

Abstract
Adipocytes produce the hormone, leptin, in proportion to fat mass to signal the status of body energy stores to the central nervous system, thereby modulating food intake and energy homeostasis. In addition to controlling satiety, leptin suppresses the reward value of food, which is controlled by the mesolimbic dopamine (DA) system. Previous results from leptin-deficient ob/ob animals suggest that chronic leptin deficiency decreases DA content in the mesolimbic DA system, thereby decreasing the response to amphetamine (AMPH). The extent to which these alterations in the mesolimbic DA system of ob/ob animals may mirror the leptin response of normal animals has remained unclear, however. We therefore examined the potential short-term modulation of the mesolimbic DA system by leptin in normal animals. We show that 4 h of systemic leptin treatment enhances AMPH-stimulated DA efflux in the nucleus accumbens (NAc) of Sprague-Dawley rats. While acute leptin treatment increased NAc tyrosine hydroxylase (TH) activity, total TH and DA content were unchanged at this early time point. Leptin also increased NAc DA transporter (DAT) activity in the absence of changes in cell surface or total DAT. Thus, leptin modulates the mesolimbic DA system via multiple acute mechanisms, and increases AMPH-mediated DA efflux in normal animals.
Keywords: leptin, dopamine, amphetamine, tyrosine hydroxylase, dopamine transporter
Multiple observations link energy balance to the function of brain reward pathways: not only are obese humans 25% less likely to develop substance abuse disorders (Simon et al. 2006), but also food restriction increases the incentive value of drugs of abuse (Carr 2002, 2007). Thus, the regulation of feeding and drug abuse likely act on shared brain pathways. Leptin, a hormone produced by adipocytes to signal the repletion of fat stores, plays a key role in regulating feeding behavior and other aspects of energy balance (Zhang et al. 1994; Halaas et al. 1995; Maffei et al. 1995; Saper et al. 2002). In addition to potentiating satiety signals, leptin modulates the incentive salience of rewards (Fulton et al. 2000; Fulton et al. 2004). While much is known about the mechanisms by which leptin influences satiety, only recently have investigators begun to explore the mechanism by which leptin regulates the incentive salience of food and drug rewards, and our understanding of this aspect of the action of leptin remains incomplete. The mesolimbic dopamine (DA) system controls the incentive salience of food (Wyvell and Berridge 2000) (as well as other natural rewards and drugs of abuse). At its core, the mesolimbic DA system contains DA neurons that project from the ventral tegmental area (VTA) to a variety of limbic targets, including the nucleus accumbens (NAc).
Leptin acts via long-form leptin receptors (LepRb) on neurons in a variety of brain regions, including several hypothalamic, midbrain, and brainstem nuclei (Elmquist et al. 1998a; Hay-Schmidt et al. 2001; Fulton et al. 2006; Hommel et al. 2006; Leshan et al. 2006). Populations of LepRb-expressing neurons in the lateral hypothalamus (LHA) (Leinninger et al. 2009) and the VTA (Fulton et al. 2006; Hommel et al. 2006) contribute to regulation of the mesolimbic DA system by leptin, and leptin may modulate the mesolimbic DA system in several ways. Leptin treatment of midbrain tissue slices slightly hyperpolarizes VTA DA neurons (Hommel et al. 2006). Also, leptin-deficient ob/ob mice exhibit decreased VTA and NAc tyrosine hydroxylase (TH; which catalyzes the rate-limiting step in DA synthesis) expression and DA content (Fulton et al. 2006; Leinninger et al. 2009). Consistent with decreased mesolimbic DA content in ob/ob mice, amphetamine (AMPH, which acts via the DA transporter, DAT, to promote DA efflux) fails to promote normal locomotor activity and sensitization in these animals, and chronic leptin treatment restores the AMPH responsiveness of ob/ob mice (Fulton et al. 2006).
While these data collectively demonstrate that leptin regulates dopaminergic reward systems, and suggest that chronic leptin deficiency attenuates both DA production and responsiveness to AMPH, it is possible that the decreased mesolimbic TH and DA content in ob/ob animals represents a secondary compensatory response to chronic leptin deficiency. Furthermore, while TH and DA content represent important variables in the function of the mesolimbic DA system, the potential leptin-mediated modulation of other determinants of mesolimbic DA function has not been carefully examined, or as in the case of the modulation of DAT, not explored at all.
To address these important outstanding issues in leptin action, our present study aimed to determine the acute regulation of the mesolimbic DA system by leptin in normal animals. We used in vivo microdialysis in freely moving animals to monitor the basal and AMPH-stimulated extracellular DA concentrations in the NAc and assayed VTA and NAc tissue for TH and DAT expression and activity 4 h after leptin treatment of Sprague-Dawley rats.
Methods
Materials
Unless otherwise noted, all materials were purchased from Sigma-Aldrich and were used without further purification.
Animals and surgery
All experiments were performed on male Sprague-Dawley (Harlan) rats 60-100 days old. Animals were maintained on a 12:12 light:dark cycle with lights on at 08:00 hr. Following surgery to implant a guide cannula, animals were housed individually. All animal procedures were approved by the University Committee for the Use and Care of Animals at the University of Michigan.
For the implantation of a guide cannula (Plastics One, Inc.), rats were anesthetized with a combination of ketamine (65 mg/kg, Fort Dodge Animal Health) and medetomidine (0.5 mg/kg, Pfizer Animal Health) i.p. Using aseptic technique, a guide cannula was implanted anterior +1.6 mm, lateral ± 1.3 mm, ventral -5.0 mm from bregma and secured in place with dental cement (A-M Systems, Inc.). The guide cannula was sealed by means of a solid metal stylet (Plastics One, Inc.). Animals were allowed to recover for 5-10 days before experimentation. Several times during the recovery period, the animals were brought to the experimental room for acclimation to the experimental environment.
Microdialysis
The extracellular DA concentration in the NAc was measured in vivo by microdialysis coupled online to capillary electrophoresis with laser-induced fluorescence (CE-LIF). Microdialysis probes were prepared as described previously (Smith and Justice 1994). Before implantation, the probes were sterilized in ethanol (Fisher Scientific) for 10 min. Between 17:00 and 20:00 hr on the day before experimentation, the animal was weighed, was briefly anesthetized with isoflurane (Baxter Healthcare Corporation), and a microdialysis probe with a 2 mm long sampling region was implanted, extending 3.4 mm past the tip of the guide cannula. The animal was placed in a Ratturn (Bioanalytical Systems, Inc.) to prevent the probe from being disturbed by normal movements of the animal. Between 08:10 and 08:50 hr on the day of experimentation, the animal received 1 mg/kg leptin or phosphate buffered saline (PBS, 20 mM phosphate pH 5.2, 0.13 M NaCl) vehicle i.p. This supraphysiological dose was selected to match the systemic doses previously given to rats by us and others (Elmquist et al. 1998b; Elias et al. 1999; Kelly et al. 2004). Four h later, the animal received 1 mg/kg D-AMPH s.c. One h before and four h after the AMPH stimulation, the extracellular DA was monitored as previously described (Shou et al. 2006). Briefly, the dialysate was mixed online with naphthalene-2,3-dicarboxaldehyde (Invitrogen) and KCN (Fisher Scientific) to derivatize the amine functionality of DA. Injections were made from the derivatized sample stream onto a separation capillary by means of a flow-gated interface. CE separations, lasting 100 to 120 s, were performed with a buffer of 30 mM phosphate, 6.5 mM sodium dodecylsulfate (SDS), and 2 mM hydroxylpropyl-β-cyclodextrin and detected on-column by laser induced fluorescence. The separation and detection were controlled using software written in Labview 5.0 (National Instruments). Following the observation period, the probe was removed and the animal was euthanized with Fatal Plus (Vortech Pharmaceuticals, Inc.). The brain was removed and preserved in 4% paraformaldehyde for histology. An external calibration was performed through the probe and the in vitro recovery was measured.
Histology
One week prior to histological study, the preserved brains were transferred to 30% sucrose in 4% paraformaldehyde. Each probe tract was visualized by cryostatic sectioning. Data from those probe tracts which fell outside of the NAc were excluded from final analysis.
Tissue collection
The expression and activity of TH and DAT were measured in VTA and NAc tissue samples. Prior to the collection of tissue from the VTA and NAc, animals experienced a mock-implant of a microdialysis probe and were housed overnight in their home cages in the experimental room. On the day of tissue harvesting, between 08:10 and 08:50, the animals received 1 mg/kg leptin or PBS vehicle i.p. Four h later, the animals were decapitated. The brain was rapidly removed and cooled for a few minutes in ice cold aCSF (artificial cerebral spinal fluid, 145 mM NaCl, 2.7 mM KCl, 1.0 mM MgSO4, 1.2 mM CaCl2, 2.0 mM phosphate pH 7.4). The VTA and NAc were dissected by means of a brain matrix. Tissue samples for qPCR were stored at -80°C until analysis. Tissue samples for other assays were treated as described below on the day of collection.
TH and DAT mRNA qPCR
RNA was prepared from microdissected VTA and NAc using Trizol (Invitrogen) and 1μg samples were converted to cDNA using the Superscript First Strand Synthesis System for RT-PCR (Invitrogen). Sample cDNAs were analyzed in triplicate via quantitative RT-PCR for Gapdh and either Th or Dat (Applied Biosystems) using an Applied Biosystems 7500. Relative mRNA expression values are calculated by the 2-ΔΔCt method, with normalization of each sample ΔCt value to the average vehicle-treated ΔCt.
Preparation of synaptosomes
The western blots for TH and DAT, the TH activity assay, the [3H]WIN 35,428 (2β-carbomethoxy-3β-(4-fluorophenyl)-tropane) ([3H]WIN35428) binding assay, the DAT biotinylation assay, and the [3H]DA uptake assay were all performed in synaptosomes. Tissue samples were homogenized on ice as follows: VTA samples in 100 μL and NAc samples in 300 μL 0.32 M sucrose for western blots; VTA samples in 150 μL and NAc samples in 400 μL 0.32 M sucrose containing a cocktail of protease inhibitors (Complete Mini, Roche Diagnostics) for the TH activity assay; and in 1.3 mL 0.32 M sucrose containing a cocktail of protease inhibitors for the [3H]WIN35428 binding, DAT biotinylation, and [3H]DA uptake assays. Homogenized samples were centrifuged at 800g for 10 min. The supernatant was centrifuged at 12,000g for 15 min. The supernatant was discarded. The pellet was resuspended as described.
TH and DAT western blot
TH and DAT protein expression were measured by quantified western blot. Synaptosomes were lysed for 1 h in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40 (Triton X-100), 1% sodium doxycholate, 1% SDS), 100 μL for VTA samples and 400 μL for NAc samples. The lysed synaptosomes were centrifuged at 12,000g for a half h. The protein concentration in the supernatant was measured with a DC protein assay kit (Bio-Rad) using bovine serum albumin (BSA, Proliant Biologicals) for calibration. Ten μg of protein for the TH western blots and 20 μg of protein for the DAT western blots together with SDS sample buffer (2% SDS, 10% glycerol, 0.1% bromophenol blue, 100 mM dithiothreitol, 50 mM Tris-Cl pH 6.8) were loaded onto a 10% polyacrylamide gel and the proteins of interest were resolved. Following protein transfer and blocking with 2.5% BSA, the membranes were cut between the 43 and 56 kDa markers. The lower molecular weight portion of each membrane was treated with actin HRP antibody (Santa Crux Biotech Corp.), diluted 1:2,000 for the TH blot and 1:5,000 for the DAT blot in 2.5% BSA, for 1 h. The higher molecular weight portion was treated overnight with monoclonal TH antibody (Zymed Laboratory), diluted 1:800 in 2.5% BSA, or MAB 16 monoclonal DAT antibody (generous gift of Dr. Roxane Vaughan), diluted 1:2,000 in 2.5% BSA, followed by goat anti-mouse HRP antibody (Santa Cruz Biotech Corp.), diluted 1:15,000 in 2.5% BSA for the TH blot and 1:20,000 in 2.5% BSA, for the DAT blot for 1 h. The membranes were imaged following treatment with the Pierce ECL Western Blotting Substrate (Thermo Scientific) for 5 min. Quantification of the bands was performed with Scion Image software (Scion Corporation).
[3H]DA uptake assay
The activity of DAT was measured by the uptake of [3H]DA into NAc synaptosomes. Synaptosomes were resuspended in 2 mL KRB. [3H]DA (specific activity 23.5 Ci/mmol, PerkinElmer Life and Analytical Sciences) uptake activity was determined in triplicate in 200 μL for 2 min at 37°C at final [3H]DA concentrations of 30 nM, 100 nM, 300 nM, and 1 μM. Non-specific activity was determined using 100 μM cocaine (Sigma) and 1 μM unlabeled DA. The reaction was stopped by the addition of 5 mL ice cold KRB and the rapid filtration through GF/C filters (Fisher Scientific). The filters were washed with two 5 mL portions of KRB and the radioactivity on the filters was measured by a Beckman LS5801 scintillation counter (Beckman Coulter). DA binding affinity for DAT (Km) and DA uptake velocity (Vmax) values were determined by nonlinear regression analysis using Prism 5 (Graphpad Software, Inc.).
[3H]WIN 35,428 binding assay
Binding of the cocaine analogue [3H]WIN35428, specific activity 85 Ci/mmol, PerkinElmer Life and Analytical Sciences) to assess DAT surface expression was measured by a method modified from Reith and colleagues (Coffey and Reith 1994; Chen et al. 1996). Synaptosomes were resuspended in 1.2 mL of the assay buffer (30 mM sodium phosphate pH 7.4 and 0.32 M sucrose). [3H]WIN 35,428 binding was measured in triplicate in 200 μL with 103 nM WIN 35,428 for 1 h at 4°C. Nonspecific binding was determined in the presence of 1 mM unlabeled [3H]WIN 35,428. The reaction was stopped by the addition of 5 mL ice cold 30 mM phosphate and the rapid filtration through GF/C filters. The filters were washed with two 5 mL portions of 30 mM phosphate and the radioactivity on the filters was measured with a liquid scintillation counter.
DAT biotinylation assay
DAT surface expression was confirmed by surface biotinylation. Synaptosomes prepared from NAc tissue were suspended in 500 μL PBS/Ca/Mg containing 1.5 mg/mL sulfo-NHS-SS-biotin (Fisher Scientific) for 2 h at 4°C. Excess biotin was quenched by incubating and washing synaptosomes with 100 mM glycine in PBS. Synaptosomes were lysed overnight in RIPA buffer containing protease inhibitors. Surface proteins (500 μg) were pulled down by streptavidin beads (Thermo Scientific) overnight, and eluded by SDS loading buffer containing 100 mM DTT. The surface protein fraction and 10 μg protein from the lysed synaptosomes (the total DAT protein) were loaded onto a 10% polyacrylamide gel and the proteins of interest were resolved. After blocking with 2.5% BSA, the membranes were treated overnight with MAB 16 monoclonal DAT antibody diluted 1:500 for the surface DAT and 1:1000 for the total DAT, followed by goat anti-mouse HRP antibody, diluted 1:15,000 for the surface DAT and 1:20,000 for the total DAT, for 1 h. The membranes were imaged following treatment of the surface DAT with Pico Enhanced Chemiluminescence (Millipore) and the total DAT with the Pierce ECL Western Blotting Substrate for 5 min. Quantification of the bands was performed with Scion Image software (Scion Corporation).
TH activity assay
TH activity was measured by a method modified from Levine et al. (Levine et al. 1984). L-[3,5-3H]tyrosine (Perkin-Elmer) was purified on a 6 cm long × 0.6 cm diameter column of Dowex 50WX4-200 beads. After application of the L-[3,5-3H]tyrosine to the column, it was washed with 10 mL H2O and 20 mL 0.5 M HCl. The L-[3,5-3H]tyrosine was eluted with 20 mL 1 M HCl. The purified solution was stored at -20°C.
Synaptosomes were lysed in 30 mM sodium acetate pH 6.0 and 0.1% Triton X-100 containing a cocktail of protease inhibitors (10 volumes in μL per mg of tissue wet weight). Ten μg protein were used for each VTA assay; 50 μg protein for each NAc assay. The blank was determined in the absence of enzyme. After purification, the activity of the L-[3,5-3H]tyrosine solution was 5,000 cpm/μL. The reaction mixture was added to 200,000 cpm of L-[3,5-3H]tyrosine dried under N2 at 50°C. In addition to L-[3,5-3H]tyrosine, the assay mixture contained 0.1 mM unlabeled tyrosine, 1 mM ascorbic acid, 0.1 mM Fe(NH4)2(SO4)2, 750 units catalase, 1 mM tetrahydrobiopterin, 40 μM potassium phosphate pH 6.6. TH was assayed in triplicate in a total volume of 100 μL at 37°C for 15 min. The reaction was stopped by the addition of 100 μL 1 M trichloroacetic acid.
The radioactive species other than [3H]H2O were removed from the assay solution by means of ion exchange chromatography. Ion exchange columns were prepared in 14.4 cm × 0.6 cm glass pipettes (Fisher Scientific) and consisted of 0.7 mL 50% slurry Dowex 50WX4-200, 80 μL 50% slurry activated charcoal, and 6 drops 50% slurry Dowex 1X2-400. One assay product mixture was purified per column. After application of the reaction product mixture to the column, the column was washed with two 0.7 mL portions of H2O. Radioactivity was measured by a liquid scintillation counter.
Total DA in tissue assay
Following rapid dissection, tissue samples were homogenized in 1.3 mL 0.32 M sucrose with a cocktail of protease inhibitors. The homogenate was mixed 1:1 with 2 M perchloric acid. The sample solution was centrifuged at 10,000g for 30 min. For analysis of DA by HPLC-EC, the supernatant was diluted 1:100 in perchloric acid containing 14.5 nM 2-aminophenol (internal standard).
DA turnover assay
Following rapid dissection, VTA tissue samples were homogenized in 2 M perchloric acid; NAc tissue samples were treated as described for the total DA assay. DA and the DA metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), and 3-methoxytyramine (3MT), were detected by HPLC-EC as described for the total DA assay. The content of each metabolite was normalized by the content of DA in each tissue sample for a metric of DA turnover (Koulu et al. 1988; Konradi et al. 1992; Pifl and Hornykiewicz 2006).
Results
Leptin enhances AMPH-evoked NAc DA efflux in normal animals
To investigate whether acute leptin treatment could modify DA signaling in normal animals, we initially examined the potential regulation of AMPH-stimulated DA efflux by leptin in the NAc of Sprague-Dawley rats. In vivo microdialysis in the NAc of freely-moving rats was coupled on-line to CE-LIF to measure the extracellular concentration of DA. Extracellular DA concentration was monitored beginning 3 h after leptin treatment and 1 h before the administration of AMPH (1 mg/kg, s.c.) (Figure 1A). We chose this duration of treatment in order to permit sufficient time for leptin to access its targets and acutely modulate neurophysiology, but to avoid potential long-term compensatory or secondary alterations in the mesolimbic DA system. While leptin treatment did not alter the basal extracellular DA prior to AMPH administration, leptin pretreatment increased the magnitude of AMPH induced DA efflux (Figure 1B). Accurate probe placement was confirmed by sectioning (Supplemental Figure 1). Thus, systemic leptin acutely potentiates AMPH-induced DA efflux in the NAc. These data reveal that leptin rapidly modulates the mesolimbic DA system of an intact rat.
Figure 1.
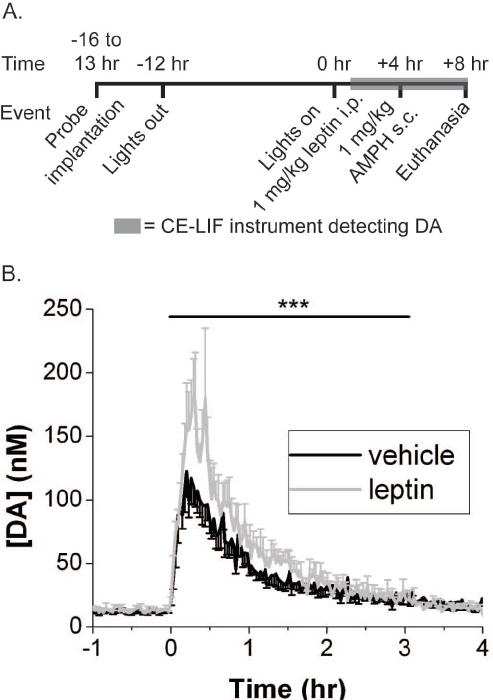
Procedure and results for microdialysis experiment. (A) Protocol for probe implantation and experimental treatments. (B) Trace of average AMPH evoked-DA efflux following treatment with leptin or vehicle 4 hours prior. Vehicle n = 6. Leptin n = 4. The [DA] is the dialysate concentration corrected for the in vitro recovery of the probe. The error bars represent SEM of [DA]. The black bar indicates the time period from time = 0-3.25 hrs which was subjected to 2 way ANOVA with repeated measures. *** indicates p value for interaction term (treatment X time) < 0.001.
Leptin acutely increases TH activity without altering TH expression or total DA in normal animals
Previous studies with ob/ob animals suggest that leptin deficiency blunts AMPH-mediated behavioral changes at least in part by decreasing NAc DA content, and that increased DA stores contribute to the reversal of this defect during chronic leptin treatment (Fulton et al. 2006). These alterations in DA stores mirror changes in TH expression, suggesting that chronic leptin treatment modulates mesolimbic DA content by controlling TH levels. We tested the ability of an acute leptin treatment to alter TH expression or DA content, by examining parameters of DA production in the mesolimbic DA system (Figure 2). We performed these experiments essentially as described for the AMPH-stimulated DA release in Figure 1, except that the animals received no AMPH treatment, but were sacrificed for tissue collection 4 h after the administration of leptin or vehicle (Figure 2A). We initially examined Th mRNA expression in the VTA, and TH protein content in the VTA and NAc (Figure 2B-D). These data demonstrated that 4 h of leptin treatment was insufficient to alter TH content by any of these measures, suggesting an alternate mechanism for the enhancement of AMPH-stimulated DA release by leptin in the normal rat. However, NAc TH activity was significantly increased by leptin treatment (Figure 2E). Despite the increase in TH activity, NAc DA content was not altered by leptin treatment (Figure 2F) although a non-significant trend toward increased DA turnover was observed in the NAc (Figure 3A). No trend regarding DA turnover was observed in the VTA (Figure 3B). These results show that a single systemic leptin injection can increase the activity of TH after 4 h but is not sufficient to promote increased expression of the enzyme or DA content. The lack of effect on DA content may be due to insufficient time to accumulate enough DA to affect the large pool of DA in the NAc or a lack of storage of local changes in DA.
Figure 2.
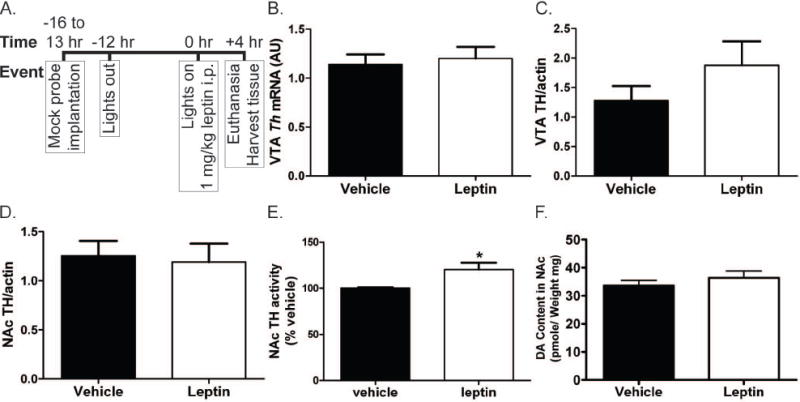
Procedure for tissue collection experiments and results for measurement of TH expression, TH activity, and total DA in tissue. All error bars represent SEM and treatment groups were compared by t test. (A) Protocol for animal treatment prior to tissue harvesting. The protocol was designed to mimic that used for the microdialysis experiments. (B) Expression of Th mRNA in the VTA as measured by qPCR. Vehicle n = 26. Leptin n = 24. (C) and (D) Expression of TH protein in the VTA(C) and NAc(D) as determined by quantified western blot. Actin band for used for standardization of the TH signal. Vehicle n = 6. Leptin n = 6. (E) Activity of TH in NAc collected from leptin or vehicle treated animals as measured by turnover of [3H]tyrosine. Vehicle n = 6. Leptin n = 5. * indicates p value < 0.05. (F) Total DA in the NAc measured by LC-EC. Vehicle n = 8. Leptin n = 9.
Figure 3.
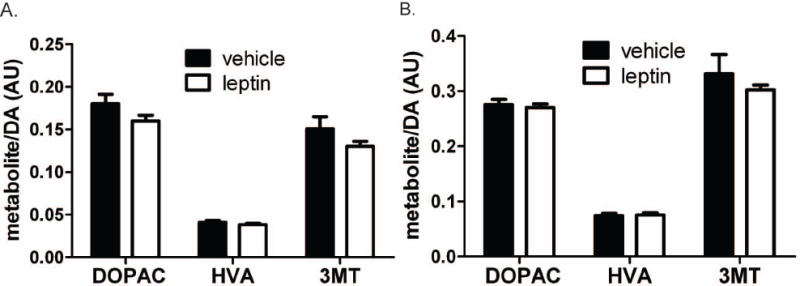
DA turnover in the NAc (A) and VTA (B). All error bars represent SEM and treatment groups were compared by t test. For all data, n = 9 except HVA in the NAc of leptin treated rats where n = 8.
Leptin increases DAT activity but not expression
Since NAc DA content is not increased following 4 h leptin treatment in Sprague-Dawley rats, some other mechanism must underlie the increase in AMPH-stimulated DA efflux observed following leptin treatment under similar conditions. As AMPH promotes the efflux of cytosolic DA stores via DAT (Sulzer et al. 2005), we examined NAc DAT activity by measuring the uptake of [3H]DA into synaptosomes following pre-treatment with leptin (Figure 4A and B). This analysis revealed a 2-fold increase in synaptosomal NAc DA uptake in leptin-treated animals compared to controls, and that the increased DAT activity reflected an increased Vmax for transport. However, this increased DAT activity was not the consequence of increased cell-surface DAT (Figure 4C-D). Nor were Dat mRNA or DAT protein expression increased by leptin treatment (Figure 4E-G). Thus, after 4 h of a single systemic leptin injection, DAT activity is increased independent of changes in total DAT content or cell surface expression. These results suggest that leptin can activate NAc DAT and this increased DAT activity may underlie the enhanced response to AMPH in acutely leptin-treated rats.
Figure 4.
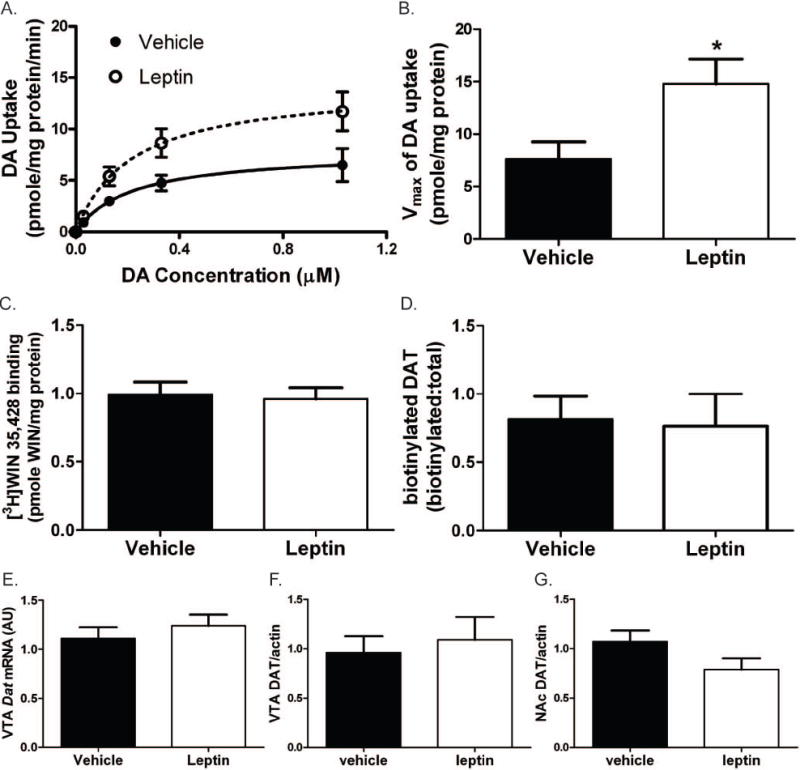
Results for measurement of DAT activity and expression. All error bars represent SEM and treatment groups were compared by t test. (A) DA uptake in the NAc as a function of [DA]. Vehicle n = 7. Leptin n = 8. DA uptake data were fit to the Michaelis-Menton equation. (B) The Vmax for DA uptake in tissue from the leptin treated animals was significantly greater than in vehicle treated animals. * indicates p value < 0.05. (C) DAT surface expression in the NAc as measured by [3H]WIN35428 binding. Vehicle n = 6. Leptin n = 6. (D) DAT surface expression as measured by biotinylation. Vehicle n = 5. Leptin n = 5. (E) Expression of Dat mRNA in the VTA as measured by qPCR. Vehicle n = 14. Leptin n = 14. (F) and (G) Expression of DAT protein in the VTA(F) and NAc(G) as determined by quantified western blot. Actin band for used for standardization of the DAT signal. Vehicle n = 6. Leptin n = 6.
Discussion
Leptin modulates behavior related to reward pathways (Fulton et al. 2000; Saper et al. 2002; Figlewicz and Benoit 2009) suggesting a partial basis for leptin effects on feeding and interaction of leptin and feeding with drugs of abuse. Studies of the neurochemical changes that underlie the effects of leptin in ob/ob mice have pointed to the mesolimbic DA pathway as a site of leptin action, specifically regulation of TH expression, DA content (Fulton et al. 2006; Leinninger et al. 2009), and somatodendritic vesicular packing (Roseberry et al. 2007). As lifelong leptin deficiency may result in adaptations that mask direct regulation of the DA system, understanding the neurochemical effects of leptin requires examination of acute leptin effects in normal animals. In this vein, leptin has been shown to decrease firing of VTA dopaminergic neurons due to hyperpolarization in anesthetized rats (Hommel et al. 2006), to decrease extracellular DA concentration as measured by microdialysis in food-deprived rats (Krügel et al. 2003), and augment the reward-potentiating effect of AMPH on lateral hypothalamic self-stimulation (Hao et al. 2006). Here we find that a single i.p. leptin injection acutely increases the activity of DAT and TH, without altering their expression or changing overall DA content or turnover in normal (fed) rats.
Previous work in leptin-deficient models, which have reduced Th mRNA and protein expression (Fulton et al. 2006; Leinninger et al. 2009), has shown that chronic leptin treatment increases TH content in mesolimbic DA neurons. Our present work in normal animals shows that acute exposure to leptin regulates TH activity rather than its expression, as well as regulating DAT activity, revealing heretofore unknown mechanisms by which leptin modulates the mesolimbic DA system. While the acute effects of leptin on the mesolimbic DA system occur independently of alteration in TH and DA content, they are consistent with the notion that leptin tends to promote pathways leading to DA synthesis, as previously shown in ob/ob animals. It is not yet clear whether the differences between the effects of acute versus chronic leptin action represent the consequences of long-term leptin action, per se, or rather differences due to compensation for the lifelong loss of leptin in ob/ob animals.
While the brief leptin treatment that we employed in this study does not alter basal concentration of extracellular DA in normal (fed) rats, AMPH treatment amplifies the effects of increased TH and DAT activity and promotes a significant increase in DA efflux in leptin-treated rats. Thus, increased activity of TH and DAT are acute downstream effects of leptin in normal rats that can lead to changes in dynamic responses to perturbations such as AMPH challenge.
The stable basal DA concentration observed following leptin treatment in our microdialysis experiments, together with increased TH activity, increased DAT activity, and suppressed dopaminergic firing rate (Hommel et al. 2006), suggest that these cellular level effects of acute leptin treatment, which would individually be predicted to cause opposing changes to the extracellular DA concentration, yield a balanced response that maintains consistent synaptic DA concentrations under baseline conditions. While leptin does not alter extracellular DA concentration under basal conditions or measurable turnover at this short time point, the dynamics of DA signaling during perturbations of the system may be altered by the change in activity of key regulatory proteins. The effect of AMPH exemplifies how these effects of leptin can give rise to a change in DA signaling.
While our present results and the previous data of others reveal no effect of leptin deficiency or treatment on baseline extracellular DA concentrations or D2-mediated IPSCs (Roseberry et al. 2007), leptin reportedly suppresses basal and feeding-related extracellular DA levels in food-restricted rats (Krügel et al. 2003). In view of the decreases in Vmax of DAT associated with food restriction (Patterson et al. 1998), it seems likely that the effect of leptin on DA concentrations after food restriction could be mediated, at least in part, by the increased DAT activity evoked by leptin. It will be of interest to determine if leptin also alters DA concentration dynamics associated with food seeking (Roitman et al. 2004) and use of abused drugs.
In agreement with our observations on AMPH-evoked DA efflux, leptin increases behavioral sensitivity to AMPH (Hao et al. 2004; Fulton et al. 2006; Hao et al. 2006). This observation has previously been considered paradoxical to the finding that food restriction increases sensitivity to drugs of abuse (Carr 2007). However, our results, showing increased activity of TH and Vmax of DAT by leptin combined with the reported hyperpolarizing effect of leptin on dopaminergic neurons (Hommel et al. 2006) provide an explanation for this effect of leptin on AMPH-evoked DA efflux. That is, the DA efflux elicited by the AMPH-induced reversal of DAT (Sulzer et al. 2005) is dependent on TH activity (Weissman et al. 1966; Chiueh and Moore 1975). Moreover, the velocity of DA uptake through DAT is enhanced at hyperpolarizing potentials (Sonders et al. 1997). Therefore, the leptin evoked changes would be expected to yield a system more responsive to AMPH.
The activation of DAT by leptin is reminiscent of the effect of insulin, another neuromodulating hormone involved in regulating energy balance and metabolism. Like leptin in this study, insulin has been shown to promote DAT activity; although in the case of insulin the regulation is believed to be via a mechanism involving trafficking of DAT (Carvelli et al. 2002). This similar site of action suggest that leptin and insulin act on the mesolimbic DA system via partially overlapping mechanisms.
This work shows that leptin acutely modulates the mesolimbic DA system in normal animals through enhanced DAT and TH activity. These effects interact in a way that does not alter the basal extracellular DA concentration but can alter DA extracellular concentration dynamics with perturbations, such as AMPH treatment. These data suggest new mechanisms for regulatory actions of leptin on the mesolimbic DA system.
Supplementary Material
Acknowledgments
Funding was provided by the National Institutes of Health R37 EB003320 (RTK), R01 DK078056 (MGM), T32 DA007267 (MLP), DA025954 (RC), the American Diabetes Association (MGM), and the University of Michigan Substance Abuse Research Center U026034 (RC) and training program (MLP). The contents of this manuscript are solely the responsibility of the authors and do no necessarily represent the official views of NIH.
Abbreviations
- aCSF
artificial cerebral spinal fluid
- AMPH
amphetamine
- BSA
bovine serum albumin
- CE-LIF
capillary electrophoresis with laser induced fluorescence detection
- DA
dopamine
- DAT
dopamine transporter
- [3H]WIN35428
[3H]WIN 35,428 (2β-carbomethoxy-3β-(4-fluorophenyl)-tropane)
- LepRb
long form of the leptin receptor
- LHA
lateral hypothalamus
- NAc
nucleus accumbens
- SDS
sodium dodecylsulfate
- TH
tyrosine hydroxylase
- VTA
ventral tegmental area
References
- Carr KD. Augmentation of drug reward by chronic food restriction: Behavioral evidence and underlying mechanisms. Physiol Behav. 2002;76:353–364. doi: 10.1016/s0031-9384(02)00759-x. [DOI] [PubMed] [Google Scholar]
- Carr KD. Chronic food restriction: Enhancing effects on drug reward and striatal cell signaling. Physiol Behav. 2007;91:459–472. doi: 10.1016/j.physbeh.2006.09.021. [DOI] [PubMed] [Google Scholar]
- Carvelli L, Moron JA, Kahlig KM, Ferrer JV, Sen N, Lechleiter JD, Leeb-Lundberg LMF, Merrill G, Lafer EM, Ballou LM, Shippenberg TS, Javitch JA, Lin RZ, Galli A. PI 3-kinase regulation of dopamine uptake. J Neurochem. 2002;81:859–869. doi: 10.1046/j.1471-4159.2002.00892.x. [DOI] [PubMed] [Google Scholar]
- Chen N-H, Xu C, Coffey LL, Reith MEA. [3H]WIN 35,428 [2[beta]-Carbomethoxy-3[beta]-(4-fluorophenyl)tropane] binding to rat brain membranes: Comparing dopamine cell body areas with nerve terminal regions. Biochem Pharmacol. 1996;51:563–566. doi: 10.1016/0006-2952(95)02208-2. [DOI] [PubMed] [Google Scholar]
- Chiueh CC, Moore KE. D-amphetamine-induced release of “newly synthesized” and “stored” dopamine from the caudate nucleus in vivo. J Pharmacol Exp Ther. 1975;192:642–653. [PubMed] [Google Scholar]
- Coffey LL, Reith MEA. [3H]WIN 35,428 binding to the dopamine uptake carrier. I. Effect of tonicity and buffer composition. J Neurosci Methods. 1994;51:23–30. doi: 10.1016/0165-0270(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin Differentially Regulates NPY and POMC Neurons Projecting to the Lateral Hypothalamic Area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998a;395:535–547. [PubMed] [Google Scholar]
- Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Aca Sci USA. 1998b;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. Am J Physiol Regul Integr Comp Physiol. 2009;296:R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton S, Woodside B, Shizgal P. Modulation of Brain Reward Circuitry by Leptin. Science. 2000;287:125–128. doi: 10.1126/science.287.5450.125. [DOI] [PubMed] [Google Scholar]
- Fulton S, Richard D, Woodside B, Shizgal P. Food restriction and leptin impact brain reward circuitry in lean and obese Zucker rats. Behav Brain Res. 2004;155:319–329. doi: 10.1016/j.bbr.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Fulton S, Pissios P, Manchon Ramon P, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS. Leptin Regulation of the Mesoaccumbens Dopamine Pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-Reducing Effects of the Plasma Protein Encoded by the Obese Gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Hao J, Cabeza de Vaca S, Carr KD. Effects of chronic ICV leptin infusion on motor-activating effects of D-amphetamine in food-restricted and ad libitum fed rats. Physiol Behav. 2004;83:377–381. doi: 10.1016/j.physbeh.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Hao J, Cabeza de Vaca S, Pan Y, Carr KD. Effects of central leptin infusion on the reward-potentiating effect of d-amphetamine. Brain Res. 2006;1087:123–133. doi: 10.1016/j.brainres.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Hay-Schmidt A, Helboe L, Larsen PJ. Leptin Receptor Immunoreactivity Is Present in Ascending Serotonergic and Catecholaminergic Neurons of the Rat. Neuroendocrinology. 2001;73:215–226. doi: 10.1159/000054638. [DOI] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu Z-W, Gao X-B, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin Receptor Signaling in Midbrain Dopamine Neurons Regulates Feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Kelly JF, Elias CF, Lee CE, Ahima RS, Seeley RJ, Bjørbæk C, Oka T, Saper CB, Flier JS, Elmquist JK. Ciliary Neurotrophic Factor and Leptin Induce Distinct Patterns of Immediate Early Gene Expression in the Brain. Diabetes. 2004;53:911–920. doi: 10.2337/diabetes.53.4.911. [DOI] [PubMed] [Google Scholar]
- Konradi C, Kornhuber J, Sofic E, Heckers S, Riederer P, Beckmann H. Variations of monoamines and their metabolites in the human brain putamen. Brain Research. 1992;579:285–290. doi: 10.1016/0006-8993(92)90062-e. [DOI] [PubMed] [Google Scholar]
- Koulu M, Lappalainen J, Pesonen U, Hietala J, Syvälahti E. Chronic treatment with SCH 23390, a selective dopamine D-1 receptor antagonist, decreases dopamine metabolism in rat caudate nucleus. European Journal of Pharmacology. 1988;155:313–316. doi: 10.1016/0014-2999(88)90521-3. [DOI] [PubMed] [Google Scholar]
- Krügel U, Schraft T, Kittner H, Kiess W, Illes P. Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin. Eur J Pharmacol. 2003;482:185–187. doi: 10.1016/j.ejphar.2003.09.047. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Jo Y-H, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S, Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Münzberg H, Myers MG., Jr Leptin Acts via Leptin Receptor-Expressing Lateral Hypothalamic Neurons to Modulate the Mesolimbic Dopamine System and Suppress Feeding. Cell Metab. 2009;10:89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshan RL, Bjornholm M, Munzberg H, Myers MG., Jr Leptin receptor signaling and action in the central nervous system. Obesity. 2006;14(Supplement):208S–212S. doi: 10.1038/oby.2006.310. [DOI] [PubMed] [Google Scholar]
- Levine RA, Pollard HB, Kuhn DM. A rapid and simplified assay method for tyrosine hydroxylase. Anal Biochem. 1984;143:205–208. doi: 10.1016/0003-2697(84)90577-3. [DOI] [PubMed] [Google Scholar]
- Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman JM. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- Patterson TA, Brot MD, Zavosh A, Schenk JO, Szot P, Figlewicz DP. Food Deprivation Decreases mRNA and Activity of the Rat Dopamine Transporter. Neuroendocrinology. 1998;68:11–20. doi: 10.1159/000054345. [DOI] [PubMed] [Google Scholar]
- Pifl C, Hornykiewicz O. Dopamine turnover is upregulated in the caudate/putamen of asymptomatic MPTP-treated rhesus monkeys. Neurochemistry International. 2006;49:519–524. doi: 10.1016/j.neuint.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM. Dopamine Operates as a Subsecond Modulator of Food Seeking. J Neurosci. 2004;24:1265–1271. doi: 10.1523/JNEUROSCI.3823-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseberry AG, Painter T, Mark GP, Williams JT. Decreased vesicular somatodendritic dopamine stores in leptin-deficient mice. J Neurosci. 2007;27:7021–7027. doi: 10.1523/JNEUROSCI.1235-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK. The Need to Feed: Homeostatic and Hedonic Control of Eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Shou M, Ferrario CR, Schultz KN, Robinson TE, Kennedy RT. Monitoring Dopamine in Vivo by Microdialysis Sampling and On-Line CE-Laser-Induced Fluorescence. Anal Chem. 2006;78:6717–6725. doi: 10.1021/ac0608218. [DOI] [PubMed] [Google Scholar]
- Simon GE, Von Korff M, Saunders K, Miglioretti DL, Crane PK, van Belle G, Kessler RC. Association Between Obesity and Psychiatric Disorders in the US Adult Population. Arch Gen Psychiatry. 2006;63:824–830. doi: 10.1001/archpsyc.63.7.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Justice JB. The effect of inhibition of synthesis, release, metabolism and uptake on the microdialysis extraction fraction of dopamine. J Neurosci Methods. 1994;54:75–82. doi: 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Sonders MS, Zhu S-J, Zahniser NR, Kavanaugh MP, Amara SG. Multiple Ionic Conductances of the Human Dopamine Transporter: The Actions of Dopamine and Psychostimulants. J Neurosci. 1997;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Weissman A, Koe BK, Tenen SS. Antiamphetamine Effects Following Inhibition of Tyrosine Hydroxylase. J Pharmacol Exp Ther. 1966;151:339–352. [PubMed] [Google Scholar]
- Wyvell CL, Berridge KC. Intra-Accumbens Amphetamine Increases the Conditioned Incentive Salience of Sucrose Reward: Enhancement of Reward “Wanting” without Enhanced “Liking” or Response Reinforcement. J Neurosci. 2000;20:8122–8130. doi: 10.1523/JNEUROSCI.20-21-08122.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.