 40±6%* at 24 h and 56±2%# at 48hrs, in the presence of empty virus at 1E7 pfu/ml [paired t-test, asterisk (*) indicates p<0.05 vs 0 h, pound sign (#) indicates p<0.05 vs 0 and 24 h]. Cells responded to CCK stimulation (10 μM, 5 min) with percentage of amylase release compared to total of 6±1.6% at 0 h and 5.4±0.7% at 24 h, while at 48 h it was only 1.7±0.7% (Student’s t-test; p = 0.015, 0.005, and 0.082, respectively).
Figure 1. Morphological appearance of freshly isolated acinar cells.
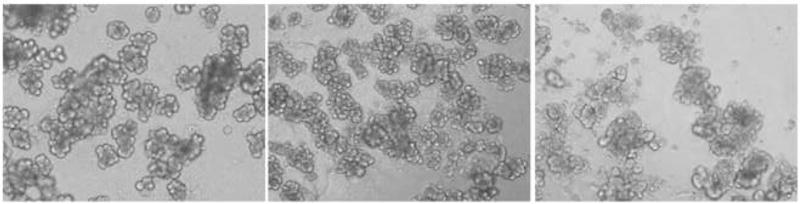
Photomicrograph of freshly isolated mouse acinar cells (magnification 60 X) shows healthy cells with the intact cell membrane discernable (left panel). The cell morphology remains essentially healthy at 24 hrs (middle panel), while at 48 hrs there is some clumping of cells and the apical darkness from zymogen granules is diminished but the cell membranes remain unbroken (right panel).
Dose response and time course of agonist stimulation
Immunoblots show that both CCK- and TNF-α-stimulation activate p38 in both a time-and dose-dependent manner in isolated acinar cells (Fig. 2), indicating that our isolation methods produce acinar cells that respond to agonists. CCK at 1 μM concentration and TNF-α at a concentration of 1 ng/ml or above showed robust p38 activation. Time course studies showed peak p38 activation at 5 minutes with CCK or TNF-α. Immunoblots of tubulin were used to evaluate protein loading.
Figure 2. Dose response and time course of p38 activation with agonist stimulation of mouse acinar cells.
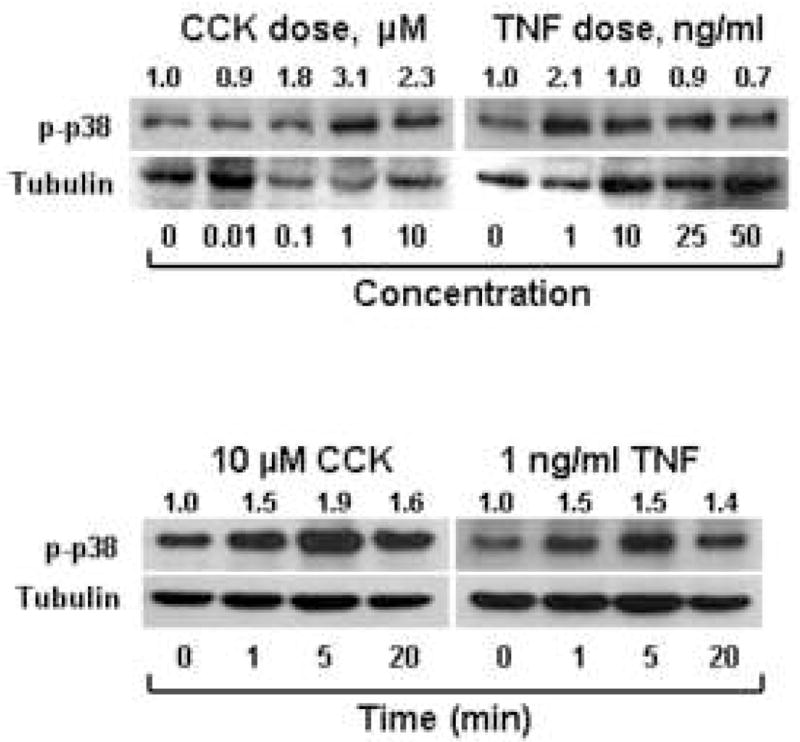
Freshly isolated acinar cultures were stimulated with increasing concentrations of either CCK for 1 minute or TNF-α for five minutes and then immunoblots were done using antibody specific to phosphorylated p38. Results show dose-dependent p38 activation with CCK peaking at 1 μM concentration, while TNF-α stimulated activation occurred at 1 ng/ml doses and higher. Similarly, when stimulated with 10 μM CCK or 1 ng/ml TNF-α for various time points, p38 activation peaked at 5 minutes. Densitometry ratios, normalized to tubulin loading, are provided above each lane with the control value represented as 1.0.
Infection efficiency
To evaluate infection efficiency, isolated acinar cells were infected with adeno-GFP. Fluorescent microscopic examination showed that essentially 100% of cells were infected, and immunoblotting of cell lysates further confirmed GFP expression (Fig. 3).
Figure 3. Infection efficiency of mouse acinar cells infected with Ad.GFP.
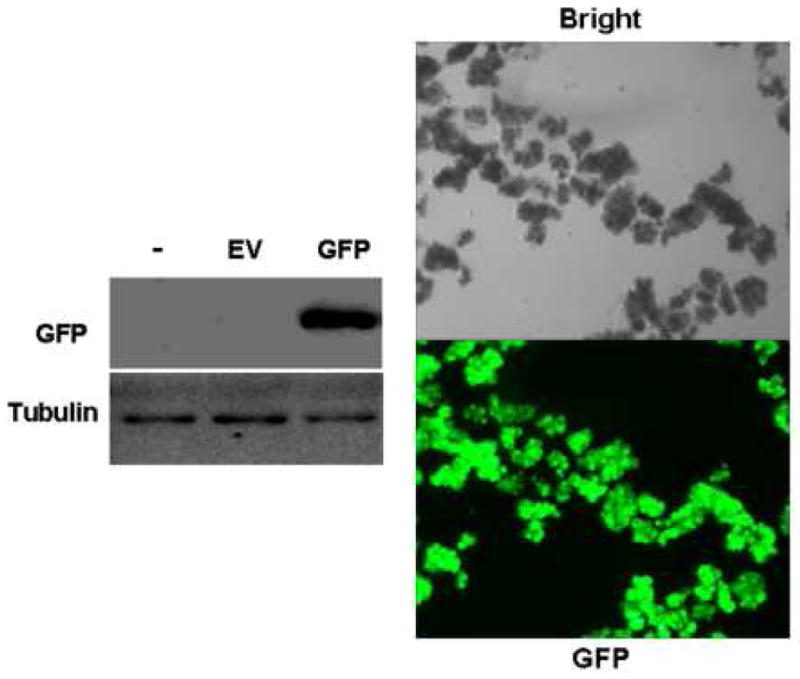
Immunoblots of acinar cells show GFP expression in cells infected with adenoviral GFP but not with empty virus (EV) or no virus controls (−). Fluorescent microscopic examination of cells infected with Ad.GFP over 24h with either bright field (top) or fluorescein isothiocyanate (FITC) filter (bottom) shows GFP fluorescence with essentially 100% infection efficiency (magnification 10 X).
Functional evidence of DN p38 expression
To evaluate the effect of Ad.DN p38 infection on agonist-stimulated increases of p38 MAP kinase activity, isolated acinar cells were infected with either Ad.DN p38 or Ad.EV prior to stimulation with 10 μM CCK or 1 ng/ml TNF-α (Fig. 4). Immunoblots showed that agonist stimulation was associated with robust activation of p38 MAP kinase in cells infected with Ad.EV. However, in cells infected with Ad.DN p38 there was evidence of markedly attenuated p38 MAP kinase activation following stimulation with CCK or TNF-α, providing evidence for the functional effect of DN p38 expression (Fig. 4).
Figure 4. Expression of DN p38 in mouse acinar cells attenuates agonist-stimulated p38 activation.

Cells infected with 1E7 pfu/ml Ad.DN p38 for 48h showed diminished activation of p38 after stimulation with 10 μM CCK or 1 ng/ml TNF-α over 5 minutes, compared to Ad.EV controls. Tubulin immunoblots show changes are not due to unequal protein loading.
Effect of DN p38 on NF-κB-dependent transcription
MAP kinases regulate the expression of inflammatory mediators at the transcriptional level in several cell types by modulation of NF-κB (3–5;10). Therefore, we evaluated the role of p38 MAP kinase in modulating transcriptional activity of NF-κB in isolated acinar cells (Figure 5). Stimulation of native CCK-A receptors or TNF-α receptors by the respective specific receptor agonist in isolated acinar cells expressing the empty vector promotes a significant increase in NF-κB-dependent gene expression, as measured by luciferase activity (Fig. 5), and this increase was markedly greater with TNF-α than with CCK stimulation. At the same time, over expression of DN p38 significantly abrogated CCK- or TNF-α-stimulated NF-κB-dependent luciferase activity. These findings indicate that CCK- or TNF-α-stimulated NF-κB-dependent gene transcription is regulated by p38 MAP kinase in isolated acinar cells. Immunoblot findings indicate that the p65 subunit of NF-κB is the target of p38 MAP kinase because CCK- or TNF-stimulated p65 activation is subdued by DN p38 over expression (Fig. 5). This corroborates our hypothesis that p38 regulates the activation of pro-inflammatory transcription factors, such as NF-κB, in pancreatic exocrine cells.
Figure 5. Expression of DN p38 in acinar cells attenuates agonist-stimulated NF-κB-dependent transcription.

Top – Rat acinar cells co-infected with Ad.NF-κB-luc and Ad.DN p38 showed diminished NF-κB luciferase activity after stimulation with 10 μM CCK or 10 ng/ml TNF-α, compared to coinfection with Ad.NF-κB-luc and Ad.EV. ANOVA, p<0.05; asterisk (*) indicates significant difference from the unstimulated empty vector control group; pound sign (#) indicates significant difference from the stimulated empty vector group; n = 3 wells/group. RLU = relative light units. Bottom – Immunoblots showed increased phosphorylation of the p65 subunit of NF-κB when mouse acinar cells were stimulated with CCK or TNF, while limited activation of p65 is evident when cells were stimulated after over expressing DN p38. Tubulin blots were used to normalize data. (EV = empty vector, DN p38 = dominant negative p38)
Discussion
This is a preliminary report using a recently developed model of isolated pancreatic acinar cells infected in culture to evaluate mechanisms of NF-κB-dependent gene transcription. Using this model, we report new findings that illustrate the important role for p38 MAP kinase in NF-κB-dependent gene expression following agonist stimulation of exocrine pancreatic cells. In addition to developing this model, we have performed preliminary characterization studies on the isolated acinar cell culture involving time course and dose response studies to important agonists such as CCK and TNF-α. Our findings suggest that without activation of p38 MAP kinase notable NF-κB-dependent transcription does not occur in acinar cells. We have also elucidated the novel finding that the p65 subunit of NF-κB is the target of p38 MAP kinase in acinar cells.
NF-κB is a transcription factor necessary for the transcription of many pro-inflammatory mediators such as cytokines, chemokines, and oxygen derived free radicals. In quiescent cells, NF-κB is present in the cytosol complexed with I-κB. The phosphorylation of I-κB on serines within the amino-terminal domain results in the dissociation and translocation of NF-κB to the nucleus and initiates gene transcription. The expression of pro-inflammatory mediators has been shown to be regulated by MAP kinases through modulation of NF-κB-dependent gene transcription in several cell types (3–5). We have previously shown that in the AR42J exocrine pancreatic malignant cell line both CCK and TNF-α activate NF-κB, and that the MAP kinase ERK plays a role in regulating NF-κB-dependent gene expression (6).
The regulation of NF-κB activation can occur at multiple levels after cell stimulation, but the mechanism by which the p38 MAP kinase regulates NF-κB-dependent gene expression in pancreatic acinar cells is not known. Studies have demonstrated that the p38 MAP kinase phosphorylated the TATA-binding protein (TBP), which is required for the interaction of TBP with the p65 subunit of NF-κB (3;4). This direct interaction is required for NF-κB transcription activity. Transcriptional activity also requires that the p65 subunit be phosphorylated. The kinases known to phosphorylate the p65 subunit are protein kinase A (PKA), p21ras, PKCζ, IKK, AKT (protein kinase B), and calmodulin-dependent protein kinase IV (CaMKIV) (11–16). It is not clear whether the p38 MAP kinase can phosphorylate the p65 subunit of NF-κB. In our study, we show that CCK- or TNF-stimulated p65 activation is subdued by DN p38 over expression suggesting that the p65 subunit of NF-κB is the target of p38 MAP kinase in acinar cells.
MAP kinases are essential in transferring signals from the cell surface receptors to the nucleus, but their role in acute pancreatitis pathogenesis is just being appreciated (17–21). Interestingly, some reports suggest that p38 activation exacerbates acute pancreatitis (18;21), while another study reports the contradictory view with findings that p38 activation actually protects against acute pancreatitis (20). Therefore, whether acute pancreatitis is exacerbated or improved by p38 remains to be clarified. Previous studies using pancreatic fragments stimulated by the CCK analog, cerulein, have shown that pro-inflammatory mediator production increased in parallel with activation of p38 and that these changes were attenuated by chemical inhibitors of p38 (22). However, chemical inhibitors of p38 MAP kinase may lack specificity as suggested by recent reports that they also inhibit the CCK-A cell surface receptor (23). Therefore, utilizing genetic manipulation via over expression of an adenoviral vector expressing DN p38 is a superior method of modulating p38 MAP kinase with respect to the non-specificity of chemical inhibitors. The isolated acinar cell model also has advantages over the pancreatic fragment preparation as the latter system includes several cell types other than exocrine cells, such as tissue macrophages, white blood cells, ductal epithelium, and vascular endothelium.
Previous studies using the rat pancreatic duct ligation-induced model of experimental acute pancreatitis show that both p38 and NF-κB are activated within a few hours of ligation (2;19;24). Bile-pancreatic juice exclusion-induced hypercholecystokininemia and subsequent acinar cell hyperstimulation in the presence of duct obstruction is implicated in the mechanism of MAP kinase activation in the duct ligation model, as duodenal bile and pancreatic juice replacement from a donor rat attenuates ligation-induced MAP kinase activation, nuclear translocation of NF-κB, and inflammatory mediator production (2;19;24). As CCK stimulation of acinar cells is associated with acinar cell overproduction of cytokines such as TNF-α, and as TNF-α has autocrine and paracrine stimulatory effects on acinar cells, we have focused our attention in the present study on the agonist effects of CCK and TNF-α on NF-κB activation (1). The most important finding in the present study is that we used gene manipulation to specifically render p38 incapable of phosphorylation by inserting a dominant negative sequence in the place of its dual phosphorylation site (Thr180/Tyr182) to show that abrogation of p38 activation in isolated acinar cells occurs with a concomitant subjugation of NF-κB-dependent gene transcriptional activity. Our current findings, taken together with previous in vivo and in vitro studies, support the view that activation of p38 by CCK, or cytokines such as TNF-α, regulates pro-inflammatory transcription factors in pancreatic exocrine cells (2;19;24;25). The potential over production by the acinar cell of pro-inflammatory mediators such as cytokines, chemokines, and oxygen-derived free radicals by NF-κB underlines the importance of the possible role of p38 MAP kinase in the pathogenesis of acute pancreatitis.
A number of studies have used isolated acinar cells as a model for elucidating agonist stimulated intracellular signaling pathways in the pancreas. For example, one study showed that CCK activates intracellular digestive zymogen release in isolated acinar cells (26). Similarly, other studies showed that increased intracellular cAMP promotes carbachol-stimulated zymogen release in isolated acinar cells (27) and that calcineurin mediates caerulein-induced intracellular pancreatic zymogen activation (28). In addition, in isolated acinar cells, phosphatidylinositol 3-kinase (PI3K) has been shown to play a potential role in regulating the severity of acute pancreatitis (29). Stimulation with calcium has been found to induce rapid changes in protein phosphorylation events in pancreatic acini (30). Adenoviral vectors were used to deliver DN forms of protein kinase C (PKC) isoforms to elucidate the specific PKC-delta isoform involved with CCK stimulated amylase secretion (31). Agonist stimulated NF-κB activation was found to be mediated by PKC-δ and –ε in isolated pancreatic acinar cells (32;33). Therefore, NF-κB may be activated by a variety of upstream signals and agonist stimulation may contribute to disease pathogenesis by zymogen activation in addition to MAP kinase activation. These previous studies support our strategy of using acinar cells for preliminary investigations prior to performing in vivo studies.
In future studies we plan to use the isolated acinar cell model to investigate the importance of MAP kinases in the production of inflammatory mediators such as cytokines, chemokines, and free radicals. It will also be useful to extend the dose response and time course studies of MAP kinase activation to acinar cells following 24 and 48 hrs in culture and to evaluate amylase secretion over a lower dose range of CCK with and without viral infection. Inhibition of MAP kinases in isolated acinar cells with the use of chemical inhibitors and RNA interference techniques will further complement our present findings. In vivo applications of viral vector-mediated MAP kinase inhibition in experimental acute pancreatitis may provide further insight into the potential clinical applications of these findings.
In summary, we have performed dose-response and time-course studies of p38 MAP kinase activation by CCK and TNF-α in isolated acinar cells, and demonstrated that Ad.DN p38 attenuates CCK- or TNF-α-stimulated p38 activation. We have shown evidence of excellent infection efficiency with adenoviral vectors in isolated acinar cells. Using a luciferase reporter construct, we have developed a novel model of isolated acinar cells that specifically detects NF-κB-dependent transcription. Using this model, we have demonstrated the role of p38 in CCK- or TNF-α-stimulated NF-κB activation and emphasize the potential role of this pathway in the pathogenesis of acute pancreatitis.
Acknowledgments
This material is based upon work supported in part by: (a) VA Merit Review Award (to Dr. I. S.), the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development (Biomedical Laboratory Research and Development), Washington D.C., (b) Grant DK-071731 (to Dr. I. S.), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health, Bethesda, MD, and (c) NIH grants ES-015981 and ES-014871 (to Dr. A.B.C.), National Institutes of Health, Bethesda, MD.
Footnotes
Abstract presented at the annual meeting of the Association of VA Surgeons, Boston, MA, April 19, 2009.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Deborah E. Williard, Iowa City Veterans Affairs Medical Center, and the Department of Surgery, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, IA 52242.
Erik Twait, Iowa City Veterans Affairs Medical Center, and the Department of Surgery, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, IA 52242.
Zuobiao Yuan, Iowa City Veterans Affairs Medical Center, and the Department of Surgery, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, IA 52242.
A. Brent Carter, Iowa City Veterans Affairs Medical Center, and the Department of Internal Medicine, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, IA 52242.
Isaac Samuel, Iowa City Veterans Affairs Medical Center, and the Department of Surgery, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, IA 52242.
Reference List
- 1.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998 Jan;175(1):76–83. doi: 10.1016/s0002-9610(97)00240-7. [DOI] [PubMed] [Google Scholar]
- 2.Samuel I, Zaheer S, Zaheer A. Bile-pancreatic juice exclusion increases p38MAPK activation and TNF-alpha production in ligation-induced acute pancreatitis in rats. Pancreatology. 2005;5(1):20–6. doi: 10.1159/000084486. [DOI] [PubMed] [Google Scholar]
- 3.Carter AB, Knudtson KL, Monick MM, Hunninghake GW. The p38 mitogen-activated protein kinase is required for NF-kappaB-dependent gene expression. The role of TATA-binding protein (TBP) J Biol Chem. 1999 Oct 22;274(43):30858–63. doi: 10.1074/jbc.274.43.30858. [DOI] [PubMed] [Google Scholar]
- 4.Carter AB, Hunninghake GW. A constitutive active MEK --> ERK pathway negatively regulates NF-kappa B-dependent gene expression by modulating TATA-binding protein phosphorylation. J Biol Chem. 2000 Sep 8;275(36):27858–64. doi: 10.1074/jbc.M003599200. [DOI] [PubMed] [Google Scholar]
- 5.Chew J, Biswas S, Shreeram S, Humaidi M, Wong ET, Dhillion MK, Teo H, Hazra A, Fang CC, Loepez-Collazo E, et al. WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat Cell Biol. 2009 Apr 19; doi: 10.1038/ncb1873. [DOI] [PubMed] [Google Scholar]
- 6.Samuel I, Tephly L, Williard DE, Carter AB. Enteral exclusion increases map kinase activation and cytokine production in a model of gallstone pancreatitis. Pancreatology. 2008;8(1):6–14. doi: 10.1159/000114850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PB, Jr, Ritchie TC, Hunninghake GW, Zandi E, Engelhardt JF. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-kappa B. J Biol Chem. 2001 Aug 10;276(32):30188–98. doi: 10.1074/jbc.M102061200. [DOI] [PubMed] [Google Scholar]
- 8.Bi Y, Page SL, Williams JA. Rho and Rac promote acinar morphological changes, actin reorganization, and amylase secretion. Am. J. Physiol Gastrointest. Liver Physiol. 2005 Sep;289(3):G561–G570. doi: 10.1152/ajpgi.00508.2004. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan R, Zaheer A, Jaye M, Lim R. Molecular cloning and expression of biologically active human glia maturation factor-beta. J Neurochem. 1991 Aug;57(2):483–90. doi: 10.1111/j.1471-4159.1991.tb03777.x. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002 Apr;109( Suppl):S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 11.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997 May 2;89(3):413–24. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell. 1998 Apr;1(5):661–71. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 13.Anrather J, Csizmadia V, Soares MP, Winkler H. Regulation of NF-kappaB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Czeta in primary endothelial cells. J Biol Chem. 1999 May 7;274(19):13594–603. doi: 10.1074/jbc.274.19.13594. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999 Oct 22;274(43):30353–6. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 15.Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol. 2000 Mar;20(5):1626–38. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang MK, Goo YH, Sohn YC, Kim YS, Lee SK, Kang H, Cheong J, Lee JW. Ca2+/calmodulin-dependent protein kinase IV stimulates nuclear factor-kappa B transactivation via phosphorylation of the p65 subunit. J Biol Chem. 2001 Jun 8;276(23):20005–10. doi: 10.1074/jbc.M010211200. [DOI] [PubMed] [Google Scholar]
- 17.Schafer C, Williams JA. Stress kinases and heat shock proteins in the pancreas: possible roles in normal function and disease. J Gastroenterol. 2000;35(1):1–9. doi: 10.1080/003655200750024443. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Murphy C, Denham W, Botchkina G, Tracey KJ, Norman J. Evidence of a central role for p38 map kinase induction of tumor necrosis factor alpha in pancreatitis-associated pulmonary injury. Surgery. 1999 Aug;126(2):216–22. [PubMed] [Google Scholar]
- 19.Samuel I, Zaheer S, Nelson JJ, Yorek MA, Zaheer A. CCK-A receptor induction and P38 and NF-kappaB activation in acute pancreatitis. Pancreatology. 2004;4(1):49–56. doi: 10.1159/000077067. [DOI] [PubMed] [Google Scholar]
- 20.Fleischer F, Dabew R, Goke B, Wagner AC. Stress kinase inhibition modulates acute experimental pancreatitis. World J Gastroenterol. 2001 Apr;7(2):259–65. doi: 10.3748/wjg.v7.i2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murr MM, Yang J, Fier A, Gallagher SF, Carter G, Gower WR, Norman JG. Regulation of Kupffer cell TNF gene expression during experimental acute pancreatitis: The role of p38-MAPK, ERK1/2, SAPK/JNK, and NF-kappa B. Journal of Gastrointestinal Surgery. 2003 Jan;7(1):20–5. doi: 10.1016/s1091-255x(02)00053-7. [DOI] [PubMed] [Google Scholar]
- 22.Samuel I, Zaheer A, Fisher RA. In Vitro Evidence for Role of ERK, p38, and JNK in Exocrine Pancreatic Cytokine Production. J Gastrointest Surg. 2006 Dec;10(10):1376–83. doi: 10.1016/j.gassur.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Morel C, Ibarz G, Oiry C, Carnazzi E, Berge G, Gagne D, Galleyrand JC, Martinez J. Cross-interactions of two p38 mitogen-activated protein (MAP) kinase inhibitors and two cholecystokinin (CCK) receptor antagonists with the CCK1 receptor and p38 MAP kinase. J Biol Chem. 2005 Jun 3;280(22):21384–93. doi: 10.1074/jbc.M408851200. [DOI] [PubMed] [Google Scholar]
- 24.Meyerholz DK, Williard DE, Grittmann AM, Samuel I. Murine pancreatic duct ligation induces stress kinase activation, acute pancreatitis, and acute lung injury. Am J Surg. 2008 Nov;196(5):675–82. doi: 10.1016/j.amjsurg.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 25.Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002 Feb;122(2):448–57. doi: 10.1053/gast.2002.31060. [DOI] [PubMed] [Google Scholar]
- 26.Leach SD, Modlin IM, Scheele GA, Corelick FS. Intracellular activation of digestive zymogens in rat pancreatic acini. J Clin Invest. 1991 Jan;87:362–6. doi: 10.1172/JCI114995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaudhuri A, Kolodecik TR, Gorelick FS. Effects of increased intracellular cAMP on carbachol-stimulated zymogen activation, secretion, and injury in the pancreatic acinar cell. Am. J. Physiol Gastrointest. Liver Physiol. 2005 Feb;288(2):G235–G243. doi: 10.1152/ajpgi.00334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Husain SZ, Grant WM, Gorelick FS, Nathanson MH, Shah AU. Caerulein-induced intracellular pancreatic zymogen activation is dependent on calcineurin. Am. J. Physiol Gastrointest. Liver Physiol. 2007 Jun;292(6):G1594–G1599. doi: 10.1152/ajpgi.00500.2006. [DOI] [PubMed] [Google Scholar]
- 29.Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J. Clin. Invest. 2001 Nov;108(9):1387–95. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner AC, Williams JA. Calcium induces rapid changes in protein phosphorylation in permeabilized pancreatic acini. Pancreas. 1995 Oct;11(3):236–40. doi: 10.1097/00006676-199510000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Li C, Chen X, Williams JA. Regulation of CCK-induced amylase release by PKC-delta in rat pancreatic acinar cells. Am. J. Physiol Gastrointest. Liver Physiol. 2004 Oct;287(4):G764–G771. doi: 10.1152/ajpgi.00111.2004. [DOI] [PubMed] [Google Scholar]
- 32.Satoh A, Gukovskaya AS, Edderkaoui M, Daghighian MS, Reeve JR, Jr, Shimosegawa T, Pandol SJ. Tumor necrosis factor-alpha mediates pancreatitis responses in acinar cells via protein kinase C and proline-rich tyrosine kinase 2. Gastroenterology. 2005 Aug;129(2):639–51. doi: 10.1016/j.gastro.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR, Jr, Shimosegawa T, Pandol SJ. PKC-delta and -epsilon regulate NF-kappaB activation induced by cholecystokinin and TNF-alpha in pancreatic acinar cells. Am J Physiol Gastrointest. Liver Physiol. 2004 Sep;287(3):G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]