

Abstract
Proteins that control the excitability of neurons, including voltage-dependent ion channels and neurotransmitter receptors, reside in a membrane lipid environment that includes sphingomyelin, but the influence of the metabolism of this lipid on excitability is unknown. Sphingomyelin in the plasma membrane can be cleaved by neutral sphingomyelinases (nSMase) to generate ceramides and sphingosine-1-phosphate (S1P) which have been shown to play a variety of roles in cellular signaling processes. We found that application of nSMase to hippocampal slices results in a selective enhancement in the population spike amplitude, resulting in ES potentiation of the CA3-CA1 schaeffer collateral synapse. Single cell recordings showed that nSMase activity increases action potential frequency in CA1 neurons in a reversible manner. Additional current clamp recordings showed that nSMase reduces the slow after-hyperpolarization after a burst of action potentials. Mass spectrometry-based measurements demonstrated that nSMase activity induces a rapid increase in the levels of ceramides and S1P in cells in hippocampal slices. The ability of nSMase to increase CA1 neuron excitability was blocked by an inhibitor of sphingosine kinase, the enzyme that converts ceramide to S1P. Moreover, direct intracellular application of S1P to CA1 neurons increased action potential firing. Our findings suggest roles for sphingomyelin metabolism and S1P in the positive regulation of the excitability of hippocampal neurons.
Keywords: Neuronal Excitability, action potentials, ceramide, lipid rafts, oxidative stress, sphingomyelinase, synaptic plasticity, sphingosin-1-phosphate
Introduction
Receptors for many cytokines, growth factors and neurotransmitters are concentrated in microdomains of the plasma membrane that are enriched in cholesterol and sphingomyelin (Mattson 2005). Receptor stimulation in such “lipid rafts” can activate sphingomyelinases (SMases) which cleave sphingomyelin to generate ceramides and sphingosine-1-phosphate (S1P), bioactive lipid mediators believed to play roles in a range of physiological and pathological conditions including cell proliferation and survival, cancer and inflammatory diseases (Hannun and Linardic 1993; Kolesnick and Krönke 1998; Hannun and Obeid 2008). In neurons, sphingomyelinases are activated in response to cytokines such as TNF and neurotrophic factors such as NGF, and may mediate the effects of these factors on neuronal survival and plasticity (DeFreitas et al. 2001; Yu et al. 2000). In addition, oxidative stress can activate SMases, a mechanism implicated in the pathogenesis of Alzheimer’s disease (Cutler et al. 2004; He et al. 2008), amyotrophic lateral sclerosis (Cutler et al. 2002), stroke (Yu et al. 2000) and HIV dementia (Bandaru et al. 2007). Neutral SMase (nSMase) and acidic SMase (aSMase), the two major SMases in mammals, are widely expressed in cells throughout the body and brain (Maruyama and Arima 1989; Tomiuk et al. 1998). nSMase localizes to the plasma membrane where it is activated by cell surface receptors, whereas aSMase is located primarily in acidic internal organelles such as lysosomes (Schissel et al. 1996; Grassme et al. 2001; Pavoine and Pecker 2009).
Ion channels that control neuronal excitability can be localized in membrane lipid rafts suggesting that sphingomyelin metabolism might influence the function of the ion channels. Biochemical analyses of lipid raft fractions isolated from excitable tissues, including heart and brain, have revealed the presence of subunits of voltage-gated Na+, K+ and Ca2+ channels (O’Connell et al. 2004; Wong and Schlichter 2004; Davies et al. 2006). Depletion of cholesterol from membranes disrupts lipid rafts and synaptic receptor signaling and plasticity (Koudinov and Koudinova 2001; Parpal et al. 2001; Pontier et al. 2008). Depletion of cholesterol also impairs AMPA receptor insertion (Hering et al. 2003; Hou et al. 2008), a principle component of synaptically-driven responses. A previous study showed that application of exogenous ceramide to cultured sensory neurons increases excitability by a mechanism involving increased Na+ currents and decreased K+ currents (Zhang et al. 2002), whereas others found that application of C2-ceramide depresses synaptic responses in the hippocampus (Yang 2000). It was also reported that NGF increases the excitability of sensory neurons by a mechanism involving S1P, a metabolite of sphingomyelin hydrolysis (Zhang et al. 2006). However, the possible effects of selective plasma membrane sphingomyelin hydrolysis by nSMase on neuronal excitability and synaptic function of CA1 hippocampal neurons, a principle target in neural degenerative diseases, are unknown.
In the present study we employed field potential and whole-cell patch clamp recordings to evaluate the influence of plasma membrane sphingomyelin metabolism on the excitability of pyramidal neurons in hippocampal slices from young adult rats. Hydrolysis of sphingomyelin by nSMase increased CA1 neuron excitability by a mechanism involving S1P production and the suppression of the K+ channel-mediated slow after-hyperpolarization.
Material and Methods
Materials
N,N-dimethylsphingosine (N,N-DMS) was purchased from Cayman Chemicals. Sphingosine-1-phosphate, D-erythro(S1P, BML-SL140) was obtained from Enzo Life Science (BML, Plymouth Meeting, PA). Endotoxin-free neutral sphingomyelinase C from Staphylococcus aureus was from Sigma-Aldrich.
Animals and brain slice preparation
Male Sprague Dawley rats (15 to 30 days old) were purchased from Charles River. The rats were anesthetized with isoflurane, and brains were removed and placed in ice cold artificial cerebrospinal fluid (ACSF; in mM: 120 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26 NaHCO3, 10 glucose). Transverse hippocampal slices 350 μm thick were cut on a vibratome.
Extracellular recordings
Slices were allowed to recuperate for at least one hour and were then placed in an 8 × 8 microelectrode array dish (MED-P515A probe, 10 mm depth, 50 μm electrodes, 150? interpolar resistance). Probes consisted of clear liquid crystal that allowed placement of hippocampal area CA1 over the 64 planar electrodes with an inverted microscope (Nikon Eclipse T300; Japan). Field CA1 responses were monitored using a 64-channel multi-electrode system that has been previously described in detail (Oka et al. 1999). Hippocampal slices were positioned to allow simultaneous recording of CA1 field excitatory postsynaptic responses (fEPSPs) and CA1 population spikes (PS). Hippocampal slices were held down using a nylon mesh (Warner Instruments), and were perfused with ACSF at room temperature with a flow rate of 1 ml/min. CA1 field responses were elicited by 64-channel integrated amplifier (SU-MED640, Panasonic) controlled by a computer running Performer 2.0 (Alpha Med Sciences) with Windows 98. Slices were stimulated at 0.05 Hz at intensities strong enough to elicit the minimum PS amplitude to avoid contamination of fEPSPs with PS. Data was analyzed using routines custom-written in Matlab (Mathworks). Population spike amplitudes were determined by subtracting the minimum peak from the average amplitude of the first and second maximums bordering the minimum. LTP experiments were carried out using 30% of maximum fEPSP responses (bipolar stimulation; 10 to 150 μA, 200 μs duration) with a 20 s inter-stimulus interval. LTP was elicited by increasing the stimulation duration to 400 μsec during the theta-burst stimulation (10 bursts at 5 Hz, consisting of 4 pulses given at 100 Hz).
For paired-pulse facilitation experiments a standard submerged recording set-up was used. fEPSPs were monitored from the stratum radiatum using a glass capillary electrode (3–5 MΩ) filled with extracellular ACSF. Responses were elicited by a nichrome bipolar stimulating electrode placed in the stratum radiatum of CA1 area to stimulate CA3 axonal fibers. Responses were recorded using an Axon 200B amplifier, digitized at 10 kHz and measured on and off line using Clampex 9. Inter-stimulus intervals are indicated in Figures 2.
Figure 2.
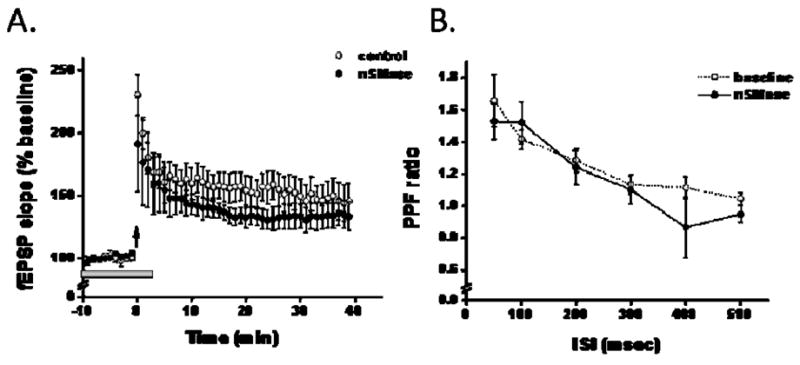
LTP and PPF are unaffected by brief applications of nSMase. A: Theta burst stimulation (TBS) elicited a similar enhancement in the fEPSP slope in the absence (open circles) and in the presence of nSMase (closed circles). nSMase (0.4 U/ml, gray bar) was perfused onto the slice 10 min prior to TBS and stopped 5 min after the termination of TBS. B: The ratio of the second pulse over the first pulse elicited by brief stimulation (30–50 μsec) given at 0.05Hz with increasing inter-stimulus intervals is similar in the absence (circle) and in the presence of nSMase (closed circles). PPF ratio was measured in the presence of nSMase (0.4 U/ml) following a 15 min pre-treatment.
Whole-cell recordings
Transverse hippocampal slices 250 μm thick were cut with sucrose dissecting buffer containing in mM: 218 sucrose, 25 NaHCO3, 3.25 potassium gluconate, 4.5 MgCl2, 0.1 CaCl2, 10 glucose, 1 sodium pyruvate, and 1.6 NaH2PO4. Slices recuperated in ACSF for 15 min at 30°C, followed by additional 1 h recuperation at room temperature. Hippocampa CA1 pyramidal neuron visualized using differential interference contrast optics, after formation of a tight seal (>4 gigaohms), gentle suction was applied to achieve whole-cell recording configuration under voltage clamp, and recordings were then made under voltage clamp or current clamp conditions. In all experiments only cells with a resting membrane potential more hyperpolarized than −55 mV were used. Series resistance was between 20–30 MΩ and cells were excluded if altered greater than 20 percent during recording. All current/voltage steps are noted in the figure legends. Data were collected using an Axopatch 200B amplifier filtered at 2 kHz, sampled at 10 kHz, captured with ClampEx and stored on a Dell Computer. Action potentials (APs) were elicited using either a series of depolarizing steps (500 ms for determination of AP frequency; 200 ms for input resistance experiments) or 1 s ramps (0.1 to 0.3 nA) which were used to determine action potential threshold. Action potential threshold was measured at the sharp rise in the action potential depolarizing phase.
The impact of S1P on action potential firing was determined in whole-cell mode with a 1 s current step. S1P was prepared as a 1 mM stock solution in ethanol, and a final concentration of 5 μM was used in the intracellular patch solution. Final ethanol concentration was 0.1% and an equal ethanol concentration was used in control experiments. S1P or equal volume of ethanol was dissolved in intracellular solution and allowed to permeate the cell for 15 min before recording.
A subset of experiments was carried out under perforated patch conditions. Micropipettes were prefilled with intracellular solution containing in mM: potassium gluconate 78, KCl 78, MgCl2 2, HEPES 10, EGTA 5, and glucose 5 (adjusted to pH 7.2 and an osmolarity of 285–290). Pipettes were then back-filled with the same solution containing 0.1 mg/ml gramicidin. In a portion of these experiments, hippocampal slices were pre-treated with10 μM of N,N-DMS for at least 30 min prior to experimentation. Action potential frequency was measured following application of nSMase in the absence or presence of N,N-DMS in response to a 1 sec current step. The other portion was used to measure AHP-mediated responses. AHP-mediated responses were measured under perforated patch conditions to minimize run-down of the AHP-mediated response. AHP-mediated responses were elicited under current clamp by 10 brief depolarizing pulses (2 ms) given at 50 Hz. The amplitude of the AHP-mediated responses was measured at the peak and 1 sec following the termination of the train.
Measurement of cholesterols, ceramides and sphingomyelins
Lipid extraction and handling of isolated lipids were performed using borosilicate-coated glass tubes, pipettes and injectors. Hippocampal slice tissue (20–30 mg wet weight) was homogenized with 10 volumes (10 μl/mg tissue) of ice cold deionized water and 30 volumes (30 μl/mg tissue) of ice cold HPLC grade methanol containing 30 mM ammonium formate, using a Pro 200 micro-homogenizer (Pro Scientific, Oxford CT) for 1 min at 15,000 rpm. Four volumes of ice cold HPLC grade chloroform was added, and the mixture was vortexed and centrifuged at 1000 g for 10 min. The bottom (chloroform) layer was removed and analyzed by direct injection into a tandem mass spectrometer. Electrospray ionization tandem mass-spectrometry (ESI/MS/MS) analyses were performed using methods similar to those used in our previous studies (Cutler et al., 2004; Haughey et al., 2004). Samples were injected using a Harvard Apparatus pump at 15 μl/min into an electrospray ionization (i.e. Turbo Ion Spray module) Sciex API 3000 triple stage quadrupole tandem mass spectrometer (ES/MS/MS) from Sciex Inc., Thornhill, Ontario, Canada, operating in the positive mode. The ion spray voltage (V) was 5,500 at a temperature of 80°C with a nebulizer gas of 8 psi, curtain gas of 8 psi, and the collision gas set at 4 psi. The declustering potential was 80 V, the focusing potential 400 V, the entrance potential −10 V, the collision energy 30V, and the collision cell exit potential was 18 V. The MS/MS scanned from 300 to 2,000 atomic mass units (amu) per second at a step of 0.1 amu. Each species of cholesterol, sphingolipid and ceramide was initially identified by a Q1 mass scan, then by precursor ion scanning or neutral loss scanning of a purified standard. Samples were injected into the ES/MS/MS for 3 min, where the mass counts accumulated and the sum of the total counts under each peak was used to quantify each species. Sphingomyelins, ceramides, cholesterol, and cholesterol ester standards C16:0, C18:0, C18:1, and cholesteryl-arachidonate (C20:0) were purchased from Sigma. Ceramides C20:0, C24:0, C24:1, phosphatidylcholine C16:0-C18:1, C18:0-C18:1, phospatidylethanolamine C16:0-C18:1, phosphatidylglycerol C16:0-C18:1, phosphatidylserine C16:0-C18:1, phosphatidylinositol C16:0-C18:1, and phosphatidic acid C16:0-C18:1 were purchased from Avanti Polar Lipids (Alabaster, AL). Palmitoyl-lactosyl ceramide C16:0-C16:0, stearoyl-lactosyl-ceramide C16:0-C18:0, lignoceryl-glucosyl-ceramide C16:0-C24:0, lignoceryl-galactosyl-ceramide C16:0-C24:0, and stearoyl-galactosyl-ceramide-sulfate C18:1-C24:0, and triacylglyceride palmitic-oleic-stearic (C52, POS) were purchased from Matreya Inc., Pleasant Gap, PA.
Results
Neutral Sphingomyelinase Reversibly Enhances Population Spike Amplitudes at CA1 Synapses
When added to the medium of cultured cells nSMase catalyzes the selective hydrolysis of sphingomyelin in the plasma membrane (Slotte et al. 1990). We therefore performed field potential recordings from CA1 striatum radiatum and CA1 stratum pyramidal layers during brief applications of nSMase. After application of nSMase the population spike (PS) was increased and the fEPSP remained unchanged (Fig. 1a). Following a 15 min application of nSMase the PS was significantly increased, and then recovered to baseline levels during a 30 min washout (Fig. 1a). The ratio of the PS/EPSP was transiently increased in the presence of nSMase (Fig. 1b). A plot of the fEPSP-population spike (ES) curve for a range of increasing stimulation intensities revealed that nSMase induced ES-potentiation (Fig 1c).
Figure 1.
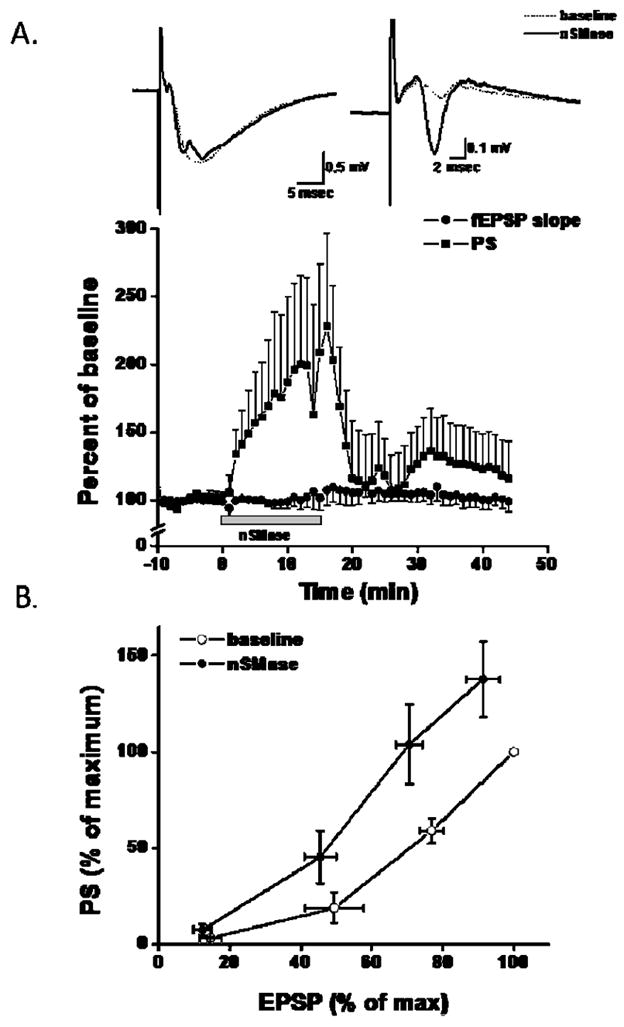
Neutral sphingomyelinase activity reversibly increases the population spike (PS) amplitudes. A: A 15 min application of nSMase (0.4 U/ml; gray bar) increases the PS (squares) while having little effect on the fEPSP (circles). The increase in PS amplitudes reverses following washout of nSMase. The data points are the mean and SEM of 5 experiments. Inset: 1 min average representative trace taken at the end of baseline and following 15 min nSMase application. B: Input-output curve generated from a series of stimulation intensities converted to an ES curve. PS and fEPSP were normalized to the maximum baseline PS and fEPSP, respectively. The PS is plotted as a function of fEPSP indicating that nSMase (closed circles) increases the PS elicited by a given fEPSP compared to baseline conditions (open circles). Values are the mean and SEM of 7 experiments.
ES-potentiation has been shown to occur with long-term potentiation (Daoudal et al. 2002), a synaptic model of learning and memory. Therefore, we investigated whether brief application of nSMase affected LTP expression. A brief exposure of hippocampal slices to nSMase did not alter LTP expression (Fig. 2a). We also investigated the impact of nSMase on paired-pulse facilitation (PPF), a measure of neurotransmitter release (Manabe et al. 1993). The PPF ratio was not altered in slices treated with nSMase (Fig. 2b). These findings suggest that sphingomyelin hydrolysis does not affect presynaptic release mechanisms and the expression of LTP at CA1 synapses. However, the alterations in the ES-curve suggest that intrinsic excitability changes occur in response to nSMase activity in CA1 pyramidal cells. We therefore employed whole-cell patch clamp technology to evaluate the possible effects of nSMase on the excitability of CA1 neurons.
Neutral Sphingomyelinase Activity Increases Hippocampal Neuron Excitability
Action potentials (APs) were recorded in hippocampal CA1 neurons in response to a depolarizing step in current clamp mode. Application of nSMase resulted in an increase in AP frequency in response to a single depolarizing current step (Fig. 3a) and to multiple current steps (Fig. 3b). To determine whether an increase in AP frequency was a consequence of dilution of secondary signaling enzymes and/or rundown of voltage-gated currents that might occur with whole-cell recording, time control experiments were performed in which recordings were made under similar conditions in neurons in untreated slices. There were no significant changes in AP frequency at 5 min (3.8 ± 0.8 APs/s) and 30 min (6 ± 2.1 APs/s) after initiation of recording. As an additional control, experiments were done using perforated patch conditions that maintain the intracellular integrity of the neuron. Perforated patch allows for longer recordings under stable conditions, and thus provided an opportunity to measure action potential firing following washout of nSMase. Over-all analysis from a subset of data taken from figure 6, in which recordings were table following a 30 min washout, ANOVA with repeated measures over drug condition was significantly different for the 6 samples (F(1,5) = 46.2, p < 0.001). Tukey post-hoc analysis showed significance between means for nSMase (10 +/− 1.4 APs/sec) versus baseline (5.5 +/− 0.8 APs/sec; p < 0.01) and washout (7.3 +/− 1.4 APs/sec; p < 0.05), but no significance for baseline versus washout (p = 0.2). Collectively, these data show that nSMase reversibly increase AP firing in CA1 hippocampal neurons.
Figure 3.
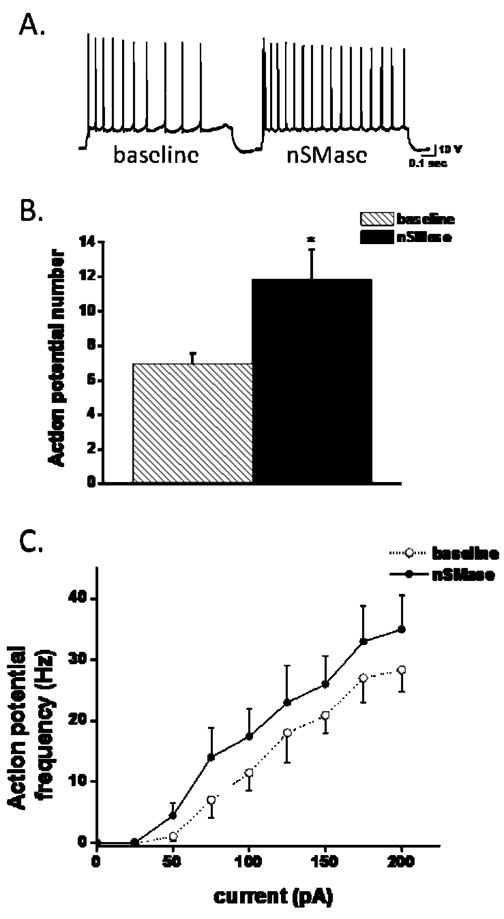
nSMase increases action potential firing frequency in CA1 pyramidal neurons. A single depolarizing step ranging from 0.05 to 0.2 nA was given to elicit at least 6–10 action potentials. A: Representative traces showing action potential firing before (baseline) and following a 15 min application of nSMase (nSMase). B: Cumulative result depicting measurement of action potential number prior to (hatch bar) and following the application of nSMase (black bar; nSMase; n=11). C: Frequency-input plot formulated from action potential frequencies determined from 50 pA current steps to 0.25 nA given for 200 msec. nSMase (closed circles) shifted the FI plot to the right when compared to baseline (open circles, n=7). Error bars indicate sem. *Student paired t-test p < 0.05 baseline versus sphingomyelinase treatment.
Figure 6.
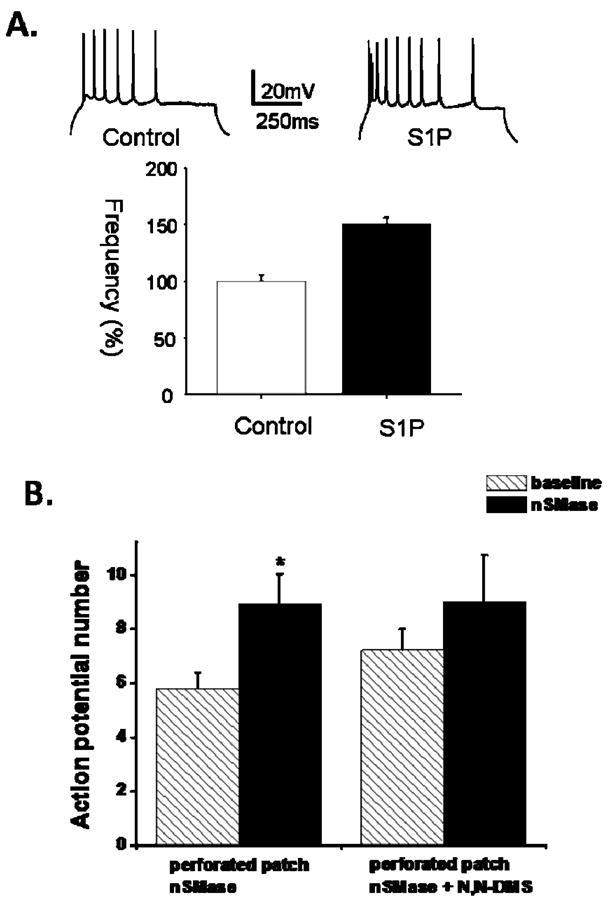
Evidence that S1P generation is sufficient and necessary for the increased neuronal excitability resulting from nSMase activation. A. S1P increases action potential firing frequency in CA1 pyramidal neurons. Upper panel): Representative traces generated from a single 50 pA depolarizing step measured 15 minutes after establishing whole cell condition in the absence (control) and presence (S1P) of S1P in the intracellular pipette. Lower panel): Cumulative data (bottom panel) showed a significant difference in the firing frequency between control (n = 8) and S1P-treated (n = 8) neurons. B. nSMase-mediated increase in action potential firing is attenuated by sphingosine-kinase inhibition. Action potential firing under perforated patch condition was increased following nSMase exposure (15 min; 0.4 U/ml) compared to baseline (n = 10). Incubation of slices with the sphingosine kinase inhibitor N,N-DMS (10 uM) for 30 – 90 min prior to nSMase application (15 min; 0.4 U/ml, n = 5) blocked the increase in action potential firing. Data are presented as the mean ± SEM. Student’s t-test was used for statistical comparisons. *p < 0.05.
To further characterize the impact of sphingomyelin hydrolysis on hippocampal CA1 neuronal excitability, we measured the AP threshold using a 1 sec ramp protocol in current clamp mode. The AP threshold was significantly decreased following nSMase treatment (Fig. 4A). The input resistance measured from IV plots of hyperpolarizing and depolarizing currents steps that did not elicit an AP was not significantly altered by nSMase (Fig 4B). Collectively, these data indicate that nSMase activity increases CA1 neuronal excitability selectively via decreasing AP threshold.
Figure 4.
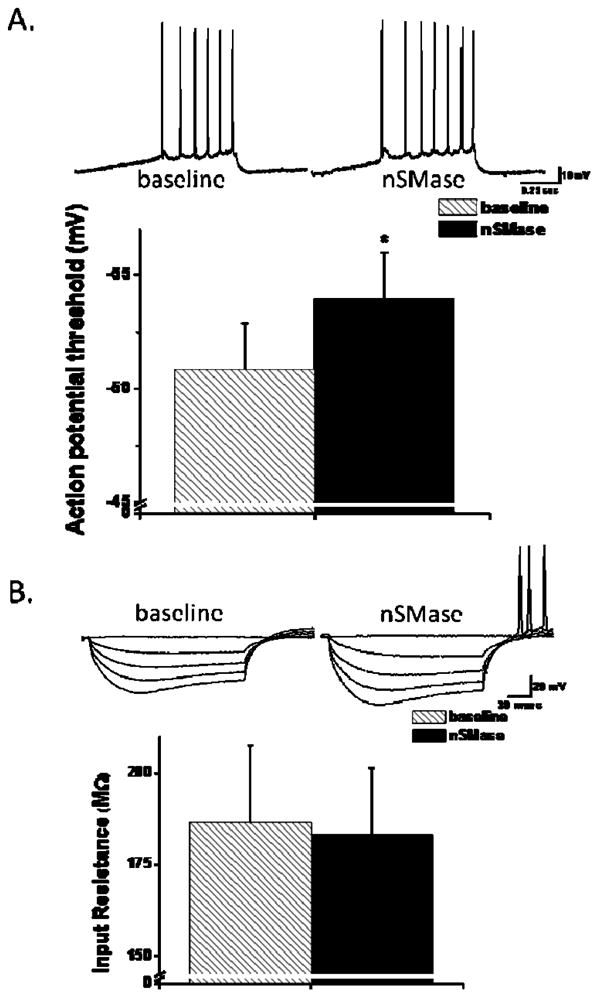
nSMase alters action potential threshold. A (top panel): Representative traces generated from a 1 sec ramp (0.1 – 0.25 nA) utilized to determine action potential threshold in the absence (hatch bar) and in the presence of nSMase (black bar). A (bottom panel): AP threshold (n = 15) was determined as a 2 mV/msec change from the ramp. B: A series of hyperpolarizing current steps were generated to determine the input resistance from an IV plot. Cumulative data (bottom panel) showed no significant difference in the input resistance in the presence (black bar) compared to that measured in the absence (hatch bar, n = 15) of nSMase. *p < 0.05; paired Student’s t-test.
Lipid Profiling of Hippocampal Slices Shows that nSMase Activation Generates Long-Chain Ceramides and S1P
Ceramides of various chain lengths and numbers of unsaturated bonds are known to be generated in the brain in pathological settings such as stroke and Alzheimer’s disease (Yu et al. 2000; Cutler et al. 2004), but the identities of the ceramides generated by plasma membrane SMase activity in hippocampal cells in physiological conditions are unknown. To elucidate the sphingomyelin metabolites that may mediate the effects of nSMase on CA1 neuron excitability we employed mass spectrometry methods to identify and quantify levels of different species of sphingomyelins, ceramides and S1P in hippocampal slices that had been exposed for 15 min to nSMase or vehicle control. There were no significant differences in the levels of phosphatidylethanolamine, phosphatidlycholine or 5 different sphingomyelins (C16:0, C18:0, C20:0, dihydro C22:0 and C24:0) in slices exposed to nSMase compared to those exposed to vehicle (Fig. 5a). However, nSMase induced large (4–6 fold) significant increases in levels of C22:0 ceramide, C24:0 dihydroceramide and glucosylceramide C16:0 (Fig. 5b). In addition, levels of S1P were significantly increased in response to nSMase. In contrast, levels of ceramides C20:0 and C24:0, galactosylceramide C24:0, and gangliosides GM1 C22 and C24 were not affected by nSMase activity (Fig. 5b).
Figure 5.

nSMase increases levels of long-chain ceramides and sphingosine-1-phosphate in hippocampal slices. A. Levels of phosphatidylethanolamine (PE), sphingomyelins of the indicated carbon chain lengths, and phosphatidylcholine (PC) were quantified by mass spectrometry in hippocampal slices that had been treated for 15 min with either vehicle (control, hatch bars, n=5) or nSMase (black bars, n=4). B. Levels of PE, sphingosine (Sph), sphingosine-1-phosphate (S1P) and the indicated ceramides were quantified by mass spectrometry in hippocampal slices that had been treated for 15 min with either vehicle (control, hatch bars, n=5) or nSMase (black bars, n=4). Values were normalized to the PE level and represent the mean and SEM. *p<0.05, **p<0.01 compared to the corresponding control value.
S1P Mediates the nSMase-Induced Enhancement of Neuronal Excitability
Studies of a range of mitotic cells have shown that S1P plays important roles in the regulation cell growth and survival, adhesion, migration and angiogenesis (Hinkovska-Galcheva et al., 2008). In addition, a recent study provided evidence that S1P mediates the enhancement of sensory neuron excitability in response to NGF (Zhang et al., 2006), suggesting roles for S1P in regulating electrochemical communication in neuronal circuits. To investigate the role of S1P on hippocampal neuron excitability, S1P was dissolved in intracellular solution. S1P in the intracellular pipette increased the firing rate of hippocampal CA1 neurons within 15 minutes compared to control experiments (Fig. 6a). In a similar experiment, hippocampal slices were pretreated with N, N-DMS, a sphingosine kinase inhibitor that prevents the generation of S1P from ceramides (Yatomi et al. 1996; Zhang et al. 2008). When hippocampal slices were pretreated with N, N-DMS to block S1P production, nSMase was unable to increase AP frequency in CA1 neurons (Fig. 6b). These findings demonstrate a requirement for S1P production in sphingomyelin metabolism-mediated enhancement of hippocampal neuronal excitability.
Calcium-Dependent After hyperpolarization is Reduced in Response to nSMase Activity
Previous studies have shown that the duration of a sustained or slow after hyperpolarization (sAHP) plays a key role in determining AP frequency in CA1 neurons, and established an important role for Ca2+-activated K+ channels in the sAHP (Hotson and Prince 1980; Disterhoft et al. 1986). A reduction in the sAHP, and the resulting increase in neuronal excitability and neurotransmitter release may play an important role in learning and memory (Tombaugh et al. 2005), and an increase in the sAHP is believed to contribute to impaired hippocampal synaptic plasticity during aging (Power et al. 2002). We therefore measured the sAHP in CA1 neurons in control and nSMase-treated hippocampal slices and found that nSMase activity significantly decreased the sAHP current amplitude in a reversible manner (Fig. 7). These findings suggest a role for reduction of the Ca2+-activated K+ current (BK channels) in the mechanism by which nSMase activation increases hippocampal CA1 neuron excitability.
Figure 7.
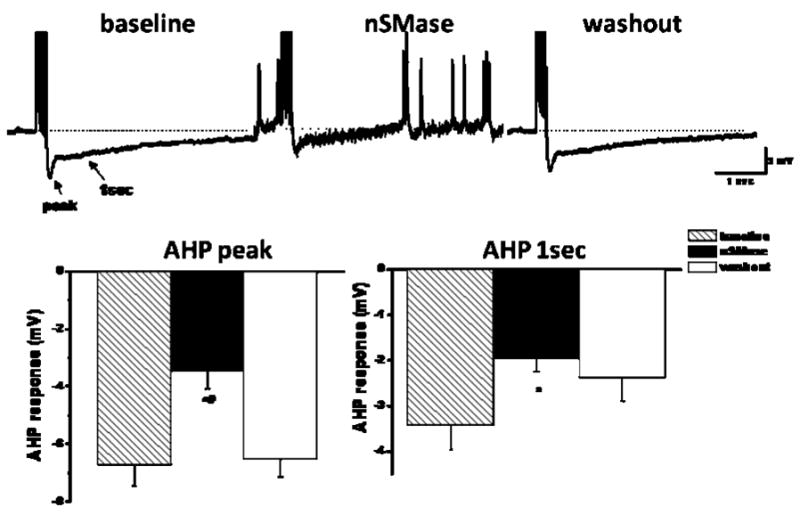
nSMase reversibly decreases the slow after hyperpolarization in CA1 neurons. Upper: Representative traces recorded prior to treatment (baseline), during exposure to nSMase, and following washout of nSMase using perforated patch recording. Note nSMase trace shows an increase in spontaneous firing following 15 min incubation of nSMase (0.4 U/ml). The increase in spontaneous firing was abolished following washout. This phenomenon occurred in 3 of 5 experiments. All responses were held at −65 mV. Lower: Cumulative plot (n = 5) of the slow AHP response measured at the peak (left panel) and 1 sec following the last AP (right panel). AHP-mediated responses were measured prior to treatment (hatch bars), immediately after a 15 min treatment with nSMase (black bars) and 30 min after washout (white). * and # denote p values < 0.05 compared to the baseline and washout values, respectively. One-way repeated ANOVA with Tukey post hoc test.
Discussion
Our mass spectrometry analysis of the relative levels of different sphingolipids in control and nSMase-treated hippocampal slices suggest that multiple pathways of ceramide metabolism are mobilized in response to ceramide generation at the plasma membrane. We found that cleavage of membrane sphingomyelin by nSMase results in the rapid generation of several long-chain ceramides (C22:0 ceramide, C24:0 dihydroceramide and glucosylceramide C16:0) and S1P in hippocampal slices. The nSMase-induced increase in the amount of ceramide C22:0 is likely the result of hydrolysis of sphingomyelin to ceramide at the plasma membrane where this particular carbon chain length of ceramide is enriched. On the other hand the increases of dihydroceramide C24:0 and glucosylceramide C16:0 levels may result from the activation of dihydroceramide desaturase and glucosylceramide synthase, respectively (Wennekes et al., 2009).
Increased ceramide from nSMase-mediated hydrolysis of sphingomyelin drives the production of S1P and glucosylceramide. However, the absence of a decrease in sphingomyelin levels may result from recruitment of sphingomyelin synthase, the signaling enzyme responsible for the conversion of ceramide to sphingomyelin, and ‘de novo’ synthesis of ceramide. This phenomenon could explain the tendency of certain sphingomyelins to be increased, and the significant increase in C24:0 dihydroceramide, in hippocampal slices exposed to nSMase. Additional studies are required to establish the recruitment of enzymes within the sphingolipid synthesis pathway following nSMase application.
nSMase-mediated production of ceramide and its metabolites are associated with increased excitability of CA1 pyramidal neruons. ES-potentiation of the field response led us to investigate intrinsic changes in CA1 pyramidal neurons. We found that AP frequency in CA1 neurons was increased by nSMase activity and this effect of nSMase was reproduced by S1P application and blocked by the S1P inhibitor N, N-DMS, demonstrating a pivotal role for S1P in the enhancement of excitability in response to sphingomyelin metabolism. Previous studies have shown that activation of receptors that are known to be coupled to nSMase can alter neuronal excitability and synaptic plasticity (see Mattson and Meffert, 2006 for review). For example, activation of tumor necrosis factor (TNF) receptors rapidly enhanced basal synaptic transmission at CA1 synapses (Tancredi et al., 1992) and LTD at CA1 synapses is impaired in mice lacking TNF receptors (Albensi and Mattson, 2000). In addition, it was recently reported that TNF initiates a rapid increase in ceramide levels that is associated with increased surface localization of NMDA receptor NR1 subunits and increased rate and amplitude of NMDA-evoked calcium bursts and enhanced excitatory post-synaptic currents in cultured neuron (Wheeler et al. 2009). Application of TNF to dorsal root ganglion sensory neurons resulted in increase excitability (Youn et al., 2008). Similarly, exposure to TNF resulted in a rapid increase in AP frequency in suprachiasmatic nucleus neurons (Nygard et al., 2009). However, the previous studies did not establish roles for sphingomyelin metabolism and S1P in the effects of TNF on neuronal excitability. In the present study, we bypassed receptors coupled to nSMase to avoid activating other pathways simultaneously engaged by these receptors. This was accomplished by simply adding nSMase to the medium bathing the hippocampal slices, thereby inducing the selective hydrolysis of sphingomyelin in the plasma membrane (Slotte et al. 1990) resulting in the production of long-chain ceramides and S1P.
Previous studies have suggested roles for S1P in regulating neuronal excitability and synaptic plasticity. Consistent with our evidence that S1P mediates the enhancement of neuronal excitability in response to nSMase activation, Kajimoto et al. (2007) found that exogenously added nanomolar concentrations of S1P elicited glutamate secretion from hippocampal cells, and that lower (picomolar) concentrations of S1P potentiated depolarization-evoked secretion in the neurons. Moreover, reducing sphingosine kinase 1 expression using small interfering RNA and sphingosine kinase 1 inhibition with N, N-DMS resulted in a strong inhibition of depolarization-evoked glutamate secretion. On the other hand, mice deficient in the S1P receptor exhibit spontaneous seizures, an abnormal electroencephalogram and increased excitability of neocortical pyramidal neurons (MacLennan et al. 2001). Further evidence for an important role for S1P signaling in synaptic plasticity comes from a study which documented an abnormality in instrumental conditioning, a process involving striatal synaptic plasticity, in mice lacking the Gpr6 S1P receptor (Lobo et al. 2006). Our findings suggest that nSMase-mediated generation of S1P may play a role in the enhanced neuronal excitability that occurs during various behaviors.
Our findings suggest the possibility that S1P acts as a second messenger and/or autocrine signal to modify neuronal excitability. Neutral SMase is known to be activated by specific receptor-coupled mechanisms (TNF, growth factors and other ligands) and also by oxidative stress (Hernandez et al., 2000; Mattson and Meffert, 2006). The mechanism by which nSMase-mediated sphingomyelin metabolism increases the excitability of CA1 hippocampal neurons appears to involve reduced activation of the Ca2+-sensitive K+ channels that mediate membrane hyperpolarization following an AP. Previous studies of CA1 neurons have shown that the duration of the AHP is a critical determinant of when a subsequent AP can occur and hence the AP frequency (Lancaster and Adams 1986; Stocker et al. 1999). Recent findings suggest that the AHP is decreased during learning (Tombaugh et al. 2005) and is increased in association with aging- and Alzheimer’s disease-related decreases in cognitive function (Disterhoft et al. 2004; Matthews et al. 2009). We found that an in increase in nSMase activity resulted in a reduction of the AHP in a rapid and reversible manner. Our findings therefore suggest possible roles for nSMase and S1P in hippocampal plasticity involved in processes such as learning and memory. Such actions of S1P may be mediated by one of the S1P receptor family members which are GTP-binding protein-coupled receptors (Lebman and Spiegel, 2008), though this remains to be determined.
Consistent with a role for sphingomyelin metabolism in synaptic plasticity are data showing abnormalities in sphingomyelin and ceramide levels in the setting of impaired cognitive function. Thus, analyses of membrane lipids in brain tissue samples from patients with Alzheimer’s disease demonstrated reductions in SM and sulphatide levels and increased accumulation of ceramides and cholesterol esters (Han et al. 2002; Cutler et al. 2004). Future studies should reveal the molecular mechanisms upstream and downstream of membrane sphingomyelin hydrolysis that mediate the effects of nSMase on neuronal excitability in health and disease.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute on Aging, National Institutes of Health. All animal related protocol was following NIH guideline and approved by NIA Animal Care & Use Committee (ACUC). The authors declare that they have no competing financial interest.
Abbreviations
- SMase
sphingomyelinase
- nSMase
neutral sphingomyelinase
- aSMase
acidic sphingomyelinase
- SIP
sphingosine-1-phosphate
- fEPSP
field excitatory postsynaptic response
- PS
population spike
- PPF
paired-pulse facilitation
- AP
action potentials
- AHP
after-hyperpolarization potential
References
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ. Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology. 2007;68:1481–1487. doi: 10.1212/01.wnl.0000260610.79853.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52:448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101:2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoudal G, Hanada Y, Debanne D. Bidirectional plasticity of excitatory postsynaptic potential (EPSP)-spike coupling in CA1 hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002;99:14512–14517. doi: 10.1073/pnas.222546399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Douglas L, Hendrich J, et al. The calcium channel alpha2delta-2 subunit partitions with CaV2.1 into lipid rafts in cerebellum: implications for localization and function. J Neurosci. 2006;26:8748–8757. doi: 10.1523/JNEUROSCI.2764-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFreitas MF, McQuillen PS, Shatz CJ. A novel p75NTR signaling pathway promotes survival, not death, of immunopurified neocortical subplate neurons. J Neurosci. 2001;21:5121–5129. doi: 10.1523/JNEUROSCI.21-14-05121.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, Alkon DL. Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci U S A. 1986;83:2733–2737. doi: 10.1073/pnas.83.8.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Wu WW, Ohno M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer’s disease. Ageing Res Rev. 2004;3:383–406. doi: 10.1016/j.arr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Grassme H, Jekle A, Riehle A, Schwarz H, Berger J, Sandhoff K, Kolesnick R, Gulbins E. CD95 signaling via ceramide-rich membrane rafts. J Biol Chem. 2001;276:20589–20596. doi: 10.1074/jbc.M101207200. [DOI] [PubMed] [Google Scholar]
- Han XM, Holtzman D, McKeel DW, Jr, Kelley J, Morris JC. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: potential role in disease pathogenesis. J Neurochem. 2002;82:809–818. doi: 10.1046/j.1471-4159.2002.00997.x. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, Turchan J, Nath A, Mattson MP. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–267. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- Hering H, Lin CC, Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci. 2003;23:3262–3271. doi: 10.1523/JNEUROSCI.23-08-03262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez OM, Discher DJ, Bishopric NH, Webster KA. Rapid activation of neutral sphingomyelinase by hypoxia-reoxygenation of cardiac myocytes. Circ Res. 2000;86:198–204. doi: 10.1161/01.res.86.2.198. [DOI] [PubMed] [Google Scholar]
- Hinkovska-Galcheva V, VanWay SM, Shanley TP, Kunkel RG. The role of sphingosine-1-phosphate and ceramide-1-phosphate in calcium homeostasis. Curr Opin Investig Drugs. 2008;9:1192–1205. [PubMed] [Google Scholar]
- Hotson JR, Prince DA. A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. J Neurophysiol. 1980;43:409–419. doi: 10.1152/jn.1980.43.2.409. [DOI] [PubMed] [Google Scholar]
- Hou Q, Huang Y, Amato S, Snyder SH, Huganir RL, Man HY. Regulation of AMPA receptor localization in lipid rafts. Mol Cell Neurosci. 2008;38:213–223. doi: 10.1016/j.mcn.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto T, Okada T, Yu H, Goparaju SK, Jahangeer S, Nakamura S. Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol Cell Biol. 2007;27:3429–3440. doi: 10.1128/MCB.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Adams PR. Calcium-dependent current generating the after hyperpolarization of hippocampal neurons. J Neurophysiol. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- Lebman DA, Spiegel S. Cross-talk at the crossroads of sphingosine-1-phosphate, growth factors, and cytokine signaling. J Lipid Res. 2008;49:1388–1394. doi: 10.1194/jlr.R800008-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Cui Y, Ostlund SB, Balleine BW, Yang XW. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nat Neurosci. 2007;10:1395–1397. doi: 10.1038/nn1987. [DOI] [PubMed] [Google Scholar]
- MacLennan AJ, Carney PR, Zhu WJ, Chaves AH, Garcia J, Grimes JR, Anderson KJ, Roper SN, Lee N. An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur J Neurosci. 2001;14:203–209. doi: 10.1046/j.0953-816x.2001.01634.x. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Maruyama EN, Arima M. Purification and characterization of neutral and acid sphingomyelinases from rat brain. J Neurochem. 1989;52:611–618. doi: 10.1111/j.1471-4159.1989.tb09163.x. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Linardakis JM, Disterhoft JF. The fast and slow after hyperpolarizations are differentially modulated in hippocampal neurons by aging and learning. J Neurosci. 2009;29:4750–4755. doi: 10.1523/JNEUROSCI.0384-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, editor. Membrane microdomain signaling: lipid rafts in biology and medicine. Humana Press; Totowa, NJ: 2005. p. 214. [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Nygård M, Lundkvist GB, Hill RH, Kristensson K. Rapid nitric oxide-dependent effects of tumor necrosis factor-alpha on suprachiasmatic nuclei neuronal activity. Neuroreport. 2009;20:213–217. doi: 10.1097/WNR.0b013e32831f1ca2. [DOI] [PubMed] [Google Scholar]
- O’Connell KM, Martens JR, Tamkun MM. Localization of ion channels to lipid Raft domains within the cardiovascular system. Trends Cardiovasc Med. 2004;14:37–42. doi: 10.1016/j.tcm.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Oka H, Shimono K, Ogawa R, Sugihara H, Taketani M. A new planar multielectrode array for extracellular recording: application to hippocampal acute slice. J Neurosci Methods. 1999;93:61–67. doi: 10.1016/s0165-0270(99)00113-2. [DOI] [PubMed] [Google Scholar]
- Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82:175–183. doi: 10.1093/cvr/cvp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Wu WW, Sametsky E, Oh MM, Disterhoft JF. Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. J Neurosci. 2002;22:7234–7243. doi: 10.1523/JNEUROSCI.22-16-07234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–18436. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- Slotte JP, Härmälä AS, Jansson C, Pörn MI. Rapid turn-over of plasma membrane sphingomyelin and cholesterol in baby hamster kidney cells after exposure to sphingomyelinase. Biochim Biophys Acta. 1990;1030:251–257. doi: 10.1016/0005-2736(90)90301-4. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992;146:176–178. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM. The slow after hyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25:2609–2616. doi: 10.1523/JNEUROSCI.5023-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiuk S, Hofmann K, Nix M, Zumbansen M, Stoffel W. Cloned mammalian neutral sphingomyelinase: functions in sphingolipid signaling? Proc Natl Acad Sci U S A. 1998;95:3638–3643. doi: 10.1073/pnas.95.7.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, Mattson MP, Geiger JD, Haughey NJ. Tumor necrosic factor-alpha-induced neutral sphingomyelinase-2-modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–1249. doi: 10.1111/j.1471-4159.2009.06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Schlichter LC. Differential recruitment of Kv1.4 and Kv4.2 to lipid rafts by PSD-95. J Biol Chem. 2004;279:444–452. doi: 10.1074/jbc.M304675200. [DOI] [PubMed] [Google Scholar]
- Yang SN. Ceramide-induced sustained depression of synaptic currents mediated by ionotropic glutamate receptors in the hippocampus: an essential role of postsynaptic protein phosphatases. Neuroscience. 2000;96:253–258. doi: 10.1016/s0306-4522(99)00582-5. [DOI] [PubMed] [Google Scholar]
- Yatomi Y, Ruan F, Megidish T, Toyokuni T, Hakomori S, Igarashi Y. N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry. 1996;35:626–633. doi: 10.1021/bi9515533. [DOI] [PubMed] [Google Scholar]
- Youn DH, Wang H, Jeong SJ. Exogenous tumor necrosis factor-alpha rapidly alters synaptic and sensory transmission in the adult rat spinal cord dorsal horn. J Neurosci Res. 2008;86:2867–2875. doi: 10.1002/jnr.21726. [DOI] [PubMed] [Google Scholar]
- Yu ZF, Nikolova-Karakashian M, Zhou D, Cheng G, Schuchman EH, Mattson MP. Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production, and neuronal apoptosis. J Mol Neurosci. 2000;15:85–97. doi: 10.1385/JMN:15:2:85. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na(+) current and delayed rectifier K(+) current in rat sensory neurons. J Physiol. 2002;544:385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Intracellular sphingosine 1-phosphate mediates the increased excitability produced by nerve growth factor in rat sensory neurons. J Physiol. 2006;575:101–113. doi: 10.1113/jphysiol.2006.111575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Chi XX, Nicol GD. Brain-derived neurotrophic factor enhances the excitability of rat sensory neurons through activation of the p75 neurotrophin receptor and the sphingomyelin pathway. J Physiol. 2008;586:3113–3127. doi: 10.1113/jphysiol.2008.152439. [DOI] [PMC free article] [PubMed] [Google Scholar]