

Abstract
We have shown previously that the plecomacrolide antibiotics bafilomycin A1 and B1 significantly attenuate cerebellar granule neuron death resulting from agents that disrupt lysosome function. To further characterize bafilomycin-mediated cytoprotection, we examined its ability to attenuate the death of naïve and differentiated neuronal SH-SY5Y human neuroblastoma cells from agents that induce lysosome dysfunction in vitro, and from in vivo dopaminergic neuron death in C. elegans. Low-dose bafilomycin significantly attenuated SH-SY5Y cell death resulting from treatment with chloroquine, hydroxychloroquine amodiaquine and staurosporine. Bafilomycin also attenuated the chloroquine-induced reduction in processing of cathepsin D, the principal lysosomal aspartic acid protease, to its mature “active” form. Chloroquine induced autophagic vacuole accumulation and inhibited autophagic flux, effects that were attenuated upon treatment with bafilomycin and were associated with a significant decrease in chloroquine-induced accumulation of detergent-insoluble α-synuclein oligomers. In addition, bafilomycin significantly and dose-dependently attenuated dopaminergic neuron death in C. elegans resulting from in vivo over-expression of human wild-type α-synuclein. Together, our findings suggest that low-dose bafilomycin is cytoprotective in part through its maintenance of the autophagy-lysosome pathway, and underscores its therapeutic potential for treating Parkinson Disease and other neurodegenerative diseases that exhibit disruption of protein degradation pathways and accumulation of toxic protein species.
Keywords: bafilomycin, autophagy, lysosome, cathepsin D, Parkinson Disease, α-synuclein
INTRODUCTION
The autophagy-lysosome pathway (ALP) is responsible for the highly-regulated recycling of intracellular contents, whereby macronutrients and damaged organelles are enclosed within double-membraned autophagic vacuoles (AVs) and delivered to lysosomes for pH-dependent degradation by lysosomal hydrolases. It is well known that intact lysosome function is critical for effective completion of the ALP (reviewed in (Pivtoraiko et al. 2009) and (Shacka et al. 2008). Lysosome dysfunction can potently inhibit autophagy completion as demonstrated by robust AV accumulation followed by the induction of cell death (Zaidi et al. 2001); (Boya et al. 2005); (Shacka et al. 2006b). While the ALP is ordinarily cytoprotective, it is unclear whether AV accumulation resulting from lysosome dysfunction contributes to cell death.
Chloroquine is an anti-malarial drug and potent lysosomotropic agent. As a weak base chloroquine accumulates in acidic vesicles and raises their pH (reviewed in Pivtoraiko et al. 2009). We and others have shown that chloroquine disrupts lysosome function and inhibits autophagy completion as demonstrated by AV accumulation, and induces both apoptotic and non-apoptotic cell death (Zaidi et al. 2001); (Boya et al. 2003); (Boya et al. 2005); (Shacka et al. 2006b); (Pivtoraiko et al. 2009).
Growing evidence suggests that the ALP is also altered in age-related neurodegenerative diseases including Parkinson disease (PD), the most common neurodegenerative movement disorder. Alterations in autophagy were reported initially by the aberrant accumulation of AVs in substantia nigra neurons of PD patients (Anglade et al. 1997). Mutations in several PD-specific genes, including α-synuclein, LRRK2, Parkin and ATP13A2, are known to adversely affect autophagy and/or lysosome function (reviewed in Shacka et al. 2008 and Pan et al. 2008). α-Synuclein is a major component of Lewy bodies in PD brain and α-synuclein accumulation is thought to play an important causal role in the onset and progression of PD. Lysosomes are important for α-synuclein clearance, and cathepsin D (CD), the principal lysosomal aspartic acid protease, is the main lysosomal enzyme involved in the degradation of α-synuclein (Sevlever et al. 2008). Consistent with this hypothesis, CD deficiency has been reported to enhance α-syn toxicity (Qiao et al. 2008); (Cullen et al. 2009), and several studies indicate the therapeutic potential for autophagy induction in promoting α-synuclein clearance in PD (Webb et al. 2003); (Spencer et al. 2009); (Yang et al. 2009a); (Yu et al. 2009). Together, these findings suggest that ALP-targeted therapies may be effective in maintaining α-synuclein clearance and in general inhibiting neurodegenerative disease-associated neuropathology.
Bafilomycin A1 represents the plecomacrolide subclass of macrolide antibiotics and was characterized initially by its selective inhibition of vacuolar type (V)-ATPase (Bowman et al. 1988). V-ATPase maintains the low pH of acidic vesicles through its regulation of proton pumping (Forgac 2007). At concentrations ≥ 10 nM, bafilomycin A1 inhibits V-ATPase and in turn increases intravesicular pH (Yoshimori et al. 1991), thus mimicking the effects of chloroquine. However, we have shown previously that the plecomacrolides bafilomycin A1, bafilomycin B1 and concanamycin all significantly attenuate chloroquine-induced death of cerebellar granule neurons (Shacka et al. 2006b); (Shacka et al. 2006a), at low concentrations (≤ 1 nM) which do not inhibit V-ATPase (Bowman et al. 1988) or induce AV accumulation (Shacka et al. 2006b). These data suggest that bafilomycin-mediated neuroprotection is independent of its inhibition of V-ATPase. However, whether bafilomycin affects the ALP or regulates α-synuclein clearance and neurotoxicity has not been investigated.
In the current study, we extended our analysis of chloroquine -induced death and bafilomycin A1 neuroprotection to SH-SY5Y, a human neuroblastoma cell line commonly used to model dopaminergic neurons. We also assessed the cytoprotective effects of bafilomycin in the nematode C. elegans that over-express human wild-type α-synuclein, which we have previously shown to induce both age- and dose-dependent neurodegeneration in vivo (Cao et al. 2005); (Hamamichi et al. 2008). Our results indicate that bafilomycin attenuates neuronal cell death in all of these models consistent with its ability to maintain ALP function and reduce α-synuclein neurotoxicity.
MATERIALS AND METHODS
Cell Culture
Naïve SH-SY5Y human neuroblastoma cells were cultured in Minimum Essential Medium Eagle (MEM) (Cellgro, Herndon, VA) and F12-K (ATCC, Manassas, VA) medium supplemented with 0.5% sodium pyruvate, 0.5% non essential amino acids (Cellgro, Herndon, VA), 1% penicillin/streptomycin (Sigma, St. Louis, MO), and 10% fetal bovine serum (FBS; HyClone, Logan, UT). Cells were plated at 200 cells/mm2 and grown for 24h in media containing 1% FBS prior to treatment. Cells were treated from 0-48h in media containing 1% FBS. SH-SY5Y cells were differentiated for 7-8 days in complete media supplemented with 10μM retinoic acid (Sigma). Retinoic-acid-supplemented media was replaced every 2-3 days. Unless otherwise noted, differentiated cells were plated at 400/mm2 in differentiation media containing 2% B-27 (Invitrogen, Carlsbad, CA) and were treated in this media for up to 48h.
Cells were treated with chloroquine, hydroxychloroquine, amodiaquine, staurosporine or 3-methyladenine (Sigma), and either bafilomycin A1 (Sigma) or bafilomycin B1 (A.G. Scientific, San Diego, CA).
Measurement of SH-SY5Y Cell Viability and Caspase-3-Like Activity
Cell viability was measured using calcein AM fluorogenic conversion assay (Invitrogen). Caspase-3-like activity was detected via fluorogenic DEVD cleavage assay and expressed relative to untreated controls. These assays were performed using previously published protocols (Nowoslawski et al. 2005).
Tandem fluorescent-tagged LC3 (tfLC3) Assay of Autophagic Flux
Differentiated SH-SY5Y cells were transiently transfected with ptfLC3 (mammalian expression vector in plasmid DH5a; Entrez ID of insert U05784) (Addgene, Cambridge, MA) using the Amaxa Nucleofector™ II device protocol (Lonza, Cologne, Germany). Two million cells were pelleted and resuspended in 100 μl of Lonza™ Nucleofector reagent followed by addition of ptfLC3 DNA (2 μg/reaction). Transfer of the reaction mixture was completed by electroporation in the Amaxa Nucleofector™ II device. 500 μl of neutralization media was immediately added followed by transfer of the full volume to a fresh tube, where cells recovered for 10 min, RT. Transfected cells were plated in poly-L-lysine coated glass chamber slides (LabTek) at 120,000 cells/600 μl media per well and kept at 37°C prior to treatment. Media was exchanged for fresh 4h after plating to remove transfection reagents in addition to cells that did not survive the transfection reaction. Cells were treated with 50 μM chloroquine in the presence or absence of 1 nM BafA1 for 8h. After treatment slides were fixed with 3% paraformaldehyde for 45 min at room temperature, washed with 1X PBS and cover-slipped. Cells were visualized using a Zeiss™ Observer.Z1 Laser Scanning Microscope (Thornwood, NY) equipped with a Zeiss™ 40X 1.3 Oil DIC M27 Plan-Apochromat objective and imaged using Zen™ 2008 LSM 710, V5.0 SP1.1 software. Fluorescence filters were used to observe bis benzemide (excitation 405 nm, emission 409-514 nm), EGFP (excitation 488, emission 494-572 nm), and mRFP (excitation 543, emission 585-734).
Generation and analysis of C. elegans
Isogenic C. elegans strain UA44 (baIn11; Pdat-1∷α-syn, Pdat-1∷GFP) that co-express α-synuclein and GFP in dopamine neurons and exhibit age-dependent α-synuclein-induced neurodegeneration were age synchronized by bleaching as previously described (Lewis and Fleming 1995) and placed in 1 ml of water containing 5% methanol (with or without bafilomycin B1) for 24h at 20°C with gentle agitation. After incubation, worms were washed with M9 buffer three times, transferred to NGM plates, and grown at 20°C. For each trial, 30 worms were immobilized with 3 mM levamisole, transferred onto a 2.5% agarose pad, and analyzed for neuroprotection at both day 7 and day 10 post-hatchings. Worms were considered wild-type (WT) when there were four intact CEP type DA neurons and two ADE type DA neurons without any signs of degeneration. Each bafilomycin B1 treatment was analyzed in triplicate (90 worms per concentration).
To determine if increased concentrations of bafilomycin B1 induced DA neuron cell death, the C. elegans strain BY200 (Pdat-1∷GFP) (Nass and Blakely 2003), which express GFP in the DA neurons without degeneration, was synchronized, treated with high concentrations of bafilomycin B1, and analyzed as described above. To study the effect of chloroquine on DA neurons, C. elegans strain UA44 was crossed into knockout strain NL131 [pgp-3(pk18)], shown previously to be sensitive to chloroquine (Broeks et al. 1995) to generate the isogenic strain UA146 [baIn11; pgp-3(pk18)]. This strain was synchronized, treated with chloroquine using methods similar to bafilomycin B1, and analyzed as described above.
Statistics
Significant effects of treatment were analyzed either by one-factor ANOVA (three or more groups) or by unpaired, two-tailed t test (two groups). Post hoc analysis was conducted using Bonferroni’s test. A level of p < 0.05 was considered significant.
RESULTS
Bafilomycin B1 is a structural analog of bafilomycin A1 and both compounds were shown previously at high doses and with equal potency to inhibit V-ATPase (Bowman et al. 1988), whereas we have shown that low doses of each are equally effective in attenuating chloroquine-induced death of cerebellar granule neurons (Shacka et al. 2006b). Treatment of naïve SH-SY5Y cells for 48h with bafilomycin A1 (Fig. 1A) or bafilomycin B1 (Fig. 1B) did not alter cell viability when added alone at ≤ 1 nM, whereas both significantly reduced cell viability at concentrations ≥ 6nM, effects similar to those reported in cerebellar granule neurons (Shacka et al. 2006a; Shacka et al. 2006b). To determine if bafilomycin-induced death of SH-SY5Y cells was apoptotic, caspase-3-like activity was measured following treatment for 48h with 0.1-10 nM bafilomycin A1 (Fig. 1C). Significant increases in caspase-3 like activity were observed only at bafilomycin A1 concentrations ≥ 6 nM, suggesting that cell death induced by high concentrations of bafilomycin A1 (Fig. 1A) was apoptotic. These results confirm that bafilomycin A1 and bafilomycin B1 neither induced SH-SY5Y cell death or apoptosis at concentrations ≤ 1 nM.
Fig. 1. Low-dose bafilomycin is not cytotoxic to SH-SY5Y cells.

48h treatment with bafilomycin A1 (BafA1, A) or bafilomycin B1 (BafB1, B) decreases cell viability at concentrations ≥ 6 nM for BafA1 and ≥ 3 nM for BafB1. (C) BafA1 significantly increases caspase-3-like activity at concentrations ≥ 6 nM. Results represent mean ± SD from at least three independent experiments. *p<0.05 vs. 0 μM vehicle CTL; #p<0.05 vs.0-3 nM BafA1 (A) or 0-1 nM BafB1 (B).
Naïve SH-SY5Y cells were treated with cell death stimuli 24h after plating, and cell viability was measured 48h after treatment (Supplemental Fig. 1). Chloroquine significantly decreased cell viability at concentrations ≥ 20 μM (Supplemental Fig. 1A), and cell death was maximal at 60-80 μM. Upon treatment of cells with 50 μM chloroquine, significant decreases in cell viability were observed from 24-48h following treatment (Supplemental Fig. 1B), and was maximal at 48h. The chloroquine analogs amodiaquine (Supplemental Fig. 1C) and hydroxychloroquine (Supplemental Fig. 1D) significantly reduced cell viability in a concentration-dependent manner similar to that of chloroquine, although amodiaquine was 2-3 times more potent in reducing cell viability by 50% (~15 μM) in comparison to chloroquine (~40 μM) and hydroxychloroquine (~50 μM). In addition, treatment for 48h with staurosporine, a classical apoptotic stimulus shown previously to disrupt lysosome function (Bidere et al. 2003); (Kagedal et al. 2005), significantly decreased SH-SY5Y cell viability at all concentrations tested (0.005-0.1 μM), and was maximally effective at 0.1 μM (Supplemental Fig. 1E).
When bafilomycin A1 or bafilomycin B1 were added at 0.1-1nM concurrently for 48h with 50 μM chloroquine, we observed significant attenuation of chloroquine-induced cell death (Fig. 2A-B), an effect that was maximal at 0.6-1 nM for bafilomycin A1 (Fig. 2A) and 0.3-1nM for bafilomycin B1 (Fig. 2B). Bafilomycin A1 (1nM) also significantly attenuated the loss in cell viability following 48h treatment with amodiaquine (15 μM) or hydroxychloroquine (50 μM; Supplemental Fig. 2A) or staurosporine (0.1 μM, Supplemental Fig. 2B). High-dose bafilomycin has been shown previously to directly inhibit chloroquine localization within lysosomes (Boya et al. 2003). To rule out this potential interaction of low-dose bafilomycin against chloroquine-induced cell death, cells were pre-treated with bafilomycin A1 for either 12h (Fig. 2C) or 24h (Fig. 2D) and following its wash-out post-treated with 50 μM chloroquine for 48h. Both 12h and 24h pre-treatment with bafilomycin A1 significantly attenuated cell death induced by chloroquine post-treatment, supporting our argument that the cytoprotective effects of low-dose bafilomycin are not from its inhibition of chloroquine localization to lysosomes.
Fig. 2. Low-dose bafilomycin attenuates chloroquine-induced cell death and apoptosis.

48h treatment with bafilomycin A1 (BafA1, A) or bafilomycin B1 (BafB1, B) significantly attenuates chloroquine (CQ, 50 μM)-induced cell death from 0.1-6 nM for BafA1 (A) and from 0.1-1 nM for BafB1 (B). (C-D) Pretreatment with 1 nM BafA1 for either 12h (C) or 24h (D) significantly attenuates the reduction in viability following 48h post-treatment with 50 μM CQ. (E) 24h treatment with BafA1 (1 nM) significantly attenuates CQ-induced increase in caspase-3-like activity. (F) Co-treatment with the general caspase inhibitor BOC-Asp (OMe)-FMK (Boc-FMK, 30 μM) neither attenuates CQ-induced cell death nor enhances the cytoprotective effects of BafA1 against CQ-induced cell death. *p<0.05 vs. 0 μM vehicle CTL; ** p<0.05 vs. vehicle pre-treatment/CQ post-treatment (C-D) or CQ (F); #p<0.05 vs.0-3 nM & 3-6 nM BafA1 (A), 0-0.1 nM & 3-10 nM BafB1 (B), 0-0.1 nM & 6-10 nM BafA1 (C); ##p<0.05 vs. CQ+Boc-FMK.
We have shown previously that chloroquine induces robust neuron apoptosis (Zaidi et al. 2001); (Shacka et al. 2006b) and that bax deficiency significantly attenuates chloroquine-induced death of cultured neurons, suggesting the importance of the intrinsic apoptotic pathway in regulating chloroquine-induced death (Zaidi et al. 2001); (Shacka et al. 2006b). We have also shown that low-dose bafilomycin attenuates chloroquine-induced apoptosis of cultured neurons (Shacka et al. 2006b). We next sought to determine the effects of chloroquine ± low-dose bafilomycin A1 on the induction of SH-SY5Y apoptosis. Treatment for 24h with 50 μM chloroquine significantly induced apoptosis as measured by enzymatic caspase-3-like activity (Fig. 2E), an effect that was significantly attenuated by treatment with bafilomycin A1 and was maximally protective at 0.3-3 nM. To determine if caspase activation was a commitment point for chloroquine-induced death of SH-SY5Y cells, we measured the effects of chloroquine ± low-dose bafilomycin A1 on cell viability in the presence of the general caspase inhibitor BOC-Asp (OMe)-FMK (Fig. 2F). This caspase inhibitor at 30 μM completely blocked chloroquine-induced caspase-3 activity but neither attenuated chloroquine-induced cell death nor enhanced bafilomycin-mediated cytoprotection. Together these data obtained in SH-SY5Y cells corroborate our previous findings in cerebellar granule neurons (Shacka et al. 2006b); (Shacka et al. 2006a) suggesting that 1) bafilomycins potently attenuate cell death and apoptosis induced by chloroquine and structurally similar analogs when used at concentrations ≤ 1 nM; and 2) bafilomycin regulation of chloroquine-induced cell death occurs upstream of caspase activation.
We have shown recently that chloroquine-induced death of SH-SY5Y cells correlated with a compromise in the maturation of CD (Pivtoraiko et al. 2009). Treatment for 24 hour with 50 μM chloroquine significantly induced levels of the unprocessed, 50 kDa pre-pro form of CD (Fig. 3A, B) and significantly decreased levels of the mature active 32 kDa form of CD (Fig. 3A, D). These effects were observed as early as 12h following chloroquine treatment (data not shown). The 47 kDa pro-form of CD was not significantly affected by treatment (Fig. 3C). When added by itself, 1 nM bafilomycin A1 did not alter CD processing. However, 1 nM bafilomycin A1 significantly attenuated the chloroquine-induced decrease in the mature form of CD (Fig. 3D), suggesting the ability of bafilomycin A1 to partially recover lysosomal enzyme function at concentrations that significantly attenuates chloroquine-induced cell death.
Fig. 3. Bafilomycin A1 attenuates chloroquine-induced inhibition of CD processing.

(A) Whole cell lysates of SH-SY5Y cells were collected 24h following treatment with chloroquine (CQ, 50 μM) and/or bafilomycin A1 (BafA1, 1 nM), and subjected to western blot analysis for CD (pre-pro form, 50 kDa; pro form, 47 kDa; mature “active” form, 32 kDa). Blots were stripped and re-probed for GAPDH (37 kDa) loading control. Levels of (B) pre-pro CD, (C) pro CD and (D) mature CD were normalized to levels of β-tubulin. Results represent mean ± SD from at least three independent experiments. *p<0.05 vs. 0 μM vehicle CTL; **p<0.05 vs. CQ.
To determine if neuronal differentiated SH-SY5Y cells exhibited protective effects of bafilomycin against chloroquine-induced cell death similar to that of naïve undifferentiated cells, naïve SH-SY5Y cells were differentiated for 7-8 days with retinoic acid. Chloroquine induced a concentration-dependent decrease the viability of differentiated cells (Supplemental Fig. 3A) in differentiated cells similar to that of naïve cells (Supplemental Fig. 1A), an effect that was significantly attenuated by the addition of 1 nM bafilomycin A1 (Supplemental Fig. 3B). We next sought to determine if chloroquine affects the clearance of endogenous α-synuclein oligomers in neuronal SH-SY5Y cells, since it is well known that intact lysosome function plays an important role in α-synuclein degradation (Lee et al. 2004); (Qiao et al. 2008); (Sevlever et al. 2008); (Cullen et al. 2009). Triton X-soluble vs. -insoluble fractions were prepared from lysates of differentiated SH-SY5Y cells following 48h treatment with chloroquine (50 μM) in the presence or absence of low-dose bafilomycin A1 (1 nM). Treatment with chloroquine significantly increased levels of endogenous detergent-insoluble α-synuclein oligomers (Fig. 4A-B). By itself, 1 nM bafilomycin A1 did not significantly alter levels of insoluble α-synuclein oligomers (Fig. 4A-B). In contrast, 1 nM bafilomycin A1 significantly attenuated the chloroquine-induced increase in detergent-insoluble α-synuclein oligomers present in the detergent-insoluble fraction (Fig. 4A-B). Chloroquine and/or bafilomycin had no effect on endogenous levels of detergent-soluble α-synuclein (data not shown). Together, these data indicate that low-dose bafilomycin regulates the clearance of detergent-insoluble forms of endogenous α-synuclein oligomers.
Fig. 4. Bafilomycin A1 attenuates chloroquine-induced increase in detergent-insoluble endogenous α-syn oligomers.

(A) Representative western blot analysis of endogenous α-syn high molecular weight oligomers (34 & 51 kDa) in detergent–insoluble fractions, prepared from lysates of differentiated SH-SY5Y cells collected 48h after treatment with chloroquine (CQ, 50 μM) and/or bafilomycin A1 (BafA1, 1 nM). Blots were stripped and re-probed for actin (42 kDa) loading control. (B) Levels of insoluble α-syn high molecular weight oligomers were quantified and normalized to levels of actin. Results represent mean ± SD from five independent experiments. *p<0.05 vs. 0 μM vehicle CTL; **p<0.05 vs. CQ.
We have shown previously that chloroquine-induced death of cerebellar granule neurons was accompanied by the robust accumulation of AVs (Shacka et al. 2006b). To determine the effects of chloroquine ± low-dose bafilomycin on AV accumulation in neuronally differentiated SH-SY5Y cells, western blot analysis was performed on detergent-soluble vs. –insoluble protein fractions (Fig. 5). Levels of LC3-II, which were used as a selective marker of AVs, were not increased following treatment with bafilomycin A1 (1 nM) or in vehicle-treated control cells. Treatment with chloroquine (50 μM) for 48h significantly induced AV accumulation as measured by LC3-II/actin ratios vs. control-treated cultures (Fig. 5A-C). In contrast, co-treatment with chloroquine and bafilomycin A1 significantly decreased AV accumulation compared to chloroquine treatment alone (Fig. 5A-C).
Fig. 5. Bafilomycin A1 attenuates chloroquine-induced AV accumulation.

(A) Representative western blot analysis for LC3-I (cytosolic, 16 kDa) vs. LC3-II (AV membrane-specific, 14 kDa) in detergent-soluble (LEFT) vs. –insoluble (RIGHT) fractions prepared from differentiated SH-SY5Y cells 48h after treatment with chloroquine (CQ, 50 μM) and/or bafilomycin A1 (BafA1, 1 nM). Blots were stripped and re-probed for actin (42 kDa) loading control. Levels of LC3-II were quantified and normalized to actin for detergent-soluble (B) and detergent-insoluble (C) fractions. Note only detergent soluble fractions exhibited LC3-I immunoreactivity. Results represent mean ± SD from at least four independent experiments. *p<0.05 vs. 0 μM vehicle CTL; **p<0.05 vs. CQ.
To further investigate whether attenuation of chloroquine-induced death by low-dose bafilomycin was associated with alterations in ALP function we utilized the tandem fluorescent-tagged LC3 (tfLC3) assay (Fig. 6), an in vitro fluorescence measure of autophagic flux (Kimura et al. 2007); (Kimura et al. 2009). Over-expression of the tfLC3 plasmid results in tandem expression of both mRFP-LC3 and GFP-LC3. Under basal conditions, GFP-LC3 loses fluorescence due to the acidic nature of lysosomes. However, a compromise in lysosome stabilizes GFP-LC3 fluorescence and in turn increases GFP-LC3 and mRFP-LC3 co-localization. Cultures of tfLC3-transfected cells were treated for 8h with chloroquine (50 μM) in the presence or absence of 1nM bafilomycin A1. Both vehicle control and low-dose bafilomycin-treated cells exhibited basal RFP and GFP fluorescence and little if any co-localization (Fig. 6A, B). Treatment with chloroquine induced the appearance of both GFP-LC3 and RFP-LC3 fluorescent punctae and their apparent co-localization, (Fig. 6C), effects that appeared to be dramatically reduced in chloroquine plus bafilomycin A1-treated cultures (Fig. 6D). Together, these data suggest that low dose bafilomycin may preserve lysosome function and thus, maintenance of autophagic flux following chloroquine exposure.
Fig. 6. Bafilomycin A1 attenuates chloroquine-induced inhibition of autophagic flux.
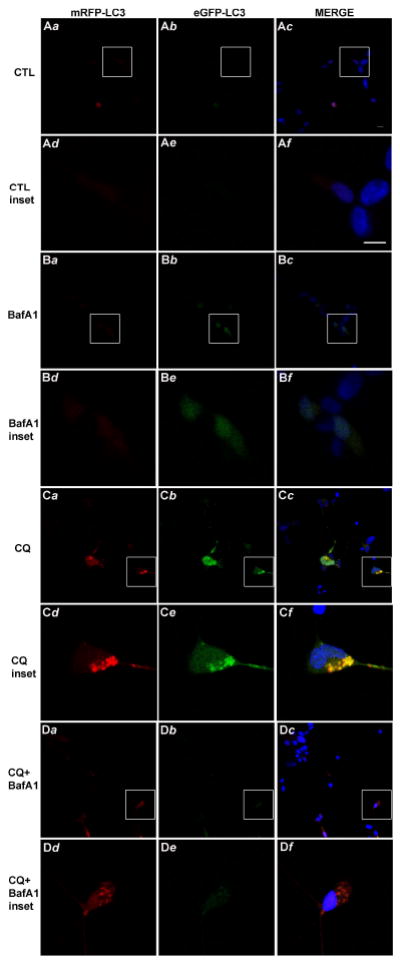
Differentiated SH-SY5Y were transiently transfected to over-express tfLC3 and following 24h recovery were treated for 8h with chloroquine (CQ, 50 μM) and/or bafilomycin A1 (BafA1, 1 μM) to observe effects of low-dose BafA1 on chloroquine-induced co-localization (merged image, right panel) of mRFP-LC3 (left panels) and eGFP-LC3 (center panels) fluorescent punctae, suggesting inhibition of autophagic flux. Cells were fixed and imaged via confocal microscopy as described in the Methods section. Panels a-c from CTL (A), BafA1 (B), CQ (C) and CQ+BafA1 are low magnification images; the box in each of these panels indicates higher magnification inset panels (d-f) for each. Images are representative of three independent experiments. Scale bar in Ac and Af = 10 μM.
We also attempted to determine if inhibition of autophagy induction by treatment of cultures with 3-methyladenine, an inhibitor of class-III PI3-K, attenuated chloroquine-induced death. Treatment of differentiated SH-SY5Y cells with either 50 μM or 5 mM 3-methyladenine did not attenuate chloroquine-induced death. However, 3-methyladenine was toxic in its own right to SH-SY5Y cells compared to vehicle control (Supplemental Fig. 4), suggesting the importance of basal autophagy induction in promoting cell survival. These data suggest that autophagy induction is not important for regulating chloroquine-induced cell death.
To determine if plecomacrolides protected against α-synuclein- induced DA neurodegeneration in vivo, isogenic worms over-expressing human wild-type α-synuclein in dopamine neurons were acutely exposed to bafilomycin B1 for 24h during larval development, and subsequently scored for dopaminergic neuron loss at either 7 or 10 days post-hatching (4 and 7 day adults, respectively). Acute exposure of animals to 50-150 μg/ml bafilomycin B1 showed significant protection against α-synuclein-induced dopaminergic degeneration (Fig. 7). Exposure of these worms to lower concentrations of bafilomycin B1 did not significantly protect against dopaminergic neuron degeneration at day 7 (Fig. 7A), whereas higher concentrations induced DA neuron death in a majority of the animals. At day 10, animals exposed to either 50 or 100 μg/ml bafilomycin B1 still exhibited significant protection against neurodegeneration (Fig. 7B). Worms lacking α-syn over-expression were also treated with bafilomycin B1and the percentage of worms exhibiting DA neurons at either 7 or 10 days following treatment was not significantly different from vehicle control at concentrations ranging from 100-300 μg/ml (Supplemental Fig. 5A). However, treatment of worms with 400-500 μg/ml bafilomycin B1 was lethal to the embryos, thus precluding neuron counts from these worms and suggesting the importance of intact V-ATPase function for worm survival. Together these results provide in vivo evidence that acute exposure of bafilomycin protects dopaminergic neurons against α-synuclein-induced neurodegeneration.
Fig. 7. Bafilomycin attenuates the death of DA neurons in C. elegans following over-expression of wild-type human α-syn.

(A, B) Worms over-expressing α-syn in DA neurons were acutely exposed to bafilomycin B1 (BafB1, 0-200 μg/ml) for 24h during larval development, and then subsequently scored for DA neuron loss at either seven days (A) or ten days (B) post-hatching (4 and 7 day adults, respectively). Results represent mean ± SD from at least three independent experiments, where 30 worms were analyzed for each experiment (n=90). *p<0.05 vs. 0 μg/ml vehicle CTL.
Since it is possible that low-dose bafilomycin exerted a “pre-conditioning effect” on DA neuron survival, we sought out to determine if treatment with low doses of chloroquine also protected against DA neuron death in worms. However, since worms are naturally resistant to some natural toxins, including chloroquine (Broeks et al. 1995), we crossed α-synuclein over-expressing worms into those mutant for pgp-3. PGP-3 encodes a P-glycoprotein transmembrane protein that has been predicted to export toxins such as chloroquine from cells and thus increase resistance to chloroquine toxicity (Broeks et al. 1995). Worms were treated with eleven concentrations of chloroquine ranging from 0.0005-10 mg/ml (as with bafilomycin-treated worms) and assayed for protection against α-synuclein- induced DA neurodegeneration (Supplemental Fig. 5B). Significant differences between treated or non-treated DA neurons were not observed at day 7 or 10, although there was a general trend of DA neurodegeneration with the highest concentrations of chloroquine. These results suggest that bafilomycin is specific in exerting a possible pre-conditioned effect in attenuating α-synuclein-induced cytotoxicity.
DISCUSSION
The function of bafilomycins was defined originally by its selective inhibition of V-ATPase (Bowman et al. 1988), which effectively increases the pH of acidic vesicles. Bafilomycin-mediated inhibition of V-ATPase results in the inhibition of lysosomal enzyme function and/or processing (Ishidoh and Kominami 2002); (Singh et al. 2006), induction of lysosome membrane permeabilization (Nakashima et al. 2003), inhibition of AV-lysosome fusion (Yoshimori et al. 1991) as well as potent inhibition of macroautophagy completion followed by induction of cell death (Shacka et al. 2006b). In contrast, results of the present study suggest that bafilomycins significantly attenuate neuronal cell death caused by agents that disrupt lysosome function and by over-expression of wild-type human α-synuclein, when used at concentrations that do not inhibit V-ATPase (Bowman et al. 1988) or affect vesicular pH (Shacka et al. 2006b). Importantly, the cytoprotective concentrations of bafilomycins used in our study (≤ 1 nM) do not disrupt the ALP when used alone, and actually attenuate markers of ALP dysfunction (decrease in CD maturation and AV accumulation; inhibition of autophagic flux; increase in detergent-insoluble α-synuclein) caused by the lysosomotropic agent chloroquine. Together, these findings delineate a potentially novel mechanism of action for bafilomycins as ALP preservation agents, and may serve to identify future therapeutics capable of delaying the onset and/or progression of neurodegenerative diseases including PD.
Bafilomycin A1 significantly and dose-dependently attenuated SH-SY5Y cell death induced by the lysosomotropic agents chloroquine, hydroxychloroquine or amodiaquine (Fig. 2; Supplemental Fig. 2). Amodiaquine exhibited a three-fold higher potency than chloroquine and hydroxychloroquine (Supplemental Fig. 1), a disparity documented previously in other cell lines (Basque et al. 2008) and may be due to structural differences in amodiaquine (phenolic substitution in its side chain) and/or its known uptake by an active transporter (Hayeshi et al. 2008). In addition, both bafilomycin A1 and bafilomycin B1 provided similar concentration-dependent protection against chloroquine-induced cell death (Fig. 2). Cytoprotective concentrations of bafilomycin A1 (≤ 1 nM) neither induced AV accumulation (Fig. 5) nor affected CD processing (Fig. 3). The effects of low-dose bafilomycin are similar to those found previously in cerebellar granule neurons, which indicated a lack of effect on pH-dependent lysotracker red fluorescence and AV accumulation (Shacka et al. 2006b).
Low-dose bafilomycin also significantly attenuated staurosporine-induced cell death (Supplemental Fig. 2), although its relative protection was modest in comparison to that of chloroquine. Staurosporine has been shown to induce lysosome dysfunction in different cell types (Bidere et al. 2003); (Kagedal et al. 2005), as indicated by lysosomal membrane permeabilization, an increase in cytosolic cathepsins and induction of apoptosis. Staurosporine’s effects on lysosome function are most likely indirect compared to chloroquine, and staurosporine has been shown previously to disrupt several different protein kinase signaling pathways (Ruegg and Burgess 1989). These reasons may explain why bafilomycin-mediated cytoprotection is less robust against staurosporine vs. chloroquine and other lysosomotropic agents. Our previous investigations indicated that low-dose bafilomycin did not attenuate staurosporine-induced death of cerebellar granule neurons (Shacka et al. 2006b). This discrepancy may be due to subtle cell-type-specific differences in the timing of and/or sensitivity to staurosporine-induced cell death.
The general caspase inhibitor BOC-Asp (OMe)-FMK neither attenuated chloroquine-induced cell death nor enhanced the attenuation of chloroquine-induced cell death by bafilomycin A1 (Fig. 2F), at a concentration that completely inhibited chloroquine-induced caspase-3-like activity. Thus bafilomycin A1 may attenuate chloroquine-induced cell death at a point either upstream and/or independent of caspase-3 activation. Our previous studies in cerebellar granule neurons also demonstrated that chloroquine-induced cell death did not require caspase-3 activation or expression, but was attenuated by the targeted deletion of bax, which regulates apoptosis upstream of caspase activation (Shacka et al. 2006b). However, Bax deficiency did not further attenuate chloroquine-induced cell death upon treatment with bafilomycin A1, suggesting that bafilomycin A1 may regulate both Bax-dependent and – independent cell death pathways resulting from lysosome dysfunction.
The nematode C. elegans is a useful model organism to study neurodegeneration in vivo caused by either chemical (Nass et al. 2002); (Cao et al. 2005) or genetic factors (Cooper et al. 2006); (Hamamichi et al. 2008). In addition, C. elegans contains only eight readily identified dopaminergic neurons, six in the anterior [two pairs of cephalic (CEP) and one pair of anterior deirid (ADE)] and two in the posterior body segments [one pair of posterior deirid (PDE)], making them a powerful model to study dopaminergic neuron degeneration in particular, allowing an unprecedented level of accuracy in quantifying effects of modifiers. Bafilomycin B1 significantly attenuated dopaminergic neuron death in C. elegans following over-expression of wild-type human α-synuclein (Fig. 7) where an inverted “U” shaped dose response curve was observed ten days after initial treatment. The maximal protective concentration of bafilomycin B1 in C. elegans was approximated at 100 μg/ml or 161 μM, a 160-fold higher concentration for optimal cytoprotection (1 nM) in cultured cells (Fig. 3); (Shacka et al. 2006b); (Shacka et al. 2006a). However, C. elegans has a protective cuticle layer that most likely compromised bafilomycin diffusion and penetration, an effect that is well characterized for other compounds (Holden-Dye and Walker 2007; Rand and Johnson 1995). In addition, the amount of active bafilomycin capable of affecting dopaminergic neurons in C. elegans may be further lowered upon metabolism within the worm, as has been demonstrated previously with other compounds (Rand and Johnson 1995), thus necessitating a higher effective concentration range than optimal for cultured cells. Over-expression of wild-type ATP13A2, a gene expressing a lysosomal ATPase and mutations in which are associated with a juvenile-onset hereditary parkinsonism (Klein and Lohmann-Hedrich 2007), attenuates neuron death induced by α-synuclein over-expression in C. elegans (Gitler et al. 2009), further implicating the importance of intact lysosome function in regulating α-synuclein-induced neurotoxicity. Thus, it is conceivable that bafilomycin B1 protected against over-expression of wild-type α-synuclein in C. elegans in part through its preservation of lysosomal function and promotion of α-synuclein clearance. Importantly, results in C. elegans suggest that bafilomycin attenuates dopaminergic neuron death following a stimulus (α-synuclein over-expression) that is, on one hand distinct from treatment with lysosomotropic agents in vitro yet may produce the same end result (disruption of the ALP).
Chloroquine treatment increased levels of high molecular weight, oligomeric forms of endogenous detergent-insoluble α-synuclein (Fig. 4), an effect that was significantly attenuated by low-dose bafilomycin A1. Aggregated α-synuclein is the most abundant protein composing Lewy bodies in PD, dementia with Lewy bodies, and a Lewy body variant of Alzheimer disease (Trojanowski and Lee 1998). Whether α-synuclein oligomerization and aggregation are cytotoxic or cytoprotective is controversial, and evidence for both has been suggested (Hasegawa et al. 2004); (Rochet et al. 2004); (Ruan et al. 2009); (Yang et al. 2009b); (Yu et al. 2009). However, it is generally accepted that enhanced α-synuclein clearance is cytoprotective (Yu et al. 2009). The ALP and the ubiquitin-proteasomal system (UPS) are both involved in α-synuclein clearance (Webb et al. 2003); (Lee et al. 2004); (Vogiatzi et al. 2008) and alterations in both are associated with α-synuclein aggregation in PD brain (Chu et al. 2009). While the relative importance of the ALP vs. the UPS on α-synuclein clearance under normal physiological conditions and during a pathological process such as PD is not completely understood, recent data suggest that ALP inhibition has a more profound role in accumulation of wild-type α-synuclein (Vogiatzi et al. 2008) and that these distinct protein degradation mechanisms can act in a compensatory manner (Pandey et al. 2007). It has also been shown that CD is the main lysosomal protease responsible for α-synuclein degradation (Sevlever et al. 2008) and that α-synuclein aggregation and toxicity are significantly impacted by relative expression levels of CD (Qiao et al. 2008). In SH-SY5Y cells, attenuation of chloroquine-induced cell death by bafilomycin A1 was associated with a partial restoration of mature CD and a decrease in endogenous, oligomeric detergent-insoluble α-synuclein. Together, our data suggest that the bafilomycin cytoprotection is mediated, at least in part, by preventing accumulation of potentially toxic oligomeric detergent-insoluble α-synuclein forms through restoration of the ALP.
Additional evidence supporting the ability of low-dose bafilomycin to “preserve” ALP function following chloroquine treatment is indicated by its ability to attenuate the chloroquine-induced decrease in the mature, “active” form of CD (Fig. 3), decrease the chloroquine-induced accumulation of AVs (Fig. 5) and attenuate the chloroquine-induced inhibition of autophagic flux (Fig. 6). CD is synthesized on ough ER and undergoes initial processing in the Golgi, in particular from the pre-pro to pro forms. However, the mature form of CD is generated from proteolytic cleavage in the low pH environment of lysosomes (Marquardt et al. 1987), thus suggesting the deleterious effects of chloroquine (and hence the protective effects of bafilomycins) on CD maturation are directly related to their respective effects on lysosome function. Chloroquine-induced inhibition of CD processing from pre-pro to pro forms (Fig. 5) may be due to a negative feedback mechanism aimed to prevent excessive CD synthesis upon inhibition of its maturation in lysosomes, a hypothesis that requires further investigation.
Our previous assessment of chloroquine-induced AV accumulation in cerebellar granule neurons (Shacka et al. 2006b) indicated little effect of bafilomycin, leading us to speculate that the cytoprotective effects of bafilomycin A1 were independent of AV accumulation. Our newest evidence however indicates that bafilomycin A1 attenuation of chloroquine-induced death correlates with a significant decrease in AV accumulation and preservation of autophagic flux in differentiated SH-SY5Y cells, differences that may be explained by cell type-specific effects of chloroquine and bafilomycin.
The molecular target for low-dose bafilomycin-mediated neuroprotection remains unresolved, since our cumulative findings predict that it is independent of V-ATPase inhibition (Shacka et al. 2006b). One potential target is hypoxia inducible factor (HIF)-1α, shown previously to compete with Von Hippel-Lindau tumor suppressor protein for binding to the c subunit of the V0 sector of V-ATPase (ATP6V0C) in a pH-independent manner (Lim et al. 2007). HIF-1α is well known to regulate trans-activation of several genes including heme oxygenase-1, an enzyme complex shown recently to regulate both the ALP (Zukor et al. 2009) and degradation of wild-type α-synuclein (Song et al. 2009). Thus bafilomycins may regulate neuronal survival through maintenance of the ALP that is in part independent of V-ATPase inhibition.
In summary, we have shown that low-dose bafilomycin protects neuronal cells against both chloroquine and wild-type α-synuclein-induced cell death in a manner consistent with a potential preservation of the ALP. The ability of bafilomycin to regulate the oligomerization of detergent-insoluble endogenous α-synuclein may be related in part to its ability to preserve CD maturation and decrease the accumulation of detergent insoluble AVs, which may ultimately correspond to an attenuation of neuronal cell death. Further study of plecomacrolide antibiotics such as bafilomycins A1 and B1 is warranted to better understand the role of the ALP in regulating neuron death in neurodegenerative diseases such as PD. Rigorous pharmacokinetics studies are also warranted to determine the relative ability of bafilomycins to cross the blood brain barrier following peripheral administration and reach therapeutic levels in the brain, and if structural analogs can be developed that effectively penetrate the CNS and exhibit a marked decrease in V-ATPase inhibitory activity.
Supplementary Material
Acknowledgments
We thank Sara L. Stone for expert technical assistance, Shawn Williams and the UAB High Resolution Imaging Facility for assistance with confocal imaging, and the UAB Neuroscience Core Facilities (NS47466 and NS57098) for technical assistance. We also thank the Caenorhabditis Genetics Center (CGC) for kindly providing the C. elegans mutant strains, as well as the C. elegans Gene Knockout Project at OMRF for generating mutant strains. Finally, we thank Rhonda Carr and Barry R. Bailey for assistance in manuscript preparation. This work is supported by grants from the National Institutes of Health (NS35107, NS41962, and CA134773) (Roth), the Howard Hughes Medical Institute and Michael J. Fox Foundation (Caldwell) and a pilot grant from the UAB Alzheimer’s Disease Research Center (Shacka). The co-authors wish to acknowledge no conflicts of interest with any aspect of this manuscript.
Abbreviations
- ALP
Autophagy-Lysosome Pathway
- AVs
autophagic vacuoles
- CD
cathepsin D
- tfLC3
tandem fluorescent-tagged LC3
- PD
Parkinson Disease
- V-ATPase
vacuolar-type ATPase
References
- Anglade P, Vyas S, Javoy-Agid F, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12(1):25–31. [PubMed] [Google Scholar]
- Basque J, Martel M, Leduc R, Cantin AM. Lysosomotropic drugs inhibit maturation of transforming growth factor-beta. Can J Physiol Pharmacol. 2008;86(9):606–612. doi: 10.1139/y08-063. [DOI] [PubMed] [Google Scholar]
- Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003 Aug 15;278(33):31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85(21):7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Gonzalez-Polo RA, Poncet D, Andreau K, Vieira HL, Roumier T, Perfettini JL, Kroemer G. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003 Jun 19;22(25):3927–3936. doi: 10.1038/sj.onc.1206622. [DOI] [PubMed] [Google Scholar]
- Broeks A, Janssen HW, Calafat J, Plasterk RH. A P-glycoprotein protects Caenorhabditis elegans against natural toxins. EMBO J. 1995 May 1;14(9):1858–1866. doi: 10.1002/j.1460-2075.1995.tb07178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci. 2005 Apr 13;25(15):3801–3812. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35(3):385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006 Jul 21;313(5785):324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen V, Lindfors M, Ng J, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2(1):5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8(11):917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA, Caldwell GA. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc Natl Acad Sci U S A. 2008 Jan 15;105(2):728–733. doi: 10.1073/pnas.0711018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa T, Matsuzaki M, Takeda A, Kikuchi A, Akita H, Perry G, Smith MA, Itoyama Y. Accelerated alpha-synuclein aggregation after differentiation of SH-SY5Y neuroblastoma cells. Brain Res. 2004 Jul 2;1013(1):51–59. doi: 10.1016/j.brainres.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Hayeshi R, Masimirembwa C, Mukanganyama S, Ungell AL. Lysosomal trapping of amodiaquine: impact on transport across intestinal epithelia models. Biopharm Drug Dispos. 2008;29(6):324–334. doi: 10.1002/bdd.616. [DOI] [PubMed] [Google Scholar]
- Holden-Dye L, Walker RJ. Anthelmintic drugs. WormBook. 2007:1–13. doi: 10.1895/wormbook.1.143.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishidoh K, Kominami E. Processing and activation of lysosomal proteinases. Biol Chem. 2002;383(12):1827–1831. doi: 10.1515/BC.2002.206. [DOI] [PubMed] [Google Scholar]
- Kagedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, Jurgensmeier JM, Ollinger K. Lysosomal membrane permeabilization during apoptosis--involvement of Bax? Int J Exp Pathol. 2005;86(5):309–321. doi: 10.1111/j.0959-9673.2005.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol. 2009;452:1–12. doi: 10.1016/S0076-6879(08)03601-X. [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Klein C, Lohmann-Hedrich K. Impact of recent genetic findings in Parkinson’s disease. Curr Opin Neurol. 2007;20(4):453–464. doi: 10.1097/WCO.0b013e3281e6692b. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004 Feb 25;24(8):1888–1896. doi: 10.1523/JNEUROSCI.3809-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- Lim JH, Park JW, Kim SJ, Kim MS, Park SK, Johnson RS, Chun YS. ATP6V0C competes with von Hippel-Lindau protein in hypoxia-inducible factor 1alpha (HIF-1alpha) binding and mediates HIF-1alpha expression by bafilomycin A1. Mol Pharmacol. 2007;71(3):942–948. doi: 10.1124/mol.106.030296. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Braulke T, Hasilik A, von Figura K. Association of the precursor of cathepsin D with coated membranes. Kinetics and carbohydrate processing. Eur J Biochem. 1987 Oct 1;168(1):37–42. doi: 10.1111/j.1432-1033.1987.tb13383.x. [DOI] [PubMed] [Google Scholar]
- Nakashima S, Hiraku Y, Tada-Oikawa S, Hishita T, Gabazza EC, Tamaki S, Imoto I, Adachi Y, Kawanishi S. Vacuolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in the human gastric cancer cell line MKN-1. J Biochem. 2003;134(3):359–364. doi: 10.1093/jb/mvg153. [DOI] [PubMed] [Google Scholar]
- Nass R, Blakely RD. The Caenorhabditis elegans dopaminergic system: opportunities for insights into dopamine transport and neurodegeneration. Annu Rev Pharmacol Toxicol. 2003;43:521–544. doi: 10.1146/annurev.pharmtox.43.100901.135934. [DOI] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, III, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002 Mar 5;99(5):3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowoslawski L, Klocke BJ, Roth KA. Molecular regulation of acute ethanol-induced neuron apoptosis. J Neuropathol Exp Neurol. 2005;64(6):490–497. doi: 10.1093/jnen/64.6.490. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131(Pt 8):1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007 Jun 14;447(7146):859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pivtoraiko VN, Stone SL, Roth KA, Shacka JJ. Oxidative stress and autophagy in the regulation of lysosome-dependent neuron death. Antioxid Redox Signal. 2009;11(3):481–496. doi: 10.1089/ars.2008.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Hamamichi S, Caldwell KA, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1(1):17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB, Johnson CD. Genetic pharmacology: interactions between drugs and gene products in Caenorhabditis elegans. Methods Cell Biol. 1995;48:187–204. doi: 10.1016/s0091-679x(08)61388-6. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23(1-2):23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Harrington AJ, Caldwell KA, Caldwell GA, Standaert DG. VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson’s disease. Neurobiol Dis. 2009 Oct 20; doi: 10.1016/j.nbd.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci. 1989;10(6):218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- Sevlever D, Jiang P, Yen SH. Cathepsin D Is the Main Lysosomal Enzyme Involved in the Degradation of alpha-Synuclein and Generation of Its Carboxy-Terminally Truncated Species. Biochemistry. 2008 Aug 15; doi: 10.1021/bi800699v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Klocke BJ, Roth KA. Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006a;2(3):228–230. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, Roth KA. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol. 2006b;69(4):1125–1136. doi: 10.1124/mol.105.018408. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- Singh CR, Moulton RA, Armitige LY, Bidani A, Snuggs M, Dhandayuthapani S, Hunter RL, Jagannath C. Processing and presentation of a mycobacterial antigen 85B epitope by murine macrophages is dependent on the phagosomal acquisition of vacuolar proton ATPase and in situ activation of cathepsin D. J Immunol. 2006 Sep 1;177(5):3250–3259. doi: 10.4049/jimmunol.177.5.3250. [DOI] [PubMed] [Google Scholar]
- Song W, Patel A, Qureshi HY, Han D, Schipper HM, Paudel HK. The Parkinson disease-associated A30P mutation stabilizes alpha-synuclein against proteasomal degradation triggered by heme oxygenase-1 over-expression in human neuroblastoma cells. J Neurochem. 2009;110(2):719–733. doi: 10.1111/j.1471-4159.2009.06165.x. [DOI] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009 Oct 28;29(43):13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol. 1998;55(2):151–152. doi: 10.1001/archneur.55.2.151. [DOI] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild Type {alpha}-Synuclein Is Degraded by Chaperone-mediated Autophagy and Macroautophagy in Neuronal Cells. J Biol Chem. 2008 Aug 29;283(35):23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003 Jul 4;278(27):25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Yang F, Yang YP, Mao CJ, Cao BY, Cai ZL, Shi JJ, Huang JZ, Zhang P, Liu CF. Role of autophagy and proteasome degradation pathways in apoptosis of PC12 cells overexpressing human alpha-synuclein. Neurosci Lett. 2009a May 1;454(3):203–208. doi: 10.1016/j.neulet.2009.03.027. [DOI] [PubMed] [Google Scholar]
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009b Jan 2;323(5910):124–127. doi: 10.1126/science.1166088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991 Sep 15;266(26):17707–17712. [PubMed] [Google Scholar]
- Yu WH, Dorado B, Figueroa HY, Wang L, Planel E, Cookson MR, Clark LN, Duff KE. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009;175(2):736–747. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi AU, McDonough JS, Klocke BJ, Latham CB, Korsmeyer SJ, Flavell RA, Schmidt RE, Roth KA. Chloroquine-induced neuronal cell death is p53 and Bcl-2 family-dependent but caspase-independent. J Neuropathol Exp Neurol. 2001;60(10):937–945. doi: 10.1093/jnen/60.10.937. [DOI] [PubMed] [Google Scholar]
- Zukor H, Song W, Liberman A, et al. HO-1-mediated macroautophagy: a mechanism for unregulated iron deposition in aging and degenerating neural tissues. J Neurochem. 2009;109(3):776–791. doi: 10.1111/j.1471-4159.2009.06007.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.