

Abstract
We report the development of IgA nephropathy (IgAN) following full myeloablative allogeneic hematopoietic cell transplantation in two patients with human leukocyte antigen (HLA) matched sibling donors, unrelated to active or chronic graft-versus-host disease. Both recipients had elevated urinary levels of galactose-deficient IgA1, and one donor–recipient pair had elevated serum levels of galactose-deficient IgA1. We propose that IgAN developed after bone marrow transplantation due to a non-graft-versus-host-disease-related multi-hit process associated with glomerular deposition of galactose-deficient IgA1. These two cases provide unique insight into the kinetics of overproduction of galactose-deficient IgA1 and its glomerular deposition and consequential renal injury in IgAN.
Keywords: bone marrow transplantation, galactose-deficient IgA1, hematopoietic cell transplantation, IgA nephropathy
Introduction
Kidney dysfunction after full myeloablative allogeneic hematopoietic cell transplantation afflicts 17–66% of adult recipients and is usually due to acute kidney injury, medication-induced nephrotoxicity, radiation therapy or thrombotic microangiopathy [1]. Glomerular disorders are unusual, with membranous nephropathy and minimal change disease found most frequently on renal biopsy [2]. IgA nephropathy (IgAN) has been reported only twice [3,4]. We present two patients who underwent hematopoietic cell transplantation and later developed IgAN unrelated to graft-versus-host disease (GVHD). We explore the role of galactose-deficient IgA1 in this hematopoietic cell transplantation-associated glomerulonephritis.
Case descriptions
Patient 1
A 54-year-old Caucasian man with acute myelogenous leukemia underwent full myeloablative allogeneic bone marrow transplantation from his 10:10 HLA antigen-matched brother in May 2003. Chimerism studies of the bone marrow detected deoxyribonucleic acid (DNA) typing of only the donor (Figure 1). Post-transplant course was complicated by acute and chronic GVHD, involving skin (grade III) with desquamative lesions and the gastrointestinal tract (grade III) as proven by biopsy during colonoscopy in June 2003. The acute GVHD was initially treated with high-dose methylprednisolone and cyclosporine A, which were successfully weaned off by March 2005 without signs of chronic GVHD. Twenty-one months after cessation of chronic immunosuppressive therapy and in the continued absence of GVHD symptoms, serum creatinine increased from 0.9 mg/dL to 5.4 mg/dL, associated with proteinuria but without overt nephrotic syndrome (urinary protein/creatinine 10.7 g/g) and new-onset microscopic hematuria. Serum IgA level was 443 mg/dL (normal 72–372 mg/dL). Renal biopsy revealed crescentic IgAN (detailed in ‘Pathology’). Glucocorticoids, cyclophosphamide and then azathioprine were given with lisinopril. Eight months later, proteinuria improved to 0.3 g/g, serum albumin normalized to 4.0 g/dL (nadir 1.5 g/dL) and serum IgA level decreased to normal, 336 mg/dL. Seventeen months after biopsy, serum creatinine was 1.4 mg/dL, off immunosuppressive therapy.
Fig. 1.
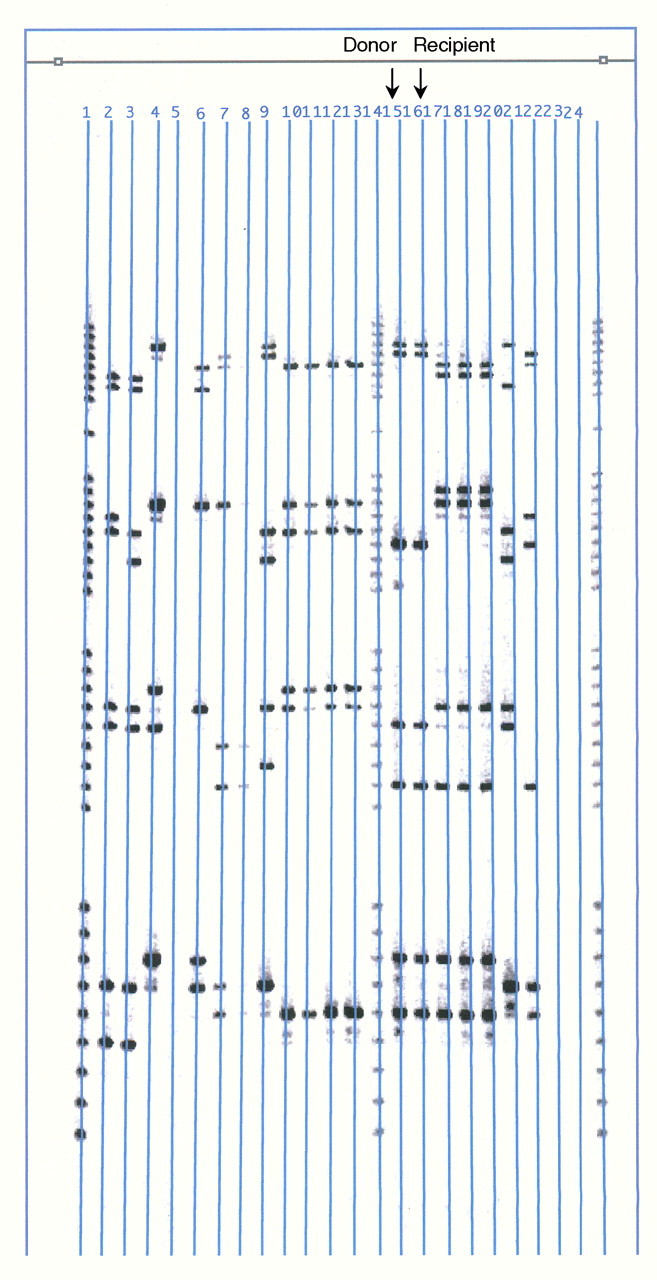
Chimerism study for patient 1: DNA typing by polymerase chain reaction technique was used to detect differences in allele types between the donor and recipient cells at several short tandem repeat loci of the bone marrow (BRT Laboratories, Inc, Baltimore, MD). Only donor types were detected as shown (black arrows) in the comparison of lane 15 (the donor sample for patient 1) and lane 16 (the post-transplant bone marrow sample of patient 1) in 2003.
The donor had no evidence of kidney dysfunction, hypertension, proteinuria or microscopic hematuria. Serum IgA level was normal, 247 mg/dL.
Patient 2
A 34-year-old Asian man with chronic myelogenous leukemia underwent 10:10 HLA-matched full myeloablative allogeneic bone marrow transplantation from his sister in August 2006. Cyclosporine A was easily tapered off over a year without GVHD. He achieved 100% donor chimerism, and the bone marrow was characterized as XX by chromosomal analysis (Figure 2). One year after cessation of immunosuppressive therapy and 24 months after transplantation in the absence of GVHD symptoms including rash, gastrointestinal symptoms or abnormal liver function tests, serum creatinine rose acutely from 1.3 mg/dL to 5.2 mg/dL, associated with substantial proteinuria 6.1 g/g (without overt nephrotic syndrome), hypoalbuminemia 2.2 g/dL and new-onset microscopic hematuria. Serum IgA level was 250 mg/dL. Renal biopsy showed crescentic and necrotizing IgAN (detailed below). Prednisone and azathioprine therapy was interrupted by sepsis after 2 months; nonetheless, serum creatinine and albumin improved to 3.5 mg/dL and 3.5 g/dL, respectively, and proteinuria improved to 2.6 g/g.
Fig. 2.

Chimerism study for patient 2: normal female chromosome complement was observed in all metaphases, without evidence for acquired clonal abnormality, by cytogenetic karyotyping of post-transplant bone marrow aspirate in 2008. The 46, XX karyotype (black arrow) is consistent with engraftment by female donor cells (Quest Diagnostics Nichols Institute, Chantilly, VA).
The donor had no evidence of kidney dysfunction, hypertension, proteinuria or microscopic hematuria. Serum IgA level was normal, 214 mg/dL.
Pathology
Renal biopsy samples of both patients exhibited moderately advanced glomerular disease (30 to 50% obsolete glomeruli) with crescents in >50% of viable glomeruli (Figure 3). Some crescents were primarily cellular, whereas others had substantial fibrin and few cells. Mild mesangial hypercellularity and sporadic matrix expansion with segmental agglutination of the capillaries were noted. Interstitial edema and multifocal collections of inflammatory cells as well as moderate arteriosclerosis were present. On immunofluorescence study, both cases showed 2–3+ diffuse mesangial staining for IgA and C3, with focal staining in the capillaries. IgG staining, similar in distribution but with less intensity of 1+ was noted for patient 1. The staining for IgG was insignificant for patient 2. On electron microscopy, numerous and large electron-dense glomerular deposits were seen in the mesangial and paramesangial areas, rare deposits in subendothelial areas of the capillary wall.
Fig. 3.
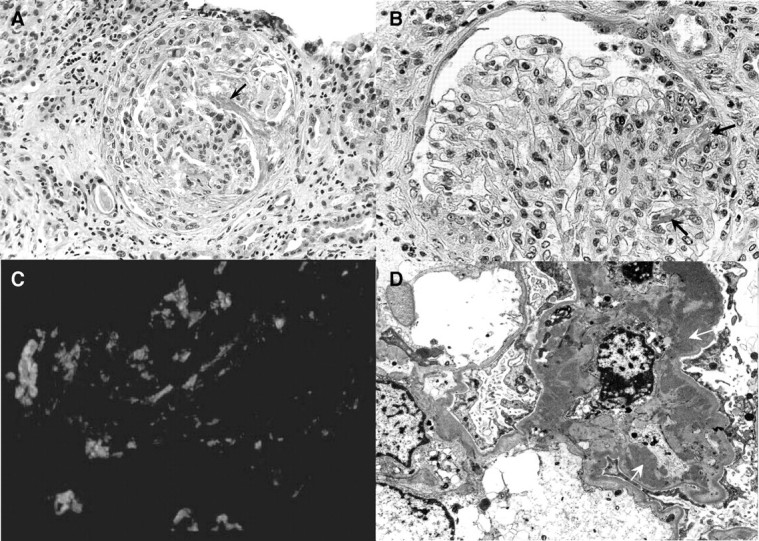
A) Light microscopy (patient 1)—a large cellular crescent with a long strand of fibrin is seen in the urinary space of this glomerulus (black arrow). The capillary tuft appears compressed and reveals mild mesangial hypercellularity (H&E, ×250). B) Light microscopy (patient 2)—glomerulus showing segmental focus of necrosis with fibrin extravasation in the capillary lumina (black arrows) and Bowman's space and epithelial cell proliferation. Note the mild increase of mesangial cellularity. The capillaries are patent and the glomerular basement membrane is unremarkable (H&E, ×400). C) Immunofluorescence (patient 1)—glomerulus stained with IgA antiserum showing abundant mesangial deposits of 3+ intensity (fluorescein isothiocyanate stained section ×400). D) Electron microscopy (patient 1)—glomerulus showing large mesangial electron-dense deposits (white arrows). The mesangial cells appear active (original magnification ×16 000).
Measurement of galactose-deficient IgA1
We measured total IgA and galactose-deficient IgA1 in samples of serum and urine collected 64 and 25 months after hematopoietic cell transplantation in cases 1 and 2, respectively, by enzyme-linked immunosorbant assay (ELISA) [5]. In case 1, the donor and recipient had elevated serum levels of galactose-deficient IgA1, comparable to those of IgAN patients, whereas the donor and recipient in case 2 had normal levels (Table 1). Notably, only the two recipients had elevated urinary excretion of galactose-deficient IgA1. Archived samples of blood or urine prior to transplantation were not available.
Table 1.
Analysis of IgA and galactose-deficient IgA1 in seruma and urine samples after hematopoietic cell transplantation.
| Serum | Urine | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IgA (μg/mL) | HAA-IgA1b (%) | Total galactose-deficient IgA1/mLc | IgA (ng/mL) | IgA/U-Cr | HAA-IgA1/U-Crd | U-prot (mg/dL) | U-Cr (mg/dL) | U-prot/Cr | |
| Recipient | |||||||||
| Case 1 | 4387 | 36 | 1575 | 11 642 | 166 | 459 | 45 | 70 | 0.6 |
| Case 2 | 1121 | 27 | 304 | 8212 | 135 | 154 | 52 | ||
| Donor | |||||||||
| Case 1 | 3335 | 40 | 1324 | 783 | 7 | ND | 11 | 106 | 0.1 |
| Case 2 | 1530 | 23 | 352 | 233 | 4 | 38 | 61 | ||
| Reference | |||||||||
| IgANe | 5194 ± 2337 | 35 ± 12** | 1669 ± 527**** | 10 315 ± 14 692** | 104 ± 122** | 127 ± 86**** | 125 ± 108** | 119 ± 66* | 1.2 ± 1.0*** |
| Controlse | 3730 ± 2141 | 24 ± 7 | 766 ± 227 | 466 ± 505 | 3 ± 3 | 17 ± 12 | 14 ± 9 | 170 ± 86 | 0.1 ± 0.1 |
Abbreviations: Cr, creatinine; HAA, Helix aspersa agglutinin; HAA-IgA1, HAA binding IgA1; IgAN, IgA nephropathy; ND, not detectable; U, urinary; prot, protein.
Serum IgA concentration was measured first, and then a normalized amount of IgA (100 ng) was added to the wells of ELISA plates coated with F(ab′)2 fragment of goat IgG specific to human IgA (Jackson Labs, West Grove, PA). The captured IgA was treated with neuraminidase (Roche, Indianapolis, IN) to remove sialic acid, and reactivity with biotin-labeled N-acetylgalactosamine-specific lectin, HAA (Sigma-Aldrich, St. Louis, MO), was measured to determine levels of galactose-deficient IgA1.
HAA binding to 100 ng serum IgA expressed as % of HAA binding to the same amount of standard galactose-deficient myeloma IgA1 protein (Ale).
Total galactose-deficient IgA1, expressed in units per mL serum, was calculated as follows: (% × μg IgA/mL) / 100.
HAA binding (optical density 490 nm) to IgA1 in 10-fold diluted urine samples normalized to urinary creatinine.
For reference, data for patients with IgAN and healthy controls are shown (average ± SD); IgAN (n = 26), Controls (n = 18).
P < 0.05.
P < 0.01.
P < 0.001.
P < 0.0001.
Discussion
Glomerulopathy after hematopoietic cell transplantation, specifically membranous nephropathy and minimal change disease, has been attributed to GVHD [2] as shown in murine models [6]. Proximity of disease onset to cessation of immunosuppression and a high prevalence of concomitant GVHD suggest that the glomerular injury is immune-mediated [2], possibly through interaction of recipient B cells and dysfunctional donor T cells [6]. However, development of IgAN is not closely associated with active GVHD, which suggests mechanisms of pathology independent of those of GVHD. Most of the cases (the present two and those reported in the literature) exhibited no symptoms of active GVHD at onset of glomerular disease [3,4] and had a rapidly progressive course with crescentic IgAN. Pathologic processes other than GVHD should be considered.
Specific clones among transplanted stem cells may induce immune-mediated tissue injury in the recipient, including IgAN [7,8]. In a murine model, hematopoietic cell transplantation from a strain with no renal disease attenuated the glomerular injury of IgAN; conversely, transplantation of marrow from mice with aggressive IgAN into a ‘disease-quiescent’ strain induced greater glomerular injury [7]. Furthermore, bone marrow transplantation in humans has been shown to dampen the injury of IgAN [8]. Transfer of donor immune characteristics after hematopoietic cell transplantation has also been documented for patients with psoriasis, rheumatoid arthritis, autoimmune thyroiditis and immune thrombocytopenia [9]. The recipient in case 1 also newly manifested psoriasis, like his donor.
IgAN likely results from glomerular deposition of circulating immune complexes comprised of galactose-deficient IgA1 bound by an IgG or IgA antibody that recognizes the galactose-deficient hinge-region glycans of IgA1 [10]. About three-quarters of patients with IgAN have elevated levels of galactose-deficient IgA1 in the circulation [5] and urine [11]. In this report, only the donor and recipient in case 1 had increased serum galactose-deficient IgA1 levels. However, both recipients had increased urinary excretion of galactose-deficient IgA1. Presumably, a clone of donor stem cells produced the galactose-deficient IgA1 in both recipients, leading to nephritogenic circulating immune complexes that deposited in the glomeruli and later entered the urine. The modest mesangial staining for IgG for patient 2 may reflect a predominance of IgA versus IgG as the isotype of anti-glycan-specific antibody [10]. The pronounced renal injury in both patients was striking and is probably due to explicit features of the immune complexes that influence nephritogenicity, such as size and composition [10,12].
The serum levels of galactose-deficient IgA1, intensity of IgG staining on immunofluorescence and gender of the donor differentiate the two cases. However, for both patients, the urinary excretion of galactose-deficient IgA1 was high. Despite differing serum levels between the recipients, both had galactose-deficient IgA1 levels corresponding to that of their donor, which may imply that donor immune characteristics are responsible for the variable findings. Moreover, the 100% chimerism (Figures 1 and 2) in both recipients shortly after transplantation suggests that their immune systems were derived entirely from donor marrow.
Our findings suggest that donor stem cells are likely integral to disease expression, possibly through modification or transfer of disease-inducing cells, although the lack of archived serum or urine prior to bone marrow transplantation precludes further investigation.
These two cases provide unique insight into the kinetics of overproduction of galactose-deficient IgA1 and its glomerular deposition and consequent renal injury in IgAN. We propose that IgAN developed after hematopoietic cell transplantation due to a non-GVHD-related multi-hit process associated with glomerular deposition of galactose-deficient IgA1.
Acknowledgments
The study was supported in part by National Institutes of Health grants DK078244, DK075868, DK080301, DK082753, DK077279, DK071802 and DK077279.
Conflict of interest statement. None declared.
Footnotes
The original version was incorrect. The title has been amended.
References
- 1.Hingorani S. Chronic kidney disease in long-term survivors of hematopoietic cell transplantation: epidemiology, pathogenesis, and treatment. J Am Soc Nephrol. 2006;17:1995–2005. doi: 10.1681/ASN.2006020118. [DOI] [PubMed] [Google Scholar]
- 2.Brukamp K, Doyle AM, Bloom RD, et al. Nephrotic syndrome after hematopoietic cell transplantation: do glomerular lesions represent renal graft-versus-host disease? Clin J Am Soc Nephrol. 2006;1:685–694. doi: 10.2215/CJN.00380705. [DOI] [PubMed] [Google Scholar]
- 3.Kimura S, Horie A, Hiki Y, et al. Nephrotic syndrome with crescent formation and massive IgA deposition following allogeneic bone marrow transplantation for natural killer cell leukemia/lymphoma. Blood. 2003;101:4219–4221. doi: 10.1182/blood-2002-07-2290. [DOI] [PubMed] [Google Scholar]
- 4.Chan GS, Lam MF, Au WY, et al. IgA nephropathy complicating graft-versus-host disease, another nephropathy causing nephrotic syndrome after bone marrow transplantation. Histopathology. 2004;45:648–651. doi: 10.1111/j.1365-2559.2004.01958.x. [DOI] [PubMed] [Google Scholar]
- 5.Moldoveanu Z, Wyatt R, Lee J, et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71:1148–1154. doi: 10.1038/sj.ki.5002185. [DOI] [PubMed] [Google Scholar]
- 6.Otani M, Shimojo H, Shiozawa S. Renal involvement in bone marrow transplantation. Nephrology. 2005;10:530–536. doi: 10.1111/j.1440-1797.2005.00478.x. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki H, Suzuki Y, Aizawa M, et al. Th1 polarization in murine IgA nephropathy directed by bone marrow-derived cells. Kidney Int. 2007;72:319–327. doi: 10.1038/sj.ki.5002300. [DOI] [PubMed] [Google Scholar]
- 8.Imasawa T. Roles of bone marrow cells in glomerular diseases. Clin Exp Nephrol. 2003;7:179–185. doi: 10.1007/s10157-003-0248-9. [DOI] [PubMed] [Google Scholar]
- 9.Cooley H, Snowden J, Grigg A, et al. Outcome of rheumatoid arthritis and psoriasis following autologous stem cell transplantation for hematologic malignancy. Arthritis Rheum. 1997;40:1712–1715. doi: 10.1002/art.1780400923. [DOI] [PubMed] [Google Scholar]
- 10.Tomana M, Novak J, Julian B, et al. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki H, Suzuki Y, Lyas CN, et al. Urinary polypeptide biomarkers of IgA nephropathy. J Am Soc Nephrol. 2008;19:665A. [Google Scholar]
- 12.Novak J, Tomana M, Matousovic K, et al. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–513. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]