

Abstract
Background
Human immunodeficiency virus (HIV) infection continues to increase at alarming rates in drug abusers, especially in women. Drugs of abuse can cause long-lasting damage to the brain and HIV infection frequently leads to a dementing illness.To determine how these drugs interact with HIV to cause CNS damage, we used an in vitro human neuronal culture characterized for the presence of dopaminergic receptors, transporters and estrogen receptors. We determined the combined effects of dopaminergic drugs, methamphetamine, or cocaine with neurotoxic HIV proteins, gp120 and Tat.
Results
Acute exposure to these substances resulted in synergistic neurotoxic responses as measured by changes in mitochondrial membrane potential and neuronal cell death. Neurotoxicity occurred in a sub-population of neurons. Importantly, the presence of 17beta-estradiol prevented these synergistic neurotoxicities and the neuroprotective effects were partly mediated by estrogen receptors.
Conclusion
Our observations suggest that methamphetamine and cocaine may affect the course of HIV dementia, and additionally suggest that estrogens modify the HIV-drug interactions.
Introduction
HIV infection is currently the third leading cause of death in women ages 25-44 years [1] and manifestations of HIV infection show important gender-dependent differences. Women frequently develop menstrual abnormalities with amenorrhea, and manifestations of AIDS occur at higher CD4 counts and lower viral load as compared to men [2]. Plasma estradiol levels are also lower in HIV-infected women [3]. It remains unclear if dementia associated with HIV infection occurs more frequently in women than men. A large multicenter European study showed that women were twice as likely to develop HIV dementia compared to men [4]; however, another study failed to confirm these differences [5].
Drug abuse accounts for nearly half of the HIV infections in women in United States[1], however, the effect of drug abuse on incidence, rate of progression or severity of HIV dementia is not entirely clear. Although no major differences were noted in cognitive functioning amongst HIV-infected asymptomatics with or without a history of drug abuse [6], a subsequent study showed that a history of injection drug use and presentation with prominent psychomotor slowing was associated with more rapid neurologic progression [7]. Other neuropathological studies show marked sever ity of HIV encephalitis in drug abusers [8] particularly involving loss of dopaminergic neurons [9]. Long term methamphetamine use has also been associated with neuronal damage as determined by magnetic resonance spectroscopy brain imaging studies [10]. Autopsy studies also confirm injury to dopaminergic neurons in cocaine as well or methamphetamine abusers [11, 12]. Interestingly, some investigators have proposed the use of psychostimulants in the treatment of HIV dementia [13], however the effects of these drugs on cerebral function in the setting of HIV infection has not been well studied.
HIV proteins gp120 and Tat have been implicated in the neuropathogenesis of HIV dementia. Both proteins are released from HIV infected cells and are present in the brains of HIV infected patients with dementia or encephalitis [14]. Recent studies from our laboratory have shown that these proteins cause synergistic neurotoxicity that involves excitatory amino acid receptors and oxidative pathways [15].
Estrogen deficiency has been implicated as a risk factor in the development of several neurodegenerative diseases [16,17,18] and estrogen replacement may result in improvement of cognitive function [19]. The mechanisms by which estrogens protect neurons is currently under intense investigation and may involve, receptor-mediated mechanisms or non-receptor-mediated antioxidative effects. For these reasons, we assessed the combined effects of HIV proteins and drugs of abuse, methamphetamine and cocaine on neuronal function and determined to what extent estrogen may protect against these neurotoxic substances.
Results
Detection of estrogen receptors and dopaminergic neurons in human fetal brain cells
Estrogen receptors were localized in the neuronal cultures by immunostaining and by mRNA analysis by RT-PCR. We found that 5-10% cells immunostained for estrogen receptors. Estrogen receptors could be localized in both neurons and astrocytes (Figures 1A, B and 2D-F). The immunostaining was noted in the cytoplasm and nucleus of these cells. mRNA for estrogen receptor-α but not estrogen receptor-β could be detected in these cultures (Figure 3). Dopaminergic neurons were detected by immunostaining for dopamine and dopamine transporter which could be co-localized in nearly 60% neurons (Table 1, Figures 2A-C) as well as dopamine receptors: D1A (50% cells) and D2 (40% cells) (Figures 2E, and 2F). Estrogen receptor colocalized with cells staining for dopamine (Figure 1A) as well as D1A and D2 receptor containing neurons (Figures 2E and 2F).
Figure 1.
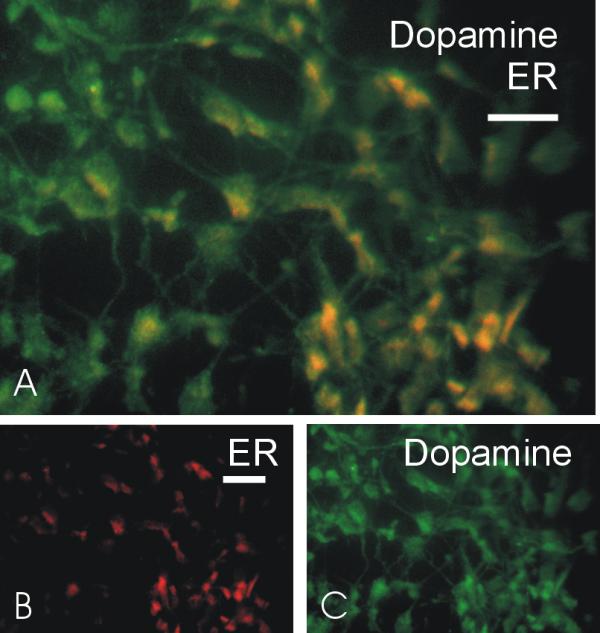
Immuonolocalization of estrogen receptor and dopamine: Mixed neuronal cultures were immunostained as described in methods. (A) co-localization of dopamine (green) and estrogen receptor(ER) (red) in neurons. Dual staining cells appear yellow. (B) Immunostaining for dopamine. (C) immunostaining for ER. Scale bar in A = 30 μm and the scale bar in B and C = 50 μm.
Figure 2.

Immuonolocalization of dopamine and estrogen receptors: showing immune reactivity in neuronal cultures (95% neurons and 5%astrocytes) for A - dopamine B - dopamine transporter (DAT) C - dopamine and DAT D - estrogen receptor (ER) E - Dopamine D1A receptor with ER F - Dopamine D2 receptor with ER Scale bars in A&B = 25 μm, C&D = 15 μm; E&F = 25 μm.
Figure 3.

Estrogen receptor mRNA in neuronal cultures. Estrogen receptor-α mRNA (ER-α) was detected by RT-PCR and Southern blotting. Lane 1: lymphoid cells; Lane 2: human neuronal cultures; Lane 3: monocytic cells. ER-α is present only in human neuronal cultures.
Table 1.
| Antigen | Primary | Dilution | Antibody | Secondary | Specificity & |
| antibodies/ | Source | antibodies/antisera | Subcellular distribution | ||
| antisera | (dilutions) | ||||
| Estrogen | Mouse | 1:10 | Chemicon | Goat anti-mouse IgG | Neurons and astrocytes / |
| receptor | IgG1 | (MAB428) | (H+L)conjugated to Cy3 | Punctate nuclear and | |
| (ER) | monoclonal | (1:250) | perinuclear distribution | ||
| Dopamine | Rabbit | 1:1000 | Chemicon | • Goat anti-rabbit IgG | Neuronal processes and cell |
| (DA) | IgG | (AB122S) | DTAF (1:500) or | body | |
| polyclonal | • Goat anti-rabbit IgG | Cytoplasmic distribution | |||
| Cy3 (1:250) | |||||
| Dopamine | Rat | 1:100 | Chemicon | Goat anti-rat IgG | Neuronal processes and cell |
| transporter | Ig type | (MAB369) | conjugated to biotin- | body; astrocytes | |
| (DAT) | monoclonal | avidin-Cy2 (1:250) | Cell membrane | ||
| Dopamine | Rabbit | 1:100 | Chemicon | Goat anti-rabbit IgG | Neurons and astrocytes; |
| D1A | polyclonal | (AB1784P) | DTAF (1:500) | punctate distribution | |
| receptor | |||||
| Dopamine | Rabbit | 1:500 | Chemicon | Goat anti rabbit IgG | Neuronal processes and cell |
| D2 | polyclonal | (AB1558) | DTAF (1:500) | bodies: perinuclear | |
| receptor | distribution, astrocytes, | ||||
| microglia | |||||
| Co-localization | |||||
| Combined | • Goat anti-rabbit IgG | Colocalized in neurons | |||
| • DA + | as above | as above | as above | DTAF + | and/or astrocytes |
| • ER | • Goat anti-mouse | ||||
| Cy3 | |||||
| Combined | • Goat anti-rabbit IgG | Colocalized in | |||
| • DA + | as above | as above | as above | Cy3 | subpopulations of neurons |
| • DAT | • Goat anti-rat IgG | and/or astrocytes | |||
| biotin-avidin Cy2 | |||||
| Combined | as above | as above | as above | • Goat anti-rabbit IgG | Colocalized in |
| • D1A + | DTAF (1:500) | subpopulations of neurons | |||
| • ER | • Goat anti-mouse | and/or astrocytes | |||
| Cy3 (1:250) | |||||
| Combined | as above | as above | as above | • Goat anti-rabbit IgG | Colocalized in |
| • D2 + | DTAF (1:500) | subpopulations of neurons | |||
| • ER | • Goat anti-mouse | and/or astrocytes and | |||
| Cy3 (1:250) | microglia | ||||
Blocking of neurotoxicity of supernatants from HIV infected monocytes with antisera to gp120 and Tat
Cell culture supernatants from HIV infected monocytic cells were collected periodically and analyzed for their neurotoxic potential. Maximum neurotoxicity was noted at two weeks post infection (Figure 4A). Complete obliteration of neurotoxicity was noted when the supernatants were immunoabsorbed against antisera to gp120 and Tat (Figure 4B). However, when immunoabsorbed against each of the antisera alone, the effect on neurotoxicity of the culture supernatants was not significant (not shown).
Figure 4.

Neurotoxic factors from HIV infected cells: Monocytic cells (THP-1) were infected with HIV-AdaM and culture supernatants were periodically collected, diluted 1:1 in Locke's buffer and analyzed for neurotoxicity on mixed human neuronal cell cultures. (A) Maximal neurotoxicity was noted in supernatants 2 weeks post infection (*P < 0.05). (B) Culture supernatants from days 13 and 15 when immunoabsorbed with antisera to gp120 and Tat show nearly complete blocking of neurotoxicity. Uninfected culture supernatants from corresponding days in both sets of experiments showed <2% neurotoxicity. Each data point represents mean ± SEM from three different cultures.
Neurotoxicity of HIV proteins with methamphetamine or cocaine
To determine the synergistic neurotoxic properties of the viral proteins and drugs of abuse, we initially conducted time and dose response curves for each compound individually and then in combination of one another. We determined that dosages for causing neuronal cell death or changes in mitochondrial potential paralleled one another, however changes in mitochondrial potential could be seen as early as 4-8 hours (Figure 5), significant cell death occurred at 16 hrs post treatment. No significant toxicity was noted with Tat <80 nM, gp120 <40 pM, or Tat (40 nM) + gp120 (30 pM) (Figure 5).
Figure 5.

Synergistic neurotoxicity of HIV proteins, cocaine and methamphetamine. Human fetal neuronal cultures were treated with HIV proteins, gp120 and Tat alone or in combination with varying concentrations of either methamphetamine or cocaine. Cell death and mitochondrial membrane potentials were monitored, at 16 hours and 6 hours respectively, in these cultures as described in methods. Subtoxic concentrations of gp120 and Tat synergize with methamphetamine (A) and cocaine (B) in a dose responsive manner to cause neuronal cell death. (C) Tat (80 nM) when combined with subtoxic concentrations of gp120, methamphetamine or cocaine causes significant decreases in mitochondrial membrane potential. Synergism is also noted when the combined subtoxic concentrations of gp120 (30 pM) + Tat (40 nM) are co-incubated with either methamphetamine or cocaine. *P < 0.05, **P < 0.005
To determine the specificity of Tat action, we used a two Tat deletion mutants (TatΔ31-61 and TatΔ48-56). No changes in neuronal cell death were noted at maximal concentrations of TatΔ31-61 tested (500 nM) and partial obliteration of neurotoxicity was noted with TatΔ48-56 (Figure 6). These observations are consistent with our previous experience with synthetic Tat peptides demonstrating that Tat31-61 contains the neurotoxic epitope [20]. To determine the specificity of gp120 action we used a non-specific glycoprotein α2-HS glycoprotein. No neurotoxicity was detected at the highest concentrations tested (1 μM).
Figure 6.

Specificity of HIV protein neurotoxicity. Tat1-72 induced neurotoxicity was compared with two deletion mutants TatΔ48-56 and TatΔ31-61. While TatΔ48-56 showed partial attenuation of neurotoxicity, TatΔ31-61 showed complete obliteration. Similarly no neurotoxicity was noted when gp120 was substituted with alpha-2 HS-glycoprotein (a2-HS-gp).
Similarly, no significant neurotoxicity was noted with methamphetamine <500 μM (Figure 5A). In addition, all concentrations of cocaine tested were non-toxic by these assays. The maximal concentration tested was 16 uM. 40 nM Tat + 30 pM gp120 with cocaine (1.6 or 16 μM) or methamphetamine (250 or 500 μM) produced significant (P < 0.05) neuronal cell death (Figure 5A and 5B) and decreases in mitochondrial potential in each case (Figure 5C). When 80 nM Tat was combined with either 30 pM gp120, 500 μM methamphetamine or 1.6 μM cocaine, significant decreases in mitochondrial potential (P < 0.05) were observed demonstrating a synergistic neurotoxic response between the viral proteins and drugs of abuse. The synergism for both methamphetamine and cocaine showed a steep dose response suggesting an "all or none" phenomenon (Figure 5).
Protection against HIV protein-induced mitochondrial toxicity by 17β-estradiol
We determined the ability of 17β-estradiol to protect against the synergistic neurotoxicity induced by the viral proteins alone or in combination with cocaine or methamphetamine. Nearly complete attenuation of neuronal cell death induced by Tat+gp120 was noted in a concentration dependent manner (Figure 7). 1 nM 17β-estradiol could also protect against the combined synergistic neurotoxicity of the viral proteins (Tat 40 nM+gp120 30 pM) with cocaine (1.6 μM) or methamphetamine (500 μM) (Figure 8). 1 nM 17β-estradiol protected against both, changes in mitochondrial membrane potential (Figure 8A and 8B) and neuronal cell death (Figure 8C). However, this toxicity of higher concentration cocaine (16 μM) could be only partially reversed by 1 nM 17β-estradiol (Figure 8B). A lower concentration of 17β-estradiol (0.1 nM) could also completely reverse the changes in mitochondrial membrane potential (Figure 8A and 8B) but not cell death (Figure 7). To determine the mechanism of estrogen action, we pretreated the cells with either ICI 182,780 (1 μM or 10 μM) or tamoxifen (100 pM, 1 nM). We tried several different combinations and concentrations of viral proteins and methamphetamine or cocaine. Although protection with 17β-estradiol was consistently present, in each case the estrogen antagonists failed to reverse the protective effects of 17β-estradiol on mitochondrial membrane potential (Figure 8A and 8B). Interestingly, however, ICI 182,780, a prototypic estrogen antagonist, blocked the protective effect of 17β-estradiol on neuronal cell death (P < 0.05) while tamoxifen had only a partial effect that was not significant (Figure 8D). Both ICI 182,780 and tamoxifen alone did not produce neurotoxicity at the above concentrations (Figure 8D).
Figure 7.

Neuroprotection against HIV proteins with 17β-estradiol. 17β-estradiol protects human fetal neurons against neuronal cell death induced by Tat+gp120 in a concentration dependent responsive manner. (*P < 0.05; **P < 0.005).
Figure 8.

Neuroprotection with 17β-estradiol against synergistic neurotoxic responses of HIV proteins, methamphetamine and cocaine. Neuroprotective properties of 17β-estradiol were assessed using a mitochondrial toxicity assay (A and B) and a neuronal cell death assay (C and D). 17β-estradiol at concentrations of >100 pM prevented mitochondrial membrane potential changes caused by HIV proteins combined with (A) methamphetamine or (B) cocaine. In each case estrogen antagonists tamoxifen and ICI 182,780 failed to reverse the effects of 17β-estradiol. 17β-estradiol had no neuroprotective effect (A). (C) Neuronal cell death was prevented when concentrations of 17β-estradiol were ≥ 1 nM. (D) The neuroprotective effects of 17β-estradiol could be partially reversed by ICI 182,780 and tamoxifen. (*P < 0.05)
Discussion
Although it is clear that patients with HIV infection may develop dementia and non-HIV infected drug abusers develop evidence for neuronal dysfunction, it is not clearly established if patients exposed to both HIV and drugs of abuse are at increased risk for neurological complications. In this study, we show that HIV proteins may synergize with drugs such as methamphetamine and cocaine to cause neurotoxicity. These findings are supported by recent observations suggesting that HIV infected drug abusers have a more severe encephalitis at autopsy [7,8,9] and at least in some HIV infected drug abusers an accelerated form of HIV dementia may be observed [21]. One possible cause of this acceleration may be the synergistic neurotoxic properties of HIV proteins and the drugs of abuse. Our observations also suggest that an acute exposure may be sufficient to cause neurotoxicity. The mechanism by which these substances synergize remains elusive. As demonstrated in these studies, at least in part the mechanism involves mitochondrial dysfunction. Since both gp120 and Tat have been shown to cause toxicity to dopaminergic cells and similarly methamphetamine and cocaine also cause toxicity to dopaminergic systems, we characterized the dopaminergic cell types and receptors in our culture system. We found that only a small proportion of these cells underwent cell death when exposed to the proteins and drugs of abuse suggesting that there must be another susceptibility factor that characterizes these cells which needs to be identified.
We demonstrate that HIV proteins gp120 and Tat are important components in supernatants of HIV infected cells that cause neurotoxicity. This does not exclude the role of other substances such as cytokines and other soluble cellular products implicated in neuropathogenesis of HIV infection. In fact previous studies have shown that most of these substances can be induced by HIV proteins in uninfected monocytes and astrocytes [14]. Although further studies are necessary to determine the relative amounts and kinetics of HIV proteins released from different HIV infected cells, previous studies have shown that both gp120 and Tat are present in the brains of patients with HIV dementia and encephalitis [15, 22]. Gp120 and Tat may also synergize to cause neurotoxicity [15].
Recently, much attention has been given to the neuroprotective properties of estrogens for both chronic neurodegenerative diseases and acute insults such as stroke. In vitro studies also show that estrogens can protect against a number of neurotoxic compounds [23]. The mechanisms by which estrogens protect cells can be broadly classified into two categories, receptor mediated and non-receptor-mediated effects. Estrogen receptors are widely expressed in the brain with some regional differences [24]. We initially demonstrated the presence of estrogen receptors in the neuronal cultures by immunocytochemistry and determined their subtypes by mRNA analysis. We found that estrogen receptors are localized both in neurons and astrocytes. We also found that some dopaminergic neurons co-expressed estrogen receptors.
We next determined that 17β-estradiol at concentrations that can either be achieved physiologically or easily obtained pharmacologically protected against the combined insult of HIV proteins and drug abuse. Protection was noted against both cell death and mitochondrial toxicity. The protection was specific since no protection was noted with 17α-estradiol. In subsequent experiments we determined if this protection was mediated via estrogen receptors. Estrogen receptor antagonist, ICI 182,780 completely, and tamoxifen partially reversed the neuroprotective effects of 17β-estradiol using cell death as an end point. However these compounds were unable to reverse the neuroprotective effects of estrogen on mitochondrial potential, suggesting that the mitochondrial effects of estrogen are non-receptor mediated. Thus the toxic effects on mitochondria and neuronal cell survival seems to be independently regulated. However, it seems that 17β-estradiol can protect against both these effects by receptor and non-receptor mediated mechanisms. These observations are consistent with recent reports showing that 17β-estradiol interacts directly with a subunit of mitochondrial F0F1-ATP synthase/ATPase required for the coupling of a proton gradient across the F0 sector of the enzyme in the mitochondrial membrane to ATP synthesis in the F1 sector of the enzyme [25]. This accounts for the antioxidant properties of 17β-estradiol. Due to its multiple sites of action, its wide spectrum of protection against multiple neurotoxic substances at easily obtainable concentrations and the ease of administration of estrogen, this compound is a very attractive candidate for consideration for therapeutic intervention in HIV-infected drug abusing women. Clinical trials are now necessary to confirm these observations. Caution may be necessary in the use of estrogens in this population, because estrogens have been associated with a small but real risk of ischemic heart disease and cerebral infarcts [26]. Some patients with HIV infection may have a hypercoagulable state and may be at increased risk of developing cerebral infarcts [27].
We conclude that methamphetamine and cocaine may synergize with HIV proteins to cause neurotoxicity and resulting in increased dysfunction in HIV infected drug users. Caution should also be used in the use of other structurally and functionally similar psychotropic drugs in HIV infected patients. Estrogen may be neuroprotective in this population acting through receptor and non-receptor mediated mechanisms.
Methods
Cultures of human brain cells
Brain specimens were obtained from human fetuses of 12-14 weeks gestational age with consent from women undergoing elective termination of pregnancy and approval by the University of Kentucky Institutional Review Board. Neuronal cultures were prepared as described previously [20, 28]. Briefly, the cells were mechanically dissociated, suspended in Opti-MEM with 5% heat-inactivated fetal bovine serum, 0.2% N2 supplement (GIBCO) and 1% antibiotic solution (penicillin G 104 units/ml, streptomycin 10 mg/ml and amphotericin B 25 μg/ml) and plated in flat bottom 96 well plates. The cells were maintained in culture for at least one month before conducting the neurotoxicity and mitochondrial experiments.
Immunostaining
Cells were plated and cultured on coverslips for immunostaining. The media was removed and neuronal cultures were fixed for 30 minutes with Zamboni's fixative [29] at room temperature. Receptor immunoreactivity was determined using monoclonal or polyclonal antisera as listed in Table 1. Cells were incubated in blocking buffer consisting of 4% casein (w/v) and 0.1% bovine serum albumin (w/v) in phosphate buffered saline (PBS) at pH 7.4 for 30 min. Cells were then incubated in primary antibody diluted in blocking buffer for at 24 h at 4°C. Following three washes in PBS the cells were incubated with fluorochrome conjugated secondary antisera diluted in blocking buffer (Table 1) for one hour at room temperature. To determine specificity of staining, some cells were incubated in blocking buffer without primary antibody. In addition, the specificity of particular antibodies was further confirmed by demonstrating appropriate patterns of subcellular immunoreactivity in positive controls, as well as the absence of immunoreactivity in negative controls. After the final wash, the coverslips containing cells were air dried in the dark and mounted on slides with ProLong™ Antifade mounting media (Sakura Finitek USA, Torrence, CA). They were maintained in the dark and at 4°C until examined.
Combined detection of estrogen receptors and dopaminergic systems
Neuronal cultures plated on coverslips were initially treated as described above to detect estrogen receptors. Cell were fixed in Zamboni's solution, rinsed 3-fold in PBS, and incubated with mouse anti-ERα or ERβ monoclonal antibodies (1:10 dilution) for at least 1 h at room temperature. Anti mouse IgG conjugated to Cy3 fluorochrome (1:250 dilution, 1 h at room temperature) was used to visualize ER antigenicity (Jackson ImmunoResearch, West Grove, PA). To detect dopamine (DA), dopamine receptors D1A and D2, cells were incubated in rabbit polyclonal antibodies (see table) overnight at 4°C. For detection of dopamine transporter (DAT) rat monoclonal antisera were used. Cells were then incubated in goat anti rabbit IgG (1:250 dilution) conjugated to DTAF for 1 h at room temperature. Cells were washes three times with PBS. After the final wash, the coverslips containing cells were air dried in the dark and coverslipped with ProLong™ Antifade mounting media (Sakura Finitek USA, Torrence, CA). They were maintained in the dark and at 4°C until examined.
The reaction was visualized using fluorescence microscopy (Nikon Diaphot) and images acquired using a Spot 2 digital camera (Diagnostic Instruments, Sterling Heights, MI). Digital images incorporating multiple fluorochromes (red and green) were overlaid in Photoshop 5.0 (Adobe).
RT-PCR for estrogen receptors
For each sample, we reverse transcribed 0.5 μg of total RNA to produce cDNA in a final reaction volume of 40 μl, containing 2.5 μM random hexamers (Perkin Elmer, Branchburg, NJ), 100 U MuLV Reverse Transcriptase (Perkin Elmer), 1 mM dNTP mix (Life Technologies, Gaithersburg, MD), 80 U RNAsin (Promega, Madison, WI), 5 mM MgCl2 (Life Technologies), and 1X reaction buffer (Life Technologies). Each sample was incubated for reverse transcription at room temp. for 15 min, 37°C for 2 min, 42°C for 1 hr, and 99°C for 5 min. The same procedure was performed on samples utilizing a reaction solution without reverse transcriptase to check for genomic contamination.
PCR amplification
We utilized well characterized RT-PCR methods to determine relative changes in gene expression at the mRNA level. For each gene examined, we generated standard curves of input RNA and cycle number to determine the optimum cycle number within the linear range for PCR amplification (data not shown); for all genes examined, this was determined to be between 25-30 cycles. These methods have been validated in studies showing that, within the optimal range of amplification, yields of PCR product are linear with respect to input RNA [30].
For each gene, stock solutions were prepared containing 1.5 mM MgCl2, 1X reaction buffer, 10 μCi of 32P- dCTP (3000 Ci/mmol) (NEN, Boston, MA), 1 μM each primer, and 1.5 U of Taq polymerase (Life Technologies). For ERα and ERβ PCR, 1.5 U of Taq Ab (Life Technologies) was included in each reaction. The stock solution was aliquoted (49 μl/tube), and 1/30 of synthesized cDNA (from reverse transcription reaction) was added to each sample tube. Samples were then thermocycled for PCR amplification (Hybaid, Touchdown Thermocycler, Middlesex, United Kingdom) according to the following reaction conditions: 1 min 95°C, 1 min 55°C, 2 min 72°C (25-30 cycles). After amplification, PCR products were resolved by polyacrylamide gel electrophoresis. The gels were dried and the products were visualized and quantified using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
All the oligonucleotide sequence pairs used for gene amplification in this study generated PCR products of expected sizes that have been sequenced to verify their identities: L27A sense primer: 5'-ATGCTAACTGTCCAAGTCTA-3' and antisense primer 5'-GGGAGCAACTCCATTCTTGT-3' (214 bp) [31]; ERα sense primer 5'-AATTCTGACAATCGACGCCAG-3' and antisense primer 5'-GTGCTTCAACATTCTCCCTCCTC-3'(344 bp) [32].
Infection of monocytic cells with HIV
THP-1 cells were cultured in RPMI+10% fetal bovine serum and infected with HIV-AdaM strain at an MOI of 100. The high MOI was chosen to ensure maximal production of viral proteins. Culture supernatants were collected every four days by half media exchange and frozen at -80°C till used for neurotoxicity assays. For immunoabsorption of viral proteins, the culture supernatants were incubated for 60 min with protein A beads conjugated with polyclonal antisera to either Tat or gp120 with constant shaking at room temperature. Following centrifugation, the supernatants were removed and immediately used for neurotoxicity assays.
Recombinant Tat and gp120 proteins
Recombinant Tat was prepared as described previously [33] with minor modifications. The tat gene encoding the first 72 amino acids were amplified from HIVBRU obtained from Dr. Richard Gaynor through the AIDS repository at the NIH and inserted into an E. coli vector PinPoint Xa-2 (Promega). Two deletion mutants from this plasmid were also prepared. One by deleting the sequence encoding amino acids 31-61 of Tat previously shown to contain the neurotoxic epitope [20]. Another one was prepared by deleting the sequences 48-56 encoding the basic region of Tat. These constructs allowed the expression of the Tat proteins as a fusion protein naturally biotinylated at the N-terminus. The biotinylated Tat proteins were purified on a column of soft release avidin resin, cleaved from the fusion protein using factor Xa and eluted from the column followed by desalting with a PD10 column. All purification steps contained dithiothreitol to prevent oxidation of the proteins. Tat proteins were >95% pure as determined by SDS-PAGE followed by silver staining, and analysis by HPLC using a C4 column showed a single symmetrical peak. Western blot analysis showed that these preparations contained both monomeric and dimeric forms of Tat1-72 but monomeric form only for TatΔ31-61 and TatΔ48-56. The functional activity of Tat1-72 was confirmed using a transactivation assay in HL3T1 cells containing an HIV-1 LTR-CAT construct [33]. gp120 from HIVSF2 was obtained as a gift from Chiron Corporation. Recombinant gp120 was made in a chinese hamster ovary cell line and purification yielded a product that was 95% gp120 with the remainder being break down products of gp120 as determined by western blot analysis. The Tat and gp120 preparations contained <1 pg/ml endotoxin as determined using a Pyrochrome Chromogenic test kit (Associates of Cape Cod Inc., Falmouth, MA.). The Tat proteins were stored in a lyophilized form and gp120 as a stock solution in water at -80°C in endotoxin-free siliconized microfuge tubes until taken for experimentation. Tat and gp120 were highly susceptible to degradation and loss of biological activity with each freeze-thaw cycle. Therefore, single aliquots were used for each experiment with the remaining solutions discarded.
Neurotoxicity Assay
At the time of experimental treatment, the culture media was replaced with Locke's buffer containing (in mM) 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 glucose, 5 N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (pH 7.2) and neurons were incubated with culture supernatants from HIV infected monocytic cells, subtoxic concentrations of Tat (40 nM) + gp120 (30 pM), various concentrations of methamphetamine or cocaine, or viral proteins + methamphetamine and cocaine. To determine the specificity of action of the viral proteins, Tat1-72 was compared to TatΔ31-61 and TatΔ48-56 at 500 nM concentration each. Similarly, gp120 (500 pM) was compared to a non-specific glycoprotein α2-HS glycoprotein (Sigma; 1 uM). Cell death in each case was monitored by trypan blue exclusion 15 hours later as described previously [20, 28]. Neuronal cell counts were determined from five fields at predetermined coordinate locations. Each field was photographed, coded and counted. At least 200 cells were counted in each field. Each experiment was conducted in triplicate wells and at least three independent experiments were conducted with each pharmacological agent. The means and standard errors of the mean were calculated and data were analyzed by ANOVA with Tukey-Kramer post-hoc comparisons. To investigate the neuroprotective properties of estrogen, the cells were incubated with 17β-estradiol (Sigma) at various concentrations followed by exposure to the viral proteins and/or the drugs of abuse in Locke's buffer and cell death monitored as described above. Preincubation with 17β-estradiol in each case was <5 min. To determine the mechanism of estrogen protection, the cultures were incubated with either ICI 182,780 (100 nM) (Tocris) or tamoxifen (10 nM) (Sigma).
Measurement of mitochondrial membrane potential activity
To determine the effect of the viral proteins on mitochondrial function, we used a fluorescent dye JC-1 to measure changes in mitochondrial membrane potential. Once loaded into the mitochondria, JC-1 undergoes aggregate formation in the regions of high potential. The resulting spectral shift of the dye can be used to detect changes in mitochondrial activity. The green fluorescent JC-1 (5,5', 6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolylcabocyanine iodine, T-3168) exist as a monomer at low membrane potential. However, at higher potentials, JC-1 forms red fluorescent "J-aggregates" that exhibit broad spectrum and an emission maximum at 590 nm. Optical filters designed for fluorescein and tetramethylrhodamine were used to separately visualize the monomer and J-aggregate forms, respectively. Mitochondrial function was monitored following treatment with the above compounds at different concentrations and time points. Briefly, after experiment treatment cells were incubated for 30 minutes at 37°C in a 5% CO2 incubator in the presence of 10 μM of the JC-1 and then washed in Locke's solution.
Optical measurements were acquired with excitation at 485 nm and emission at 527 nm, and 590 nm. The levels of fluorescence at both emission wavelengths were quantified and ratio of measurements was assessed. The pertinent data are given as mean ± S.E.M. for mean optical measurements. The values are expressed as percent of the mean control values ± S.E.M. and analyzed using ANOVA.
Acknowledgments
Acknowledgements
This manuscript was supported by NIH grants awarded to AN, KH, PW, RB, and WM. We also thank Melinda Wilson for assistance.
Contributor Information
Jadwiga Turchan, Email: jturc2@pop.uky.edu.
Caroline Anderson, Email: cfmart@pop.uky.edu.
Kurt F Hauser, Email: khauser@pop.uky.edu.
Qinmiao Sun, Email: qsun2@pop.uky.edu.
Jiayou Zhang, Email: jzhan1@pop.uky.edu.
Ying Liu, Email: yliu6@pop.uky.edu.
Phyllis M Wise, Email: pmwise1@pop.uky.edu.
Inna Kruman, Email: krumanln@grc.nia.nih.gov.
William Maragos, Email: maragos@pop.uky.edu.
Mark P Mattson, Email: mattsonm@grc.nia.nih.gov.
Rosemarie Booze, Email: booz@pop.uky.edu.
Avindra Nath, Email: anath@pop.uky.edu.
References
- CDC (1996) Center for Disease Control and Prevention: Update: Mortality attributable to HIV infection among persons aged 25-44 years - United States 1994. MMWR. 1996;45:121–125. [PubMed] [Google Scholar]
- Farzadegan H, Hoover DR, Astemborski J, Lyles CM, Margolick JB, Markham RB, Quinn TC, Vlahov D. Sex differences in HIV-1 viral load and progression to AIDS. Lancet. 1998;352:1510–4. doi: 10.1016/S0140-6736(98)02372-1. [DOI] [PubMed] [Google Scholar]
- Grinspoon S, Corcoran C, Miller K, Biller BM, Askari H, Wang E, Hubbard J, Anderson EJ, Basgoz N, Heller HM, Klibanski A. Body composition and endocrine function in women with acquired immunodeficiency syndrome wasting [published erratum appears in J Clin Endocrinol Metab 1997 Oct;82(10):3360]. J Clin Endocrinol Metab. 1997;82:1332–7. doi: 10.1210/jcem.82.5.3907. [DOI] [PubMed] [Google Scholar]
- Chiesi A, Vella S, Dally LG, Pedersen C, Danner S, Johnson AM, Schwander S, Goebel FD, Glauser M, Antunes F, et al. Epidemiology of AIDS dementia complex in Europe. AIDS in Europe Study Group. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;11:39–44. doi: 10.1097/00042560-199601010-00005. [DOI] [PubMed] [Google Scholar]
- Robertson K, Vaughn B, Kapoor C, Robertson W, Kalkowski J, Fiscus S, Helms R, Hall C. Gender and Neurological Functioning in HIV Infection. Conference on Human Retroviruses and Related Infections. 2000;Abstract #297 [Google Scholar]
- Concha M, Selnes OA, Vlahov D, Nance-Sproson T, Updike M, Royal W, Palenicek J, McArthur JC. Comparison of neuropsychological performance between AIDS-free injecting drug users and homosexual men. Neuroepidemiology. 1997;16:78–85. doi: 10.1159/000109674. [DOI] [PubMed] [Google Scholar]
- Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC. Variable progression of HIV-associated dementia. Neurology. 1998;50:1814–20. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain. 1998;121:2043–52. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R. Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathol. 1991;82:39–44. doi: 10.1007/BF00310921. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chang L, Leonido-Yee M, Speck O. Evidence for long-term neurotoxicity associated with methamphetamine abuse: A 1H MRS study. Neurology. 2000;54:1344–1349. doi: 10.1212/wnl.54.6.1344. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Bergeron C, Kalasinsky K, Ang L, Peretti F, Adams VI, Smialek J, Anderson W.R, Shannak K, Deck J, Niznik HB, Kish SJ. Striatal dopamine, dopamine transporter, and vesicular monoamine transporter in chronic cocaine users. Ann Neurol. 1996;40:428–39. doi: 10.1002/ana.410400312. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Brown GR. The use of methylphenidate for cognitive decline associated with HIV disease. Int J Psychiatry Med. 1995;25:21–37. doi: 10.2190/72U2-FQ94-RVK5-6J5C. [DOI] [PubMed] [Google Scholar]
- Nath A. Pathobiology of HIV dementia. Sem Neurol. 1999;19:113–128. doi: 10.1055/s-2008-1040830. [DOI] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. doi: 10.1002/1531-8249(200002)47:2<186::AID-ANA8>3.3.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Manly JJ, Merchant CA, Jacobs dM, Small sA, Bell K, Ferin M, Mayeux R. Endogenous estrogen levels and Alzheimer's disease among postmenopausal women. Neurology. 2000;54:833–837. doi: 10.1212/wnl.54.4.833. [DOI] [PubMed] [Google Scholar]
- Slooter AJ, Bronzova J, Witteman JC, Van Broeckhoven C, Hofman A, van Duijn CM. Estrogen use and early onset Alzheimer's disease: a population-based study. J Neurol Neurosurg Psychiatry. 1999;67:779–81. doi: 10.1136/jnnp.67.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders-Pullman R, Gordon-Elliott J, Parides M, Fahn S, Saunders HR, Bressman S. The effect of estrogen replacement on early Parkinson's disease. Neurology. 1999;52:1417–1421. doi: 10.1212/wnl.52.7.1417. [DOI] [PubMed] [Google Scholar]
- Asthana S, Craft S, Baker LD, Raskind MA, Birnbaum RS, Lofgreen CP, Veith RC, Plymate SR. Cognitive and neuroendocrine response to transdermal estrogen in postmenopausal women with Alzheimer's disease: results of a placebo- controlled, double-blind, pilot study. Psychoneuroendocrinology. 1999;24:657–77. doi: 10.1016/S0306-4530(99)00020-7. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Maragos W, Avison M, Schmitt F, Berger J. Accelerated HIV dementia with methamphetamine and cocaine use. J Neurovirol. [DOI] [PubMed]
- Hudson L, Liu J, Nath A, Narayan O, Male D, Jones M, Everall I. Detection of human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–358. doi: 10.1016/S0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- Gundlah C, Kohama SG, Mirkes SJ, Garyfallou VT, Urbanski HF, Bethea CL. Distribution of estrogen receptor beta (ERbeta) mRNA in hypothalamus, midbrain and temporal lobe of spayed macaque: continued expression with hormone replacement. Brain Res Mol Brain Res. 2000;76:191–204. doi: 10.1016/S0006-8993(99)02475-0. [DOI] [PubMed] [Google Scholar]
- Zheng J, Ramirez VD. Purification and identification of an estrogen binding protein from rat brain: oligomycin sensitivity-conferring protein (OSCP), a subunit of mitochondrial F0F1-ATP synthase/ATPase. J Steroid Biochem Mol Biol. 1999;68:65–75. doi: 10.1016/S0960-0760(98)00161-7. [DOI] [PubMed] [Google Scholar]
- Gillum LA, Mamidipudi SK, Johnston SC. Ischemic stroke risk with oral contraceptives: A meta-analysis. JAMA. 2000;284:72–78. doi: 10.1001/jama.284.1.72. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Berger JR, Nath A, Rayens M. Cerebrovascular disease in young, HIV-infected, black Africans in the KwaZulu Natal province of South Africa. J Neurovirol. 2000;6:229–36. doi: 10.3109/13550280009015825. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995;37:373–380. doi: 10.1002/ana.410370314. [DOI] [PubMed] [Google Scholar]
- Zamboni L, De Martino C. Buffered picric acid formaldehyde: a new rapid fixative for electron microscopy. JCell Biol. 1967;35:148A. [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes : A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Hoshimaru M, Ray J, Sah DW, Gage FH. Differentiation of the immortalized adult neuronal progenitor cell line HC2S2 into neurons by regulatable suppression of the v-myc oncogene. Proc Natl Acad Sci U S A. 1996;93:1518–1523. doi: 10.1073/pnas.93.4.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–70. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- Ma M, Nath A. Molecular determinants for cellular uptake of Tat protein of human immunodeficiency virus type 1 in brain cells. J Virol. 1997;71:2495–2499. doi: 10.1128/jvi.71.3.2495-2499.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]