

Summary
Acyl-CoA synthetase-1 (ACSL) contributes 80% of total ACSL activity in adipose tissue and was believed to be essential for the synthesis of triacylglycerol. We predicted that an adipose-specific knockout of ACSL1 (Acsl1A−/−) would be lipodystrophic, but, compared to controls, Acsl1A−/− mice had 30% greater fat mass when fed a low fat diet, and gained weight normally when fed a high fat diet. Acsl1A−/− adipocytes incorporated [14C]oleate into glycerolipids normally, but fatty acid oxidation rates were 50–90% lower than in control adipocytes and mitochondria. Acsl1A−/− mice were markedly cold intolerant, and β3-adrenergic agonists did not increase oxygen consumption, despite normal adrenergic signaling in brown adipose tissue. The reduced adipose FA oxidation and marked cold intolerance of Acsl1A−/− mice indicate that normal activation of FA for oxidation in adipose tissue in vivo requires ACSL1. Thus, ACSL1 has a specific function in directing the metabolic partitioning of fatty acids towards β-oxidation.
Keywords: fatty acid oxidation, cold thermogenesis, brown adipose, triacylglycerol
Introduction
Dysfunctional lipid metabolism underlies the development of obesity and obesity-related complications such as hepatic steatosis, diabetes, and heart disease. Although the mechanisms that regulate the cellular uptake, activation, and metabolism of fatty acid (FA) are not fully understood, nearly all pathways of FA metabolism require the acyl-CoA synthetase–mediated conversion of FAs to acyl-CoAs (FA + CoA + ATP → acyl-CoA + AMP + PPi). Acyl-CoAs have multiple fates, including use in complex lipid formation and lipid remodeling, effects on signal transduction, activation of transcription factors, and oxidation to provide cellular energy. Long-chain acyl-CoAs are formed by a family of five acyl-CoA synthetases (ACSL), ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6, and by several fatty acid transport proteins (FATP). ACSL isoenzymes are intrinsic membrane proteins whose active sites face the cytosol to produce acyl-CoAs which are either partitioned into the facing membrane monolayer where they can encounter downstream metabolic enzymes (Coleman and Bell, 1978; Mannaerts et al., 1982), or transported to different organelles by the cytosolic acyl-CoA binding protein (Stremmel et al., 2001). The ability of amphipathic acyl-CoAs to move freely within a single membrane monolayer or to be transported to distant membranes should render all acyl-CoAs metabolically equivalent, no matter which ACSL isoenzyme catalyzes their formation. Yet, gain-of-function studies suggest that each of the ACSL isoform has a distinct function in directing acyl-CoAs to one or more specific downstream pathways (Li et al., 2006; Mashek et al., 2006b).
The functions of the ACSLs are of particular interest in adipose tissue because of the central role that white adipose tissue (WAT) plays in energy storage and that brown adipose tissue (BAT) plays in energy dissipation. In WAT, Acsl1 mRNA is the most abundantly expressed isoform, with relatively lower expression of Acsl3, -4, and -5 (Mashek and Coleman, 2006). Acsl1 mRNA in liver is upregulated by PPARα ligands (Martin et al., 1997), whereas its mRNA in WAT is increased by PPARγ agonists (Gerhold, 2002; Shimomura et al., 1993). In differentiating 3T3-L1 adipocytes, ACSL specific activity and mRNA expression increase 100- and 160-fold, respectively (Coleman et al., 1978; Marszalek et al., 2004), whereas the expression of the other Acsl isoenzymes remains unchanged (Oikawa et al., 1998). This induction of Acsl1 expression suggested that ACSL1 is upregulated as part of the program of adipocyte differentiation culminating in triacylglycerol (TAG) synthesis and storage. Consistent with this interpretation, an expression cloning strategy designed to find cDNAs that augment FA uptake and incorporation into TAG cloned ACSL1 (Schaffer and Lodish, 1994). Further, overexpressed ACSL1 in 3T3-L1 adipocytes increases FA uptake and incorporation into TAG (Gargiulo et al., 1999; Richards et al., 2006; Souza et al., 2002). In order to examine the putative role of ACSL1 in TAG synthesis, we created a mouse deficient in adipose ACSL1, and predicted that these mice would be unable to synthesize and store TAG in adipocytes. Surprisingly, however, the Acsl1A−/− mice had normal fat stores, but severely impaired adipose FA oxidation, suggesting not only that ACSL1 catalyzes the initial required step in FA oxidation, but also that it specifically directs FA towards mitochondrial β-oxidation.
Results
Generation of mice that lack ACSL1 specifically in adipose tissue
Mice with Loxp sequences inserted on either side of exon 2 in the Acsl1 gene (Li et al., 2009) and backcrossed to the C57Bl/6 strain six times were crossed with mice in which Cre expression is driven by the aP2 promoter [B6.Cg-Tg (Fabp4-cre) 1Rev/J; Jackson Laboratories, Bar Harbor ME] to generate adipose-specific Acsl1 knockout mice (Acsl1A−/−). Littermates lacking the Cre gene (Acsl1flox/flox) were used as controls.
ACSL1 protein was virtually absent in brown, gonadal, and inguinal adipose tissue from Acsl1A−/− mice (Fig. 1A). ACSL specific activity was 80% lower than in controls, confirming that ACSL1 is, functionally, the major ACSL in adipose tissue (Fig. 1B), with the remaining 20% of ACSL activity catalyzed by other ACSL isoenzymes in adipocytes and all ACS enzymes in stromovascular tissue (Mashek et al., 2006a). Confirming the tissue-specificity of the knockdown, ACSL1 protein and total ACSL specific activity were unaffected in Acsl1A−/− liver (Fig. 1A, B). Real-time PCR showed nearly absent expression of Acsl1 mRNA in Acsl1A−/− gonadal and brown adipose tissue, but unchanged expression of the other four Acsl isoenzymes and a 50% reduction in Fatp1 mRNA, a related ACS (Fig. 1C, D). Thus, other acyl-CoA synthetases did not compensate for the loss of ACSL1.
Figure 1. Generation of mice that lack ACSL1 specifically in adipose tissue.
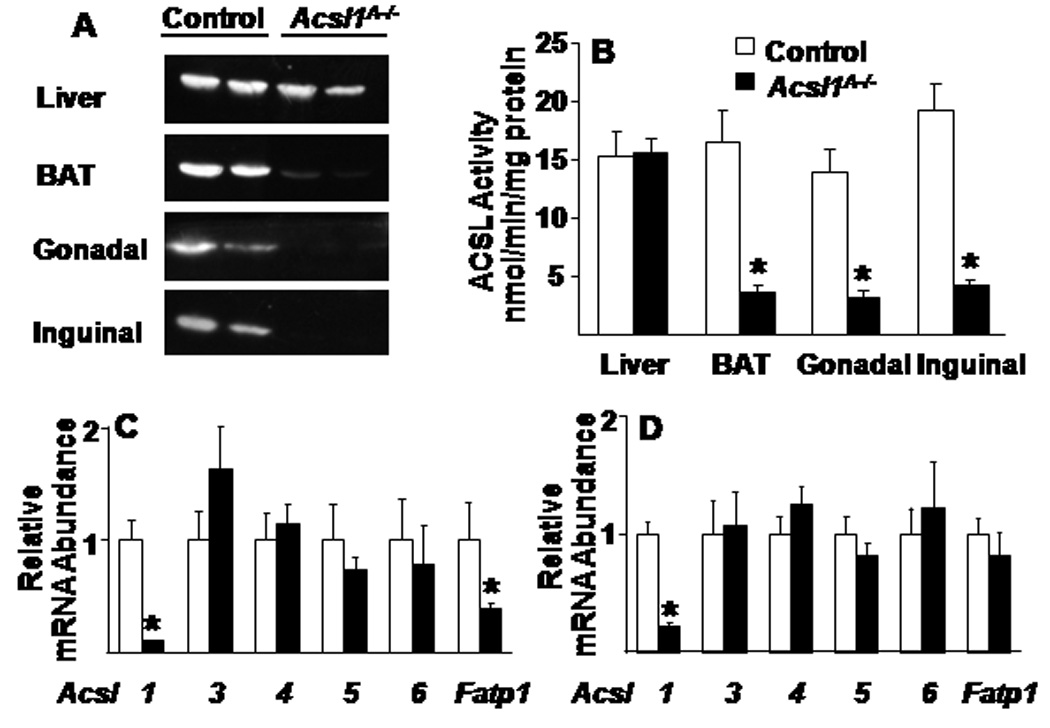
A) Representative immunoblot against ACSL1 protein and B) ACSL activity in control and Acsl1A−/− liver, brown, gonadal, and inguinal adipose of 20 wk old mice, n=5–7. ACSL isoenzyme mRNA expression in C) gonadal adipose and D) brown adipose from 20 wk old control and Acsl1A−/− mice, n=6. Data represent mean ± SEM; *, P≤0.05 versus control.
Acsl1A−/− mice have increased fat mass and synthesize TAG normally
Despite the 80% loss of adipose ACSL activity, Acsl1A−/− mice had increased adiposity. When fed a 10%-fat diet, Acsl1A−/− body weights were similar to controls (Fig. 2A), but MRI analysis showed that the mice had 30% more body fat (Fig. 2B). Consistent with the MRI data, gonadal fat depot weights were ~40% greater in mice fed chow or 10%-fat diets (Fig. 2C). Gonadal adipose tissue histology, adipocyte size, GPAT specific activity, and AMPK phosphorylation were similar between genotypes (Fig. 2D–G). No genotype differences in liver or BAT weights were observed.
Figure 2. Acsl1A−/− mice have increased fat mass.
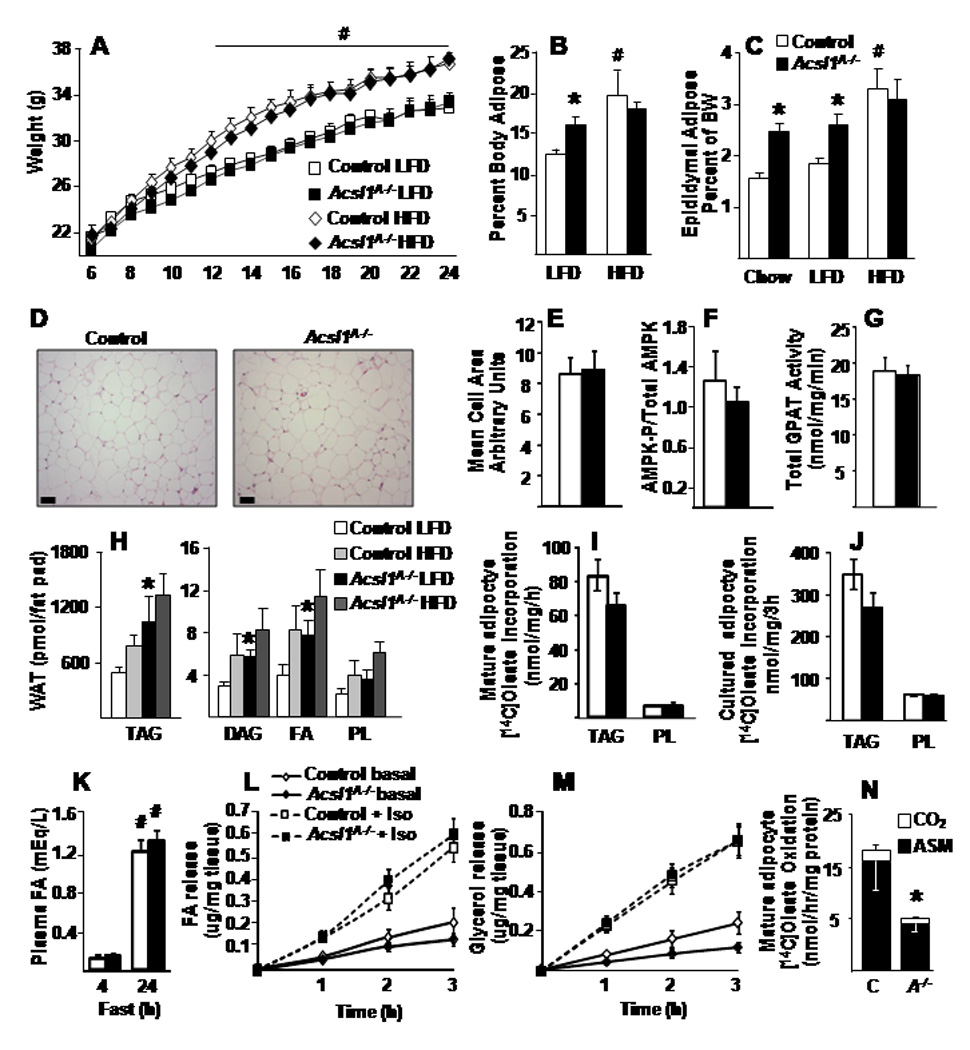
A) Body weight from 6 to 24 weeks of age in mice fed LFD or HFD, n=10–15. B) Total body fat measured by MRI in 26 wk old control and Acsl1A−/− male mice, n=5–7. C) Fat depot weight as a percent of body weight (BW) in 20–26 wk old control and Acsl1A−/− male mice, n=10–15. D) Representative sections from control and Acsl1A−/− epididymal WAT and E) mean cells size from sections stained for hematoxylin and eosin, scale bars = 50 µM. F) AMPK phosphorylation, relative to total AMPK, from control and Acsl1A−/− inguinal adipose, n=4–5. G) Total GPAT activity in control and Acsl1A−/− gonadal adipose, n=6. H) Lipid content of control and Acsl1A−/− epididymal WAT in 6 mo old male mice fed LFD or HFD diet for 4.5 mo, n=5. [1-14C]Oleate incorporation into (I) glycerolipids or (N) acid soluble metabolites (ASM) and carbon dioxide (CO2), n=3–4, by control (C) and Acsl1A−/− (A−/−) mature epididymal adipocytes, n=6. J) [1-14C]Oleate incorporation into (I) glycerolipids by control and Acsl1A−/− cultured primary adipocytes. K) Plasma FA in control and Acsl1A−/− mice fasted for 4 or 24 hours, n=6–8. Release of (L) FA or (M) glycerol from control and Acsl1A−/− gonadal adipocyte explants basally or with 10µM isoproterenol (Iso), n=8. HFD, high-fat diet; LFD, low-fat diet; TAG, triacylglycerol; DAG, diacylglycerol; PL, phospholipids; FA, fatty acids. Data represent mean ± SEM *, P≤0.05 versus control; # P≤0.05 chow or LFD versus HFD within genotype.
No diet-related differences were observed between genotypes in plasma glucose, FA, cholesterol, or insulin concentrations (Table S1). However, plasma TAG concentrations were 17% lower in Acsl1A−/− mice. A 45%-fat diet resulted in weight gain, adiposity, and insulin resistance that was similar in both genotypes (Fig. 2A, Table S1). Loss of ACSL1 could theoretically change the cellular content and ratio of its FA substrate and acyl-CoA product, thereby alter (Berger and Moller, 2002; Hostetler et al., 2005), however target genes of these transcription factors did not change significantly in Acsl1A−/− gonadal WAT (Table S2).
When fed a low-fat diet, the amount of phospholipid extracted from epididymal WAT was similar in both genotypes, but Acsl1A−/− WAT contained more TAG, DAG, and FA per fat pad (Fig. 2H). With the high-fat diet, no differences were present between control and Acsl1A−/− lipid classes in epididymal WAT. BAT from Acsl1A−/− and control mice fed either diet also contained similar amounts of all lipid classes (Fig. S1A, B). Thus, lack of ACSL1 did not impair adipose TAG accumulation.
Confirming normal TAG synthesis in both isolated white gonadal adipocytes and cultured primary white adipocytes from control and Acsl1A−/− mice, the rates of [1-14C]oleate incorporation into TAG and phospholipid were similar, indicating that the lack of ACSL1 did not impair FA incorporation into complex lipids (Fig. 2I,J). Thus, despite greater than 100-fold increases in Acsl1 mRNA and ACSL specific activity during fat cell differentiation (Coleman et al., 1978; Marszalek et al., 2004), TAG synthesis does not require FA activation by ACSL1. The increased fat mass did not result from impaired lipolysis because in 24h fasted mice and in media from white adipose tissue explants with or without exposure to lipolytic stimuli (Fig. 2K–M, Table S3) FA and glycerol release were similar between genotypes.
Differences in FA species content in adipose tissue
We did not expect to find major changes in FA species because each of the purified ACSL isoenzymes in vitro can activate a wide range of long-chain, saturated and unsaturated FA (Oikawa et al., 1998). However, compared to controls, Acsl1A−/− mice fed both the low-fat and high-fat diets contained 20–30% less 18:2n6 in TAG from brown and gonadal white adipose (Fig. S1C, D). The TAG fraction of BAT from Acsl1A−/− mice was composed of 15% more 16:0 than controls fed the low-fat diet (Fig. S1E). In Acsl1A−/− BAT, the PL fraction from mice fed the high-fat diet contained 30% more 16:0 and 15%, 22%, and 14% less 18:0, 18:2n6, and 20:4n6, respectively, than controls (Fig. S1F). No other differences in the major fatty acid species were observed. These small, but statistically significant, alterations in lipid composition of adipose tissues suggest that ACSL1 does contribute to the relative content of esterified FA species in neutral lipids and phospholipids, despite the minimal differences in FA preference observed in vitro.
Fatty acid oxidation was impaired in white adipocytes from Acsl1A−/− mice
Compared to mature adipocytes isolated from control mice, the rate of [1-14C]oleate incorporation into CO2 and acid soluble metabolites (ASM) was 50% lower in Acsl1A−/− adipocytes (Fig. 2N). CO2 and ASM represent complete and incomplete products of fatty acid oxidation; thus, the 50% reduction of FA incorporation into these oxidative products reflects reduced β-oxidation and suggests that impaired FA catabolism could have caused the larger WAT depots in the Acsl1A−/− mice.
Decreased FA oxidation rendered Acsl1A−/− mice cold intolerant
FA oxidation increases markedly in BAT during cold exposure. To generate heat and maintain normal body temperature, ATP production is reduced when activated uncoupling protein (UCP1) increases the flux of protons through the electron transport chain and dissipates the mitochondrial proton gradient (Lowell and Spiegelman, 2000). Fasted wildtype mice can maintain their body temperature above 35°C for as long as 24 h at 4°C (Cohen et al., 2005). In striking contrast, however, the body temperature of the Acsl1A−/− mice fell below 30°C within 3 h (Fig. 3A). During cold exposure, both genotypes shivered similarly, and gastrocnemius total ACSL activity (Fig. S4A) and Acsl1 mRNA abundance (data not shown) did not change in Acsl1A−/− mice. In shivering muscle, fuel preference and the contribution of muscle heat to total body heat remain unclear (Haman, 2006); however, plasma FFA and VLDL were not limiting for either Acsl1A−/− skeletal muscle or BAT (Fig. 6C, D).
Figure 3. Acsl1A−/− mice are cold intolerant due to impaired FA oxidation.
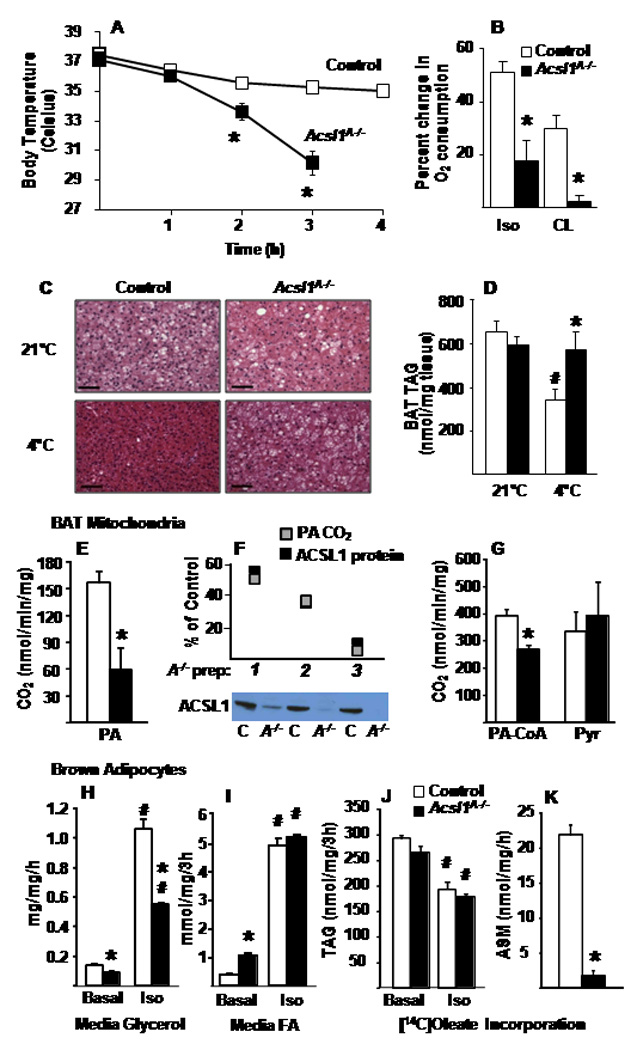
A) Body temperature of fasted control and Acsl1A−/− male mice exposed to a 4°C environment for 4 h, n=14–16. B) Control and Acsl1A−/− male mouse O2 consumption was monitored in a metabolic chamber for 3 h before and after injection of isoproterenol (Iso) or CL-316243 (CL) (10 mg/kg), n=6–8. C) Representative BAT histology stained for hematoxylin and eosin in control and Acsl1A−/− mice before and after cold exposure for 2–4 h when Acsl1A−/− body temperature reached 30°C; scale bars = 50 µm. D) TAG content of BAT from control and Acsl1A−/− mice fasted at 21°C or at 4°C for 4 h, n=5–6. BAT mitochondrial (E,F) [1-14C]palmitate and (G) [1-14C]palmitoyl-CoA and [1-14C]pyruvate oxidation into carbon dioxide (CO2) from control and Acsl1A−/− mice, n=3. F) Control (C) and Acsl1A−/− (A−/−) ACSL1 immunoblot and protein, and CO2 production from [1-14C]palmitate by 3 mitochondrial preparations (AC), relative to control. Control and Acsl1A−/− cultured primary brown adipocyte release of (H) glycerol and (I) FA with or without 10µM isoproterenol, and incorporation of [1-14C]oleate into (J) TAG and (K) ASM, n = 3. Data represent mean ± SEM *, P≤0.05 versus control; # P≤0.05 21°C versus 4°C within genotype.
Figure 6. Plasma metabolites and BAT acyl-CoAs in Acsl1A−/− mice after adrenergic stimuli.
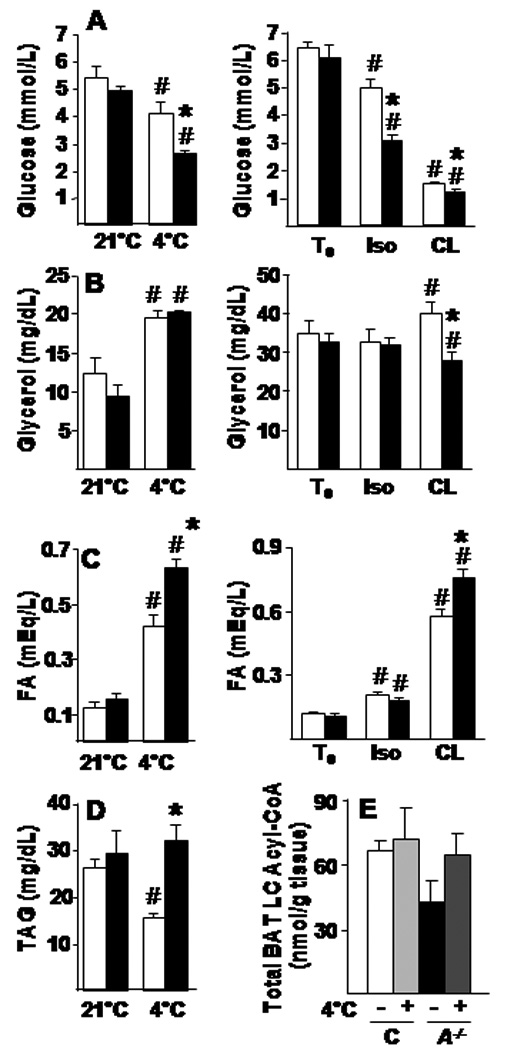
Plasma glucose (A), glycerol (B), FA (C), and TAG (D) from control and Acsl1A−/− mice fasted for 4 h at 21°C or 4°C, or before (T0) and 3 h after isoproterenol (Iso) or CL-316243 (CL) injection (10 mg/kg), n=6–8. E) Sum of long-chain acyl-carnitines (C14–C20) in control (C) and Acsl1A−/− (A−/−) with or without cold exposure for 2–4 h. Data represent mean ± SEM; *, P≤0.05 versus control; # P≤0.05 21°C or T0 versus treated within genotype.
Direct measurements of FA oxidation rates in BAT homogenates from Acsl1A−/− mice showed that the rate of [1-14C]palmitate incorporation into CO2 and ASM was 36% lower than controls (Fig. S5C). Because label dilution occurs when fatty acid oxidation is measured in cells with a high lipid content, we confirmed this oxidation defect by monitoring O2 consumption in conscious Acsl1A−/− and control mice in Oxymax chambers. Whole body O2 consumption normally increases in response to adrenergic stimuli and reflects an increase in electron transport chain activity. Isoproterenol, a non-specific β-adrenergic receptor agonist, increased O2 consumption 51% in control mice, but only 18% in Acsl1A−/− mice (Fig. 3B). This blunted response indicates that whole body oxidation is diminished. When CL-316243, an adipose-specific, β3-adrenergic receptor agonist (Yoshida et al., 1994) was injected, O2 consumption increased 30% in control mice, but only 2% in Acsl1A−/− mice, showing that the block in adrenergic-mediated oxidation was adipose-specific (Fig. 3B,S5B).
In mitochondria from Acsl1A−/− BAT, the production of CO2 from [1-14C]palmitate oxidation was reduced 62% (Fig. 3E,G), and this reduction in mitochondrial FA oxidation correlated directly with the amount of mitochondrial ACS activity and ACSL1 protein present in each of three mitochondrial preparations (Fig. 3F,S4F,E). In contrast, CO2 production from [1-14C]pyruvate oxidation was similar to that of control mitochondria, showing that Acsl1A−/− BAT mitochondria are functional for substrates other than FA. The 31% reduction in [1-14C]palmitoyl-CoA oxidation (Fig. 3G) suggests that FA oxidation may require the generation of acyl-CoA at the mitochondrial membrane, and/or that the mechanism by which ACSL1 directs its acyl-CoA product towards mitochondrial oxidation may be partially disrupted by homogenization. The rate of oxygen consumption in control and Acsl1A−/− BAT mitochondria was similarly increased by UCP1-dependent pyruvate stimulation and, in both genotypes, GDP effectively reduced oxygen consumption by inhibiting UCP1 (Fig. S4). These data show that ACSL1 deficient BAT mitochondria have full UCP1 activity and that Acsl1A−/− cold intolerance is not due to impaired UCP1 function. Taken together, these data from live mice, BAT homogenates, and isolated mitochondria indicate that cold intolerance of the Acsl1A−/− mice resulted from impaired FA oxidation and show that ACSL1 is absolutely required for cold-adaptive thermogenesis in BAT.
After 4 h at 4°C, BAT from control mice depleted its lipid droplets, but lipid droplets were retained in Acsl1A−/− BAT (Fig. 3C). Control mice lost ~50% of BAT lipid stores after 4 h of cold exposure, whereas the TAG content in Acsl1A−/− BAT did not change (Fig. 3D). Liver and WAT histology was similar at 21°C and 4°C in both genotypes (data not shown). Although TAG retention in Acsl1A−/− BAT after cold exposure could be due to impaired lipolysis, the increase in plasma FA and glycerol in cold-exposed Acsl1A−/− mice is consistent with active TAG lipolysis in WAT (Fig. 6B,C), and in differentiated primary brown adipocytes from Acsl1A−/− mice, the release of FA into the media was greater than controls under basal conditions and similar after adrenergic stimuli, demonstrating that Acsl1A−/− brown adipocytes lipolyzed intracellular triacylglycerol normally (Fig. 3I). Surprisingly, compared to control cells, glycerol release in Acsl1A−/− brown adipocytes was blunted by ~50% after adrenergic stimulation (Fig. 3H). Reduced release of glycerol by Acsl1A−/− brown adipocytes could result from increased conversion to glycerol-3-P for lipid synthesis or because the glycerol was oxidized as an alternative substrate. After 3 h of isoproterenol exposure, both Acsl1A−/− and control differentiated brown adipocytes had similar ~33% reductions in [14C]oleate-labeled TAG (Fig. 3J), indicating that both lipolysis and FA re-esterification after lipolysis were unaffected by ACSL1 deficiency. [1-14C]Oleate incorporation into ASM was 90% lower in Acsl1A−/− brown adipocytes than in controls, consistent with the marked decrease in FA oxidation (Fig. 3K). Thus, in cold-exposed Acsl1A−/− BAT, the retention of lipid droplets probably reflects an influx of FA that cannot be oxidized without ACSL1 and are, therefore, incorporated into neutral lipid.
Thermogenic regulation of adrenergic and oxidative genes was normal in Acsl1A−/− mice
β-adrenergic signaling during cold exposure induces the expression of genes that activate mitochondrial biogenesis, heat production, and oxidative capacity. At 4°C both control and Acsl1A−/− mice upregulated mRNA levels of Ucp1 and PPAR-γ coactivator-1α (Pgc1α) 2- and 30- fold, respectively (Fig. 4A,B). These increases indicate that adrenergic-stimulated signaling was unimpaired in Acsl1A−/− BAT (Bachman et al., 2002).
Figure 4. Thermogenic regulation of genes was normal in Acsl1A−/− mice.
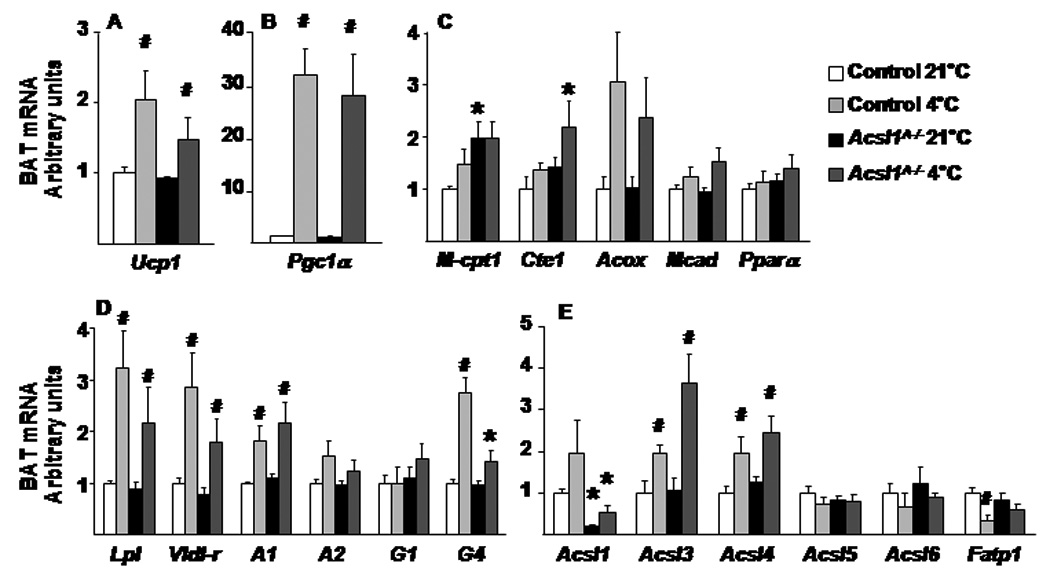
BAT mRNA levels were quantified by real-time PCR for genes of A,B) adrenergic regulation C) FA oxidation D) lipogenesis and E) acyl-CoA synthetases in BAT from male control and Acsl1A−/− mice without or with 4°C exposure, relative to cyclophilin A expression, n=6. Data represent mean ± SEM; *, P≤0.05 Acsl1A−/− versus control; # P≤0.05 21°C versus 4°C within genotype. A, Agpat; G, Gpat
Compared to controls, the mRNA expression of muscle-type carnitine palmitoyltransferase-1 (mCpt1) was significantly higher in the Acsl1A−/− BAT at 21°C, and cytosolic thioesterase-1 (Cte1) was significantly higher after cold exposure, suggesting that the lack of ACSL1 might increase cellular FA to activate PPARα-mediated transcription (Fig. 4C). However, the expression of other PPARα-regulated genes, medium-chain acyl-CoA dehydrogenase (Mcad), acyl-CoA oxidase (Aox), and Ppara, did not differ between the genotypes, suggesting that the lack of ACSL1 does not affect all PPARα target genes with or without 4 h of cold exposure.
Because Acsl1A−/− BAT retained lipid droplets after cold exposure, we measured genes that encode lipogenic and substrate-producing proteins that would normally increase during cold exposure. Acsl1A−/− BAT should have been able to lipolyze and/or endocytose circulating TAG-rich lipoproteins and to make use of these exogenous FA sources, because the lipogenic genes lipoprotein lipase (Lpl) and VLDL-receptor (Vldl-r) had similar expression patterns with or without cold exposure in both genotypes (Fig. 4D). Similarly, 1-acylglycerol-3-phosphate O-acyltransferase- 1 (Agpat1) increased in both genotypes, as did Gpat4, a major GPAT in BAT (Vergnes et al., 2006). These data suggest that the retained lipid droplets in Acsl1A−/− BAT were not caused by abnormal regulation of genes required for TAG synthesis.
BAT expression of Acsl3 and Acsl4 (but not Acsl1, Acsl5, or Acsl6) was upregulated in both genotypes in response to cold (Fig. 4E), and long-chain acyl-CoA content in cold exposed Acsl1A−/− BAT did not differ from controls (Fig. 6E). These data indicate that acyl-CoAs synthesized by other ACSL isoenzymes were unavailable for β-oxidation and that maintenance of normal body temperature requires acyl-CoAs synthesized by ACSL1.
Acyl-carnitines were reduced in Acsl1A−/− BAT
To gain further insight into the consequences of ACSL1 deficiency, we used mass spectrometry to profile a targeted array of intermediary metabolites. Long-chain acyl-carnitines accumulate when their production by mitochondrial CPT1 exceeds their flux through β-oxidation (Makowski et al., 2009). In wild type mice, cold exposure lowered major long-chain acyl-carnitine species and increased β-hydroxybutyryl-carnitine (4-OH), a strong marker of increased β-oxidation and ketone metabolism (Figs. 5A–C, S2B). This profile is consistent with activation of mitochondrial uncoupling and a shift in flux limitation from long-chain acyl-CoA dehydrogenase to short-chain β-hydroxy-acyl-CoA dehydrogenase. In contrast, under thermoneutral conditions, BAT concentrations of most long-chain acyl-carnitine species were markedly lower in Acsl1A−/− mice compared to controls. Taken together with our measures of FA oxidation, this finding suggests impaired delivery of long-chain acyl-CoAs to CPT1. During cold exposure, although Acsl1A−/− BAT had increases in several long- and short-chain acyl-carnitines, these remained substantially lower than in controls (Figs. 5A–C, S2B,C). BAT levels of acetyl-carnitine (C2), which is derived from acetyl-CoA, were similar between genotypes, suggesting that increased catabolism of glucose and/or amino acids in the Acsl1A−/− mice partially compensated for impaired FA oxidation.
Figure 5. Long-chain acyl-carnitines were reduced in Acsl1A−/− brown adipose tissue.
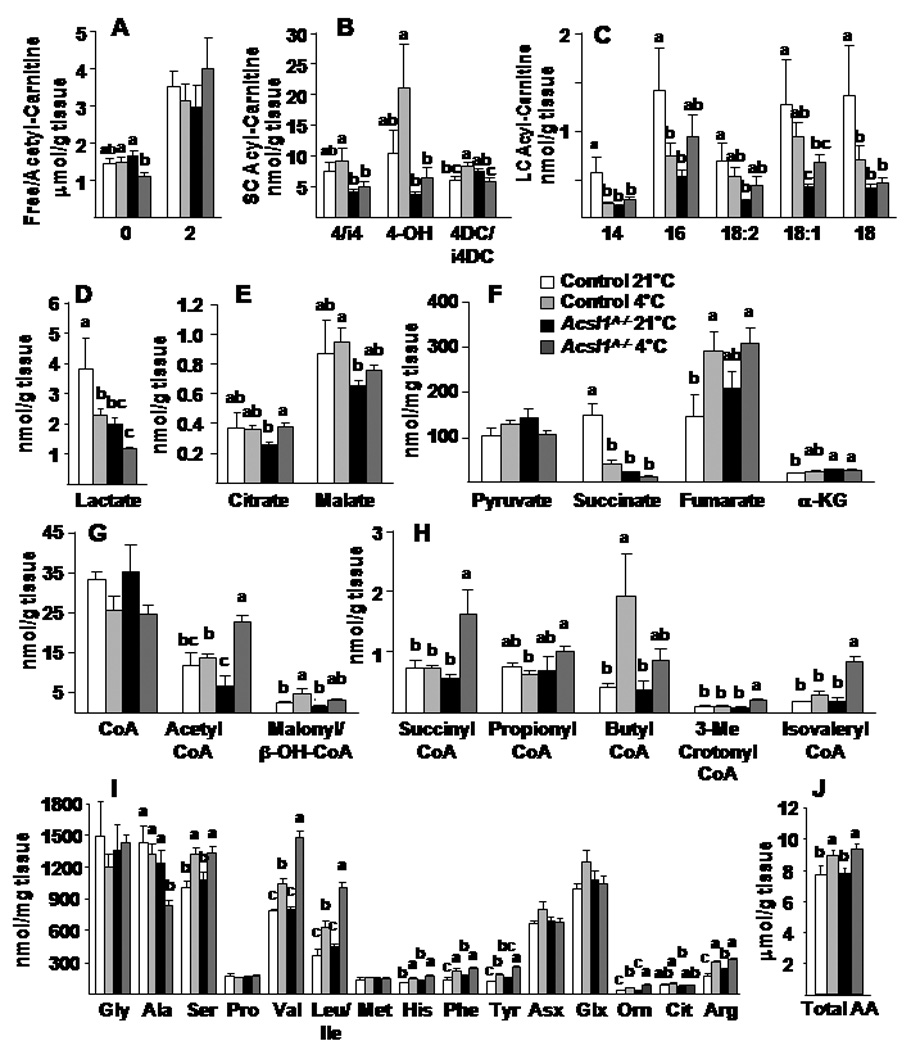
A,B,C) Free, acetyl-, and acyl-carnitine content in BAT with and without cold exposure at 4°C. D) Lactate and, E,F) Citric acid cycle intermediates, G,H) short-chain acyl-CoA, I) amino acids, and J) total amino acid content in BAT with and without 4°C exposure, n=7. DC, dicarboxylated acyl-carnitines; OH, hydroxylated acyl-carnitines; Asx, aspartate/asparagine; Gsx, glutamate/glutamine. Data represent mean ± SEM *, P≤0.05 versus control; # P≤0.05 21°C versus 4°C within genotype (n=6–10).
In control mice the BAT content of most organic acids was maintained during cold exposure, with the exceptions of lactate and succinate which declined (Fig. 5D–F). Compared to controls, lactate was lower in Acsl1A−/− mice, regardless of temperature, consistent with enhanced pyruvate oxidation and a shift in cellular redox state. The lower levels of succinate in Acsl1A−/− BAT may reflect disinhibition of succinate dehydrogenase (complex II). Notably, BAT levels of several amino acid-derived CoA species (propionyl-CoA, succinyl-CoA, 3-methylcrotonyl-CoA, isovaleryl-CoA) were elevated in Acsl1A−/− compared to control mice, principally during cold exposure (Fig. 5G,H). These elevations are consistent with a compensatory increase in protein catabolism and amino acid oxidation. Supporting this interpretation, the concentrations of branched chain and anaplerotic amino acids (valine, leucine/isoleucine, tyrosine) were elevated in the Acsl1A−/− mice, whereas alanine, the major gluconeogenic amino acid, was decreased (Fig. 5I,J).
Hypoglycemia and hyperlipidemia in Acsl1A−/− mice after adrenergic stimuli
Plasma glucose decreased in both genotypes during cold exposure, however, the plasma glucose concentration in Acsl1A−/− mice fell 58% more than in the controls (Fig. 6A). Similarly, after β-adrenergic stimulation with isoproterenol or CL-316243, Acsl1A−/− plasma glucose was 36% or 19% lower, respectively, than controls. The larger decrements in glucose in Acsl1A−/− mice after cold exposure and β-adrenergic agonists, but not after 24 h of fasting (Table S3), suggest enhanced glucose use by Acsl1A−/− BAT. Plasma glycerol increased similarly in both genotypes, both during cold exposure and after isoproterenol injection; however, in response to CL-316243, circulating glycerol increased 14% in controls, but decreased 15% in Acsl1A−/− mice, consistent with the reduced glycerol release by Acsl1A−/− primary brown adipocytes (Fig. 3H). Again, the glycerol decrease in BAT may reflect its increased use as an alternate oxidative substrate (Fig. 6B). FA concentrations after cold exposure or CL-316243 were 30% higher in Acsl1A−/− mice compared to controls (Fig. 6C); this increase in plasma FA induced by β-adrenergic stimuli, but not after 24 h of fasting, provides additional evidence for intact Acsl1A−/− lipolytic activity and is consistent with diminished use of FA for oxidation in BAT. In response to cold exposure, plasma TAG, another potential source of FA for BAT metabolism, decreased 41% in control mice, but was unchanged in Acsl1A−/− mice despite elevated Lpl mRNA (Figs. 6D, 4E), probably as a result of hepatic recycling of the elevated plasma FA into VLDL.
Discussion
After a long-chain FA enters a cell, its metabolic fate is determined by the metabolic demands of the cell, the availability of other energy substrates, PPARα- and PPARγ-mediated changes in enzymes of complex lipid synthesis and β-oxidation (Qi et al., 2000), and the relative content of intracellular AMP which controls AMP-activated kinase and, thus, CPT1 (Ruderman et al., 2003). Here we show that ACSL1 contributes critically to adipose FA metabolism by catalyzing the formation of acyl-CoAs that are used for β-oxidation. If all acyl-CoAs were metabolically equivalent, one would expect that the rates of acyl-CoA incorporation into all metabolic end products would be diminished equally and that adipose from Acsl1A−/− mice would have impairments in both FA oxidation and complex lipid synthesis. However, the role of adipose ACSL1 in catalyzing the key initial step in FA oxidation is supported by the normal rate of FA incorporation into complex lipids and the 50–90% reduction in the rate of FA oxidation in white and brown adipocytes and in BAT mitochondria from Acsl1A−/− mice, together with the inability of Acsl1A−/− BAT to oxidize FA for thermogenesis. Although the content of acyl-CoA in BAT was similar in control and Acsl1A−/− mice, these acyl-CoAs were unavailable for β-oxidation in the absence of ACSL1. The inability of the other ACSL isoenzymes to direct acyl-CoAs towards β-oxidation, especially during the physiological stress of acute cold exposure in BAT, indicates that ACSL1 has a distinct function in partitioning FA, and that the other ACSL isoenzymes in adipose tissue cannot compensate for its absence. Our data also suggest that the importance of FA oxidation in WAT has been underestimated.
Acute cold exposure stimulates norepinephrine release, which induces adipocyte TAG lipolysis via β-adrenergic receptors (Fig. 7). In BAT, lipolyzed FA activates UCP1 to dissipate the mitochondrial proton gradient; activation of UCP1 results in energy expended in the form of heat rather than ATP generation. Thermogenesis is maintained primarily through FA oxidation; glucose cannot maintain euthermia at 4°C in knockout mice deficient in short-chain-, medium-chain-, long-chain-, or very-long-chain acyl-CoA dehydrogenases (Guerra et al., 1998; Schuler et al., 2005; Tolwani et al., 2005). When Acsl1A−/− mice were exposed to a 4°C environment, their body temperature dropped below 30°C within 2 to 5 h, despite the normal increase in BAT catecholamine-regulated genes. Because no method exists to distinguish between FA and acyl-CoAs as activators of UCP1 (Strieleman et al., 1985; Strieleman and Shrago, 1985), the actual activator of UCP1 could be acyl-CoA generated by ACSL1. If this were true, Acsl1A−/− mice would be cold intolerant because UCP1 is not activated. However, impaired acyl-CoA oxidation was observed directly in BAT mitochondria, brown primary adipocytes, and BAT homogenates, and in isolated gonadal adipocytes which do not require UCP1 activation. In addition, compared to control mice, the in vivo rate of oxygen consumption of Acsl1A−/− mice at 21°C was 60% lower, and when stimulated by isoproterenol or by an adipose-specific β3 adrenergic receptor agonist, was more than 90% lower, indicating reduced flux through the electron transport chain and reduced adipose mitochondrial oxidation. Finally, Acsl1A−/− UCP1 uncoupling was activated by pyruvate and inhibited by GDP, indicating normally activated UCP1 (Fig. S4) (Shabalina et al., 2004).
Figure 7. BAT fatty acid metabolism in cold-exposed mice.
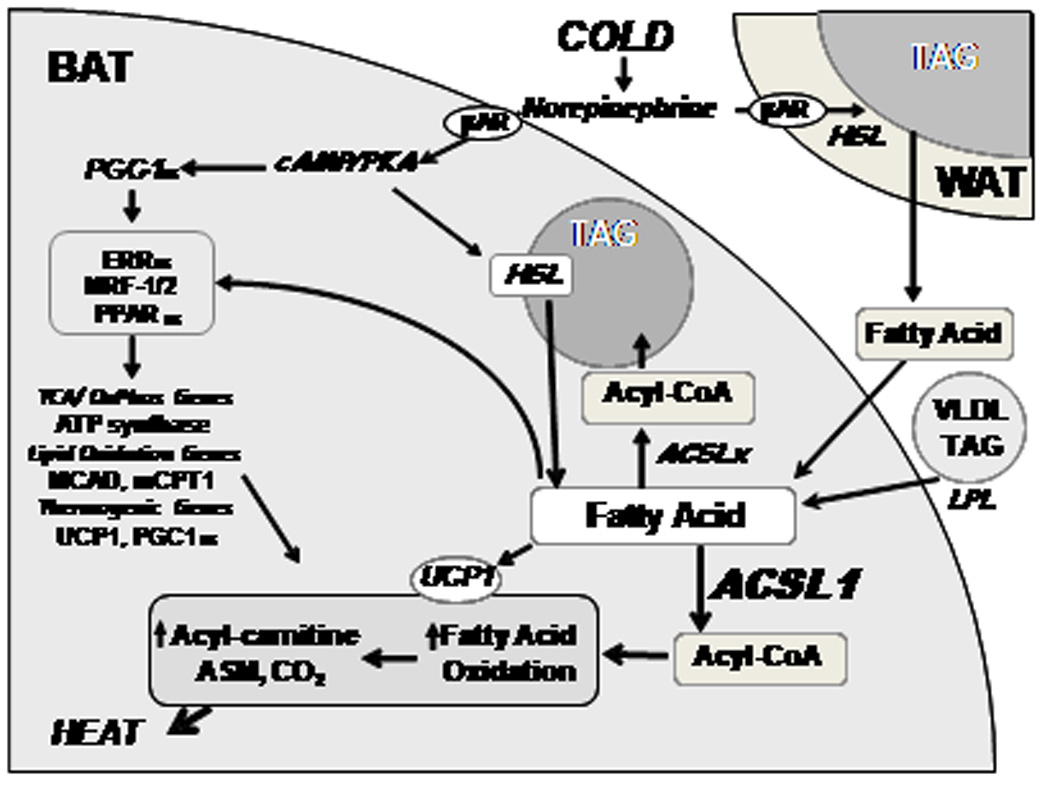
When norepinephrine activates β- adrenergic receptors, activated protein kinase A (PKA) induces PGC1α in brown adipocytes which, together with FA, activates transcription factors that upregulate genes required for mitochondrial oxidative and thermogenic functions. PKA also activates hormone sensitive lipase (HSL) which lipolyzes triacylglycerol (TAG) in white and brown adipose cells. FA released from WAT and from VLDL (via lipoprotein lipase [LPL]) enter brown adipocytes and are activated by ACSLs to form acyl-CoAs that can be esterified to form TAG or are oxidized. FA lipolyzed from TAG stores in brown adipocytes are activated by ACSL1, enter mitochondria, and are oxidized. FA also activate UCP1 which uncouples the mitochondrial proton gradient, producing heat.
The role that FA oxidation plays in WAT has not been extensively investigated (Deveaud et al., 2004). Apart from the normal demands of cellular metabolism, adipocytes require energy both to synthesize FA de novo and to activate FA to acyl-CoAs for TAG synthesis. Our data show that the loss of ACSL1 in WAT reduced FA oxidation 50% and resulted in a 30% increase in adipose mass. Although it is possible that ACSL1 is not the sole isoform in WAT that can activate the FA that is targeted towards β-oxidation, we attribute the increased WAT mass in Acsl1A−/− mice to diminished use of FA for energy generation within WAT itself, because mice that cannot use BAT for thermogenesis are not obese unless they are kept at a thermoneutral temperature (Lodhi and Semenkovich, 2009). The potential importance of FA β-oxidation in WAT is supported by the observation that mitochondrial proteins increase 20- to 30-fold in WAT after treatment with a PPARγ agonist or during 3T3-L1 adipocyte differentiation (Shi et al., 2008; Wilson-Fritch et al., 2003). The physiological stimuli that increase the rate of FA oxidation in WAT are unknown, but treating 3T3-L1 cells with an adipocyte-differentiation cocktail plus rosiglitazone increases the content of both mitochondrial ACSL1 protein and proteins required for FA oxidation, suggesting coordinated regulation (Wilson-Fritch et al., 2004).
The AMP generated by the ACSL1 reaction may activate AMP kinase in 3T3-L1 adipocytes stimulated with isoproterenol or forskolin (Gauthier et al., 2008; Lobo et al., 2009). However, AMPK phosphorylation was not altered in Acsl1A−/− WAT (Fig. 2F), and acute cold exposure does not activate AMPK in BAT (Mulligan et al., 2007). Thus AMPK activity does not mediate the cold intolerance of Acsl1A−/− mice. When RNAi was used to knockdown ACSL1 in 3T3-L1 adipocytes (Lobo et al., 2009), the cells increased FA efflux during lipolysis, suggesting that ACSL1 activates and re-esterifies FA that has been lipolyzed from intracellular TAG. Our results suggest a different function. If reesterification were impaired in Acsl1A−/− adipose, one would expect that an acute cold challenge would deplete TAG stores in BAT and that WAT would contain less TAG. However, Acsl1A−/− mice retained their BAT lipid stores at 4°C, suggesting that reesterification was not diminished. Further, in Acsl1A−/− primary brown adipocytes, loss of [14C]- labeled FA in TAG after lipolytic stimuli was similar to controls, and in white adipose explants, the release of FA and glycerol was similar between genotypes with or without lipolytic stimuli. Although the increased release of FA from brown adipocytes (Fig. 3I) could suggest diminished reesterification under basal conditions, both genotypes had similar losses of [14C]oleate that had been incorporated into TAG and in the release of FA under lipolytic conditions. These data confirm normal rates of lipolysis and, together with the cold intolerance and increased adipose mass of the Acsl1A−/− mice, are consistent with normal FA reesterification. In fact, a proteomics study demonstrates marked differences in the protein expression of mitochondria from 3T3-L1 adipocytes and those from primary mouse adipocytes, so it is not surprising to find functional differences in these two cell types (Forner et al., 2009; Lobo et al., 2009).
Based on the dramatic increase in Acsl1 mRNA and total ACSL activity in differentiating 3T3-L1 adipocytes (Coleman et al., 1978; Marszalek et al., 2004), we had predicted that adipocyte ACSL1 would catalyze the synthesis of acyl-CoAs destined for TAG synthesis. However, both mature and primary differentiated white adipocytes from Acsl1A−/− mice incorporated [14C]oleate into TAG normally, and Acsl1A−/− fat depots were larger than those of controls. Further, Acsl1A−/− mice became obese and insulin resistant when fed a high-fat diet. Although the lack of ACSL1 in white and brown adipose tissue reduced ACSL specific activity by 80%, the remaining ACSL activity, derived from other ACSL and FATP isoenzymes, was able to synthesize acyl-CoAs directed towards normal TAG biosynthesis. Thus, ACSL1 is not required for TAG synthesis in adipose tissue.
How does ACSL1 channel acyl-CoAs towards β-oxidation? In adipocytes, subcellular fractionation studies identified ACSL1 on the plasma membrane, microsomes and mitochondria (Gargiulo et al., 1999), on GLUT4 vesicles (Sleeman et al., 1998), and on lipid droplets (Brasaemle et al., 2004). In BAT and WAT ACSL1 protein has also been found in mitochondria fractions (this ms; (Forner et al., 2009)). However, even if ACSL1 were located solely on the mitochondrial outer membrane, its ability to channel its acyl-CoA product towards β-oxidation remains difficult to conceptualize. Long-chain acyl-CoAs are water-soluble, amphipathic molecules that can partition into membranes or bind to cytosolic carriers like acyl-CoA binding protein. Thus, acyl-CoAs should be able to move freely within the membrane monolayer at their site of synthesis, and to travel between different organelles. Direct physical interaction of ACSL1 with other proteins, such as CPT-1, is the most likely explanation for acyl-CoA channeling, but evidence for such interaction is lacking.
In summary, the loss of ACSL1 in adipose tissue produced mice with increased adiposity and severe cold intolerance. Adipocytes derived from Acsl1A−/− mice had normal rates of FA incorporation into TAG and normal loss of TAG stores after lipolysis, but had reduced rates of FA oxidation. Although deficient ACSL1 in liver, which expresses relatively more of the ACSL4 and ACSL5 isoenzymes, results in only a minor defect in FA oxidation (Li et al., 2009), we conclude that the function of ACSL1 in adipose tissue is to activate FA destined for β-oxidation.
Experimental Procedures
Animal Treatment
Acsl1A−/− and littermate Acsl1flox/flox control mice were housed in a pathogen-free barrier facility (12 h light/dark cycle) in accordance with the UNC IACUC, with free access to water and food (Prolab RMH 3000 SP76 chow). For the diet study, mice were fed a low-fat (10%) diet (D12450B, Research Diets) or a high-fat (45%, primarily from lard) diet (D12451) ad libitum from age 8 to 25 wk. Weights were measured weekly. Body fat mass was assessed by magnetic resonance imaging analysis (EchoMRI-100, Echo Medical Systems, LLC) in 5 mo old mice. For thermogenesis experiments, mice were fasted for 4 h and placed in a 4°C environment without food. Temperature was measured hourly with a rectal probe thermometer (Thermalert TH-5, physitemp). Mice were removed from the cold when core temperature dropped below 28°C or at 4 h. Rates of O2 consumption were determined in Oxymax chambers (Columbus Instruments) after mice were acclimated overnight. Data were collected from mice for 3 h after saline injection. After isoproterenol or CL-316243 was injected (10 mg/kg), O2 consumption was measured for an additional 3 h, and then plasma was collected in 5% 0.5 M EDTA. Plasma TAG, beta-hydroxybutyrate (Stanbio), cholesterol, FFA, glucose (Wako), and free and total glycerol (Sigma) were measured colorimetrically. Adiponectin was measured by ELISA (Linco). Oral glucose tolerance tests were performed by gavaging with glucose (2.5 mg/kg body weight) and measuring tail blood glucose at baseline, 15, 30, 60, and 120 minutes (One Touch Ultra glucometer, Lifescan, Inc).
Enzyme Assays
ACSL initial rates were measured with 50 µM [1-14C]palmitic acid, 10 mM ATP, and 0.25 mM CoA with total membrane fractions (0.5–4.0 µg) (Polokoff and Bell, 1978). GPAT was measured with 800 µM [3H]glycerol-3-phosphate and 82.5 µM palmitoyl-CoA (Nagle et al., 2008).
Adipocyte studies
Adipose explants were isolated and release of FFA and glycerol was measured (Warne et al., 2006). Epididymal adipocytes were isolated (Varma et al., 2008). Rates of CO2 and ASM production from [1-14C]oleate (Perkin Elmer) were measured in aliquots of the reaction mixture after incubating with 0.5 mL buffer containing 10 µM [1-14C]oleate for 1.5 h in a 10% adipocyte solution. The incubation chamber contained a center well filled with filter paper and was sealed with a rubber stopper. Carbon dioxide was trapped by adding 200 µl 70% perchloric acid to the reaction mixture and 300 µl of 1 M NaOH to the center well, and incubating the samples at RT for 45 min. The center well was then placed into scintillation fluid and counted. The acidified reaction mixture was incubated overnight at 4°C and centrifuged at 4,000 rpm for 30 min before aliquots of the supernatant were counted for [14C]-labeled ASM. For FA incorporation, a 10% adipocyte solution was incubated with 0.5 mL buffer containing 10 µM [1-14C]oleate and 90 µM unlabeled oleate for 2 h. Brown and white pre-adipocytes from control and Acsl1A−/− adipose tissue (Cao et al., 2001) were differentiated into adipocytes for 7–14 days and exposed to 500 µM oleate with 0.6 µCi [1-14C]oleate per 60 mm dish for 3 hours. Cells were then harvested or labeled media was removed, replaced with growth media for 21 hours and cells were harvested or 10 µM isoproterenol was added to media for an additional 3 hours followed by harvest. FA released into the media were measured colorimetrically (Wako) in unlabeled control plates. Total lipid was extracted from the cells into CHCl3 (Bligh and Dyer, 1959). The lipid extract was dried under N2, resuspended in CHCl3 and chromatographed with authentic lipid standards run in parallel on 0.25-mm silica gel G plates in hexane:ethyl ether:acetic acid (80:20:1; v/v). The 14C-labeled lipids were detected and quantified with a Bioscan 200 Image System.
Fatty acid oxidation in BAT
Freshly isolated BAT was minced and homogenized with a motor-driven Teflon pestle and glass mortar in ice-cold buffer A (100 mM KCl, 40 mM Tris·HCl, 10 mM Tris base, 5 mM MgCl2·6H2O, 1 mM EDTA, and 1 mM ATP, pH 7.4) at a 20-fold dilution (wt/vol) or mitochondria was isolated (Cannon and Nedergaard, 2008), resuspended in buffer A and the rate of [1-14C]palmitate, [1-14C]palmitoyl-CoA, or [1-14C]pyruvate, was measured (Noland et al., 2007). Isolated BAT mitochondria (20 µg protein) in buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 2 mM MgCl2, 2 mM HEPES, 1 mM EDTA, 0.1% BSA, 3 mM malate, and 1.3 µg/mL oligomycin) were loaded onto XF24 plates and rates of O2 consumption were measured according to the manufacturer’s instructions with a Seahorse X24F Analyzer (Seahorse Bioscience) during the consecutive addition at 4 min intervals of 5 mM pyruvate, 2 mM GDP, and 1 µM FCCP.
Highlights
Adipose knockout of long-chain acyl-CoA synthetase 1 decreases ACS activity 80%
Mice lacking ACSL1 are cold intolerant
ACSL1 activates fatty acids that are directed towards β-oxidation
Supplementary Material
Acknowledgements
We thank Dr. S.K. Fried for her advice about fat cell isolations and Drs. A.S. Greenberg and J.W. Perfeld for their advice about adipocyte histology. This work was supported by NIH grants DK59935 (RAC), a grant from the American Diabetes Association (DMM), a postdoctoral fellowship (LOL) and a predoctoral fellowship (JME) from the American Heart Association-Mid-Atlantic Region, a pre-doctoral training grant HL069768 (JME), P30 DK034987, and P30 DK056350.
Abbreviations
- ACSL
long-chain acyl-CoA synthetase
- ASM
acid soluble metabolites
- BAT
brown adipose tissue
- CO2
carbon dioxide
- DAG
diacylglycerol
- FA
fatty acids
- PL
phospholipids
- TAG
triacylglycerol
- UCP1
uncoupling protein one
- WAT
white adipose tissue
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information
Supplement includes 4 figures, 4 tables, and additional experimental procedures.
References
- An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, Koves TR, Stevens R, Millington DS, Newgard CB. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10:268–274. doi: 10.1038/nm995. [DOI] [PubMed] [Google Scholar]
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–845. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Studies of thermogenesis and mitochondrial function in adipose tissues. Methods Mol Biol. 2008;456:109–121. doi: 10.1007/978-1-59745-245-8_8. [DOI] [PubMed] [Google Scholar]
- Cao W, Medvedev V, Daniel W, Collins S. Beta -adrenergic activation of p38 MAP kinase in adipocytes. cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. 2001;276:27077–27082. doi: 10.1074/jbc.M101049200. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Schubert W, Brasaemle DL, Scherer PE, Lisanti MP. Caveolin-1 expression is essential for proper nonshivering thermogenesis in brown adipose tissue. Diabetes. 2005;54:679–686. doi: 10.2337/diabetes.54.3.679. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Bell RM. Submicrosomal localization of phosphatidylcholine, phosphatidylethanolamine, and triacylglycerol biosynthetic enzymes. J Cell Biol. 1978;76:245–253. doi: 10.1083/jcb.76.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Reed BC, Mackall JC, Student AK, Lane MD, Bell RM. Selective changes in microsomal enzymes of triacylglycerol, phosphatidylcholine, and phosphatidylethanolamine biosynthesis during differentiation of 3T3-L1 preadipocytes. J Biol Chem. 1978;253:7256–7261. [PubMed] [Google Scholar]
- Deveaud C, Beauvoit B, Salin B, Schaeffer J, Rigoulet M. Regional differences in oxidative capacity of rat white adipose tissue are linked to the mitochondrial content of mature adipocytes. Mol Cell Biochem. 2004;267:157–166. doi: 10.1023/b:mcbi.0000049374.52989.9b. [DOI] [PubMed] [Google Scholar]
- Forner F, Kumar C, Luber CA, Fromme T, Klingenspor M, Mann M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009;10:324–335. doi: 10.1016/j.cmet.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Gargiulo CE, Stuhlsatz-Krouper SM, Schaffer JE. Localization of adipocyte long-chain fatty acyl-CA synthetase at the plasma membrane. J Lipid Res. 1999;40:881–892. [PubMed] [Google Scholar]
- Gauthier MS, Miyoshi H, Souza SC, Cacicedo JM, Saha AK, Greenberg AS, Ruderman NB. AMP-activated protein kinase is activated as a consequence of lipolysis in the adipocyte: potential mechanism and physiological relevance. J Biol Chem. 2008;283:16514–16524. doi: 10.1074/jbc.M708177200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Koza RA, Walsh K, Kurtz DM, Wood PA, Kozak LP. Abnormal nonshivering thermogenesis in mice with inherited defects of fatty acid oxidation. J Clin Invest. 1998;102:1724–1731. doi: 10.1172/JCI4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haman F. Shivering in the cold: from mechanisms of fuel selection to survival. J Appl Physiol. 2006;100:1702–1708. doi: 10.1152/japplphysiol.01088.2005. [DOI] [PubMed] [Google Scholar]
- Hostetler HA, Petrescu AD, Kier AB, Schroeder F. Peroxisome proliferator-activated receptor alpha interacts with high affinity and is conformationally responsive to endogenous ligands. J Biol Chem. 2005;280:18667–18682. doi: 10.1074/jbc.M412062200. [DOI] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Li LO, Ellis JM, Paich HE, Wang S, Gong N, Altshuller G, Thresher RJ, Koves TR, Watkins SM, Muoio DM, et al. Liver-specific loss of long-chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and β-oxidation, and alters phospholipid fatty acid composition. J Biol Chem. 2009 doi: 10.1074/jbc.M109.022467. under revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LO, Mashek DG, An J, Doughman SD, Newgard CB, Coleman RA. Overexpression of rat long chain acyl-CoA synthetase 1 alters fatty acid metabolism in rat primary hepatocytes. J Biol Chem. 2006;281:37246–37255. doi: 10.1074/jbc.M604427200. [DOI] [PubMed] [Google Scholar]
- Lobo S, Wiczer BM, Bernlohr DA. Functional analysis of long-chain acyl-coa synthetase 1 in 3T3-L1 adipocytes. J Biol Chem. 2009 doi: 10.1074/jbc.M109.017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi IJ, Semenkovich CF. Why we should put clothes on mice. Cell Metab. 2009;9:111–112. doi: 10.1016/j.cmet.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404:652–660. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- Makowski L, Noland RC, Koves TR, Xing W, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Muoio DM. Metabolic profiling of PPARalpha−/− mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. Faseb J. 2009;23:586–604. doi: 10.1096/fj.08-119420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaerts GP, Van Veldhoven P, Van Broekhoven A, Vandebroek G, Debeer LJ. Evidence that peroxisomal acyl-CoA synthetase is located at the cytoplasmic side of the peroxisomal membrane. Biochem J. 1982;204:7–23. doi: 10.1042/bj2040017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Kitidis C, Dararutana A, Lodish HF. Acyl CoA synthetase 2 (ACS2) over-expression enhances fatty acid internalization and neurite outgrowth. J Biol Chem. 2004;279:23882–23891. doi: 10.1074/jbc.M313460200. [DOI] [PubMed] [Google Scholar]
- Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARα and PPARγ activators. J Biol Chem. 1997;272:28210–28217. doi: 10.1074/jbc.272.45.28210. [DOI] [PubMed] [Google Scholar]
- Mashek DG, Coleman RA. Cellular fatty acid uptake: the contribution of metabolism. Curr Opin Lipidol. 2006;17:274–278. doi: 10.1097/01.mol.0000226119.20307.2b. [DOI] [PubMed] [Google Scholar]
- Mashek DG, Li LO, Coleman RA. Rat long chain acyl-CoA synthetase mRNA, protein and activity vary in tissue distribution and in response to diet. J Lipid Res. 2006a;47:2004–2010. doi: 10.1194/jlr.M600150-JLR200. [DOI] [PubMed] [Google Scholar]
- Mashek DG, McKenzie MA, Van Horn CG, Coleman RA. Rat long chain acyl- CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle - RH7777 cells. J Biol Chem. 2006b;281:945–950. doi: 10.1074/jbc.M507646200. [DOI] [PubMed] [Google Scholar]
- Mulligan JD, Gonzalez AA, Stewart AM, Carey HV, Saupe KW. Upregulation of AMPK during cold exposure occurs via distinct mechanisms in brown and white adipose tissue of the mouse. J Physiol. 2007;580:677–684. doi: 10.1113/jphysiol.2007.128652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle CA, Verges L, Wang S, deJong H, Wang S, Lewin TM, Reue K, Coleman RA. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4 as the enzyme deficient in Agpat6−/− mice. J Lipid Res. 2008;49:823–831. doi: 10.1194/jlr.M700592-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland RC, Woodlief TL, Whitfield BR, Manning SM, Evans JR, Dudek RW, Lust RM, Cortright RN. Peroxisomal-mitochondrial oxidation in a rodent model of obesity-associated insulin resistance. Am J Physiol Endocrinol Metab. 2007;293:E986–E1001. doi: 10.1152/ajpendo.00399.2006. [DOI] [PubMed] [Google Scholar]
- Oikawa E, Iijima H, Suzuki T, Sasano H, Sato H, Kamataki A, Nagura H, Kang M-J, Fujino T, Suzuki H, Yamamoto TT. A novel acyl-CoA synthetase, ACS5, expressed in intestinal epithelial cells and proliferating preadipocytes. J Biochem. 1998;124:679–685. doi: 10.1093/oxfordjournals.jbchem.a022165. [DOI] [PubMed] [Google Scholar]
- Polokoff MA, Bell RM. Limited palmitoyl-CoA penetration into microsomal vesicles as evidenced by a highly latent ethanol acyltransferase activity. J Biol Chem. 1978;253:7173–7178. [PubMed] [Google Scholar]
- Qi C, Zhu Y, Reddy JK. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem Biophys. 2000 Spring;32:187–204. doi: 10.1385/cbb:32:1-3:187. [DOI] [PubMed] [Google Scholar]
- Richards MR, Harp JD, Ory DS, Schaffer JE. Fatty acid transport protein 1 and long-chain acyl coenzyme A synthetase 1 interact in adipocytes. J Lipid Res. 2006;47:665–672. doi: 10.1194/jlr.M500514-JLR200. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171. doi: 10.1210/en.2003-0849. [DOI] [PubMed] [Google Scholar]
- Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- Schuler AM, Gower BA, Matern D, Rinaldo P, Vockley J, Wood PA. Synergistic heterozygosity in mice with inherited enzyme deficiencies of mitochondrial fatty acid beta-oxidation. Mol Genet Metab. 2005;85:7–11. doi: 10.1016/j.ymgme.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. 2004;279:38236–38248. doi: 10.1074/jbc.M402375200. [DOI] [PubMed] [Google Scholar]
- Shi X, Burkart A, Nicoloro SM, Czech MP, Straubhaar J, Corvera S. Paradoxical effect of mitochondrial respiratory chain impairment on insulin signaling and glucose transport in adipose cells. J Biol Chem. 2008;283:30658–30667. doi: 10.1074/jbc.M800510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman MW, Donegan NP, Heller-Harrison R, Lane WS, Czech MP. Association of acyl-CoA synthetase-1 with GLUT4-containing vesicles. J Biol Chem. 1998;273:3132–3135. doi: 10.1074/jbc.273.6.3132. [DOI] [PubMed] [Google Scholar]
- Souza SC, Muliro KV, Liscum L, Lien P, Yamamoto MT, Schaffer JE, Dallal GE, Wang X, Kraemer FB, Obin M, Greenberg AS. Modulation of hormone-sensitive lipase and protein kinase A-mediated lipolysis by perilipin A in an adenoviral reconstituted system. J Biol Chem. 2002;277:8267–8272. doi: 10.1074/jbc.M108329200. [DOI] [PubMed] [Google Scholar]
- Stremmel W, Pohl L, Ring A, Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001;36:981–989. doi: 10.1007/s11745-001-0809-2. [DOI] [PubMed] [Google Scholar]
- Strieleman PJ, Schalinske KL, Shrago E. Fatty acid activation of the reconstituted brown adipose tissue mitochondria uncoupling protein. J Biol Chem. 1985;260:13402–13405. [PubMed] [Google Scholar]
- Strieleman PJ, Shrago E. Specific interaction of fatty acyl-CoA esters with brown adipose tissue mitochondria. Am J Physiol. 1985;248:E699–E705. doi: 10.1152/ajpendo.1985.248.6.E699. [DOI] [PubMed] [Google Scholar]
- Tolwani R, Hamm D, Tian L, Sharer J, Vockley J, Rinaldo P, Matern D, Schoeb T, Wood P. Medium-chain acyl-CoA dehydrogenase deficiency in gene-targeted mice. PLOS Genet. 2005;1:e23. doi: 10.1371/journal.pgen.0010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma V, Yao-Borengasser A, Bodles AM, Rasouli N, Phanavanh B, Nolen GT, Kern EM, Nagarajan R, Spencer HJ, 3rd, Lee MJ, et al. Thrombospondin-1 is an adipokine associated with obesity, adipose inflammation, and insulin resistance. Diabetes. 2008;57:432–439. doi: 10.2337/db07-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes L, Beigneux AP, Davis RG, Watkins SM, Young SG, Reue K. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J Lipid Res. 2006;47:745–754. doi: 10.1194/jlr.M500553-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne JP, John CD, Christian HC, Morris JF, Flower RJ, Sugden D, Solito E, Gillies GE, Buckingham JC. Gene deletion reveals roles for annexin A1 in the regulation of lipolysis and IL-6 release in epididymal adipose tissue. Am J Physiol Endocrinol Metab. 2006;291:E1264–E1273. doi: 10.1152/ajpendo.00655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SM, Reifsnyder PR, Pan HJ, German JB, Leiter EH. Lipid metabolome-wide effects of the PPARgamma agonist rosiglitazone. J Lipid Res. 2002;43:1809–1817. doi: 10.1194/jlr.m200169-jlr200. [DOI] [PubMed] [Google Scholar]
- Wilson-Fritch L, Burkart A, Bell G, Mendelson K, Leszyk J, Nicoloro S, Czech M, Corvera S. Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol Cell Biol. 2003;23:1085–1094. doi: 10.1128/MCB.23.3.1085-1094.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Sakane N, Wakabayashi Y, Umekawa T, Kondo M. Anti-obesity effect of CL 316,243, a highly specific beta 3-adrenoceptor agonist, in mice with monosodium-L-glutamate-induced obesity. Eur J Endocrinol. 1994;131:97–102. doi: 10.1530/eje.0.1310097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.