

Abstract
The HIV replication cycle offers multiple targets for chemotherapeutic intervention, including the viral exterior envelope glycoprotein, gp120; viral co-receptors CXCR4 and CCR5; transmembrane glycoprotein, gp41; integrase; reverse transcriptase; protease and so on. Most currently used anti-HIV drugs are reverse transcriptase inhibitors or protease inhibitors. The expanding application of simulation to drug design combined with experimental techniques have developed a large amount of novel inhibitors that interact specifically with targets besides transcriptase and protease. This review presents details of the anti-HIV inhibitors discovered with computer-aided approaches and provides an overview of the recent five-year achievements in the treatment of HIV infection and the application of computational methods to current drug design.
Introduction
Since AIDS was recognized 27 years ago, 25 million people have died of HIV-related causes. On a global scale, although the HIV epidemic has stabilized since 2000, unacceptably high levels of new HIV infections and AIDS deaths still occur each year. In 2007, there were an estimated 33 million (30–36 million) people living with HIV and 2.0 million (1.8–2.3 million) people died because of AIDS, compared with an estimated 1.7 million (1.5–2.3 million) in 2001 (http://www.unaids.org). After more than 20 years of research, HIV remains a difficult target for a vaccine [1]; thus, the AIDS treatment continues to focus on the search for chemical anti-HIV agents. Most currently approved anti-HIV drugs belong to nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs) or protease inhibitors (PIs). Highly active antiretroviral therapy (HAART), which combines several such drugs (typically three or four), has dramatically improved patients’ lives [2]. The therapeutic effects are limited, however, by adverse effects and toxicities caused by long-term use and the emergence of drug resistance [3]. The multiple steps of the HIV replication cycle present novel therapeutic targets other than the viral enzyme reverse transcriptase (RT) and protease (PT) for drug development (Fig. 1). Continued efforts have been made to discover new inhibitors that target not only RT and PT but also other viral targets – achievements that have been reviewed comprehensively in the literature [2, 5].
FIGURE 1.

The reproductive cycle of HIV. (a) Attachment. HIV attaches to CD4 and a chemokine receptor on the surface of a T cell. (b) Fusion. The virus fuses with the cell membrane and releases the virion core into the host cell. (c) Reverse transcription. The HIV enzyme called reverse transcriptase converts the single-stranded viral RNA to double-stranded viral DNA. (d) Integration. The viral DNA is integrated into cellular DNA by the HIV enzyme integrase. (e) Transcription. The virus uses the host enzyme RNA polymerase to create copies of the HIV genomic material and messenger RNA (mRNA). The mRNA is then used to produce long chains of viral proteins. (f) Regulator protein. These are essential for the HIV viral cycle because they dramatically increase HIV gene expression. (g) Assembly. The HIV enzyme protease hydrolyzes the long chains of viral proteins into functional small proteins. New virions are then assembled with the small viral proteins an d RNA. (h) Budding. The newly assembled virions use the cellular envelope as cover and bud off from the host cell.
Computer-aided drug design (CADD) is a rapidly evolving field that leverages new data and methods to provide approaches for tackling the needs of drug discovery. The applications of CADD now span the whole drug discovery process and contribute significantly to improve the low overall productivity of the pharmaceutical industry. By using CADD or a combination of experiments and computational approaches, a great many new compounds have been discovered that are able to inhibit HIV replication by interacting with specified target(s). The use of computational methods has not only enabled more efficient drug discovery and lead optimization but also provided insights into target–drug interactions. As the broad set of CADD approaches continues to develop, with innovative new methods continually appearing, the impact on drug discovery will undoubtedly continue to grow. In this review, we take a look at the novel anti-HIV inhibitors discovered by computer-aided approaches in the past few years. The inhibitors to be discussed are grouped in different categories according to the target(s) with which they interact.
Overview of CADD
Current drug discovery is becoming increasingly challenging, inefficient and costly. A main reason for this is that the applied science required for drug development is not able to keep pace with the tremendous advances in basic science. The estimated average cost to bring a new drug to the market is approximately US$ 802 million, according to a recent report on the price of drug development [6]. The traditional drug development strategy widely adopted by industry is the use of combinatorial chemistry and high-throughput screening, which is costly and unable to address the specific needs of many biological systems. CADD, which emerged in the 1960s, takes advantage of available scientific knowledge to guide drug discovery and has now become one of the core technologies in the drug industry. With the assistance of CADD technologies, the cost of drug development could be reduced by up to 50%. In addition, CADD makes it possible to predict the absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of potential drugs, which is a main concern for further medicinal development. According to the methodologies employed, CADD approaches fall into several natural categories: structure-based drug design (SBDD), ligand-based drug design (LBDD) and other approaches that commonly combine SBDD and LBDD.
Entry inhibitors
HIV entry into host cells is a multistep process that is yet to be fully elucidated. Advances in the research of molecular mechanism involved in the entry process have revealed at least three steps: (i) specific attachment of the viral surface glycoprotein (gp120) to the T-cell receptor CD4 on the cellular membrane [7]. This induces a conformational change in gp120 that opens up a high-affinity binding site located within the third variable loop (V3) and surrounding surfaces for the chemokine co-receptors (primarily CCR5 and CXCR4) [8]; (ii) binding of gp120 to the chemokine co-receptors. This results in further conformational rearrangements of gp120 that expose the transmembrane glycoprotein gp41 and (iii) the heptad repeat (HR) regions of the three subunits of gp41, HR1 and HR2, fold into a six-helical bundle, which leads to the fusion of the viral and cellular membranes [9]. The proteins involved in the entry process have become attractive targets for drug design. Several peptide/non-peptide inhibitors have been discovered to be able to block the HIV entry process.
Attachment inhibitors
The crystal structure of gp120 core bound to CD4 reveals specific targets for developing anti-HIV drugs. Computational methods have been used frequently to investigate the interaction of gp120 with its inhibitors [10]. These studies provide useful information for lead optimization and novel drug design. However, developing robust gp120 inhibitors remains a challenge for CADD.
CD4 plays an important part in the binding of MHC class II proteins to T-cell receptors [11]. Developing CD4 ligands, therefore, might interfere with the human immune system because the binding sites for gp120 binding to CD4 overlap with those for MHC class II proteins [12]. The contact area of the gp120–CD4 interaction is much bigger than that of CD4–MHC class II protein [13], however, and might serve as a potential drug target. Neffe and Meyer [14] modified a known CD4 binding peptide, NMWQKVGTPL (1), and attained a compound (2) that showed much better pharmacological properties (170-fold stronger binding to CD4, a four to five times higher proteolytic stability and a lower molecular weight than the lead peptide). Inspired by this result, Neffe et al. [15] designed a class of 85 peptidomimetics by changing the residues of the lead compound. The 85 compounds were docked to CD4 using the FlexiDock module in SYBYL [16] The docked model of compound I was used as starting position for docking the other ligands. By analyzing the docking results and evaluating binding affinities, 11 compounds were selected for synthesis, seven of which showed improved binding abilities compared with the lead in experiments. The most potent compound (3) has an activity of KD = 6 μM. In a further study comparing molecular docking and experimental data, Neffe et al discussed structure–activity relationships of these compounds [17]. The carboxy terminal subunit was observed to be essential for binding. Optimizations at the carboxy terminus of these peptidomimetic CD4 ligands result in much higher binding affinities than the lead peptide NMWQKVGTPL.
CCR5 inhibitors
CCR5 and CXCR4 are chemokine receptors that belong to the superfamily of human G-protein-coupled receptors (GPCRs). GRCRs regulate many physiological functions and are the targets for more than 30% of all marketed therapeutics [18]. As a co-receptor for HIV-1 and many other viruses, CCR5 enables these viruses to enter into the cells. Research has been published on different roles of CCR5 in diseases such as rheumatoid arthritis, multiple sclerosis, transplant rejection and inflammatory bowel disease. Studies on developing CCR5 antagonists could benefit humans in a wide range.
Quantitative structure–activity relationship (QSAR) studies have been reported on different classes of CCR5 antagonists, attempting to attain structural and physicochemical information for developing new drugs. Roy and Leonard [19] carried out QSAR analysis on a series of 3-(4-benzylpiperidin-1-yl)-N-phenylpropylamine derivatives (4) using the linear free-energy-related (LFER) model of Hansch. The binding affinity data of these antagonists were published by Imamura et al. The models were built in Cerius 2 4.8 and further analyzed with more specific techniques, including molecular shape analysis (MSA), receptor surface analysis and molecular field analysis. The MSA-derived models showed best statistical qualities for both the training sets and the test sets. Aher et al. [20] reported their 3D-QSAR study on analog 4 using comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) methods realized in SYBYL7.1. More recently, another 3D-QSAR study on this series of antagonists was done by Dessalew [21], using an integrated analysis package, TSAR3.3. Afantitis et al. [22] applied multiple linear regression analysis on analog 4 and developed a linear QSAR model. The elimination selection stepwise regression method was employed for the selection of molecular descriptors. The produced model was finally used to virtually screen a group of new derivatives of this class. Several guanidine derivatives were filtered out with significantly improved predicted activities. However, synthesis and biological assay must be applied to validate the result.
In a later study by Leonard and Roy [23], a QSAR study using the LFER model was presented on a set of substituted 1-(3,3-diphenylpropyl)-piperidinyl amides and ureas (5). Zhuo et al. [24] performed CoMFA and CoMSIA on 1,3,4-trisubstituted pyrrolidine-based CCR5 antagonists (6). 3D features (i.e. steric, electrostatic, hydrophobic and hydrogen bonding) for the binding of CCR5 antagonists to the target were identified in the above studies. Specific substitutional requirements for CCR5 antagonists were also attained, which provides helpful information for lead optimization and new drug discovery.
1-Amino-2-phenyl-4-(piperidin-1-yl)-butane analogs (7) were reported by Merck Research Laboratories as CCR5 antagonists [25] in 2001. Xu et al. [26] adopted a strategy that combines CoMFA and CoMSIA, molecular docking and molecular dynamic (MD) simulation to investigate detailed interactions of this class of antagonists with CCR5 co-receptor. The structure of CCR5 used in this study was constructed by homology modeling using the crystal structure of bovine rhodopsin (PDB entry 1F88) as a template. Bovine rhodopsin is the first solved structure of the GPCR family, and it has been used as a template for modeling many other GPCR drug targets. Automated Docking of the antagonists to the binding site in CCR5 modulated each ligand to the binding conformation and orientation. Docking results were further validated by MD simulation and binding energy calculation. 3D-QSAR models of the ligands were built based on an alignment of the ligands’ binding conformation. The CoMFA and CoMSIA models attained in this way show excellent predictabilities and enable an understanding of the ligand–protein interactions.
Also based on the bovine rhodopsin template, Kellenberger et al. [27] modeled the 3D structure of CCR5 and modulated an antagonist-binding site by using the data of known CCR5 antagonists. The receptor structure was further customized for structure-based virtual screening. Surflex and GOLD were used in parallel as docking tools to screen a set of 44,524 compounds. A list of 77 hits was returned, and the most potential binders were picked out to construct database queries for a follow-up ligand-based virtual screening. 83 commercial available compounds were selected from the ligand-based screening hits list. The most potent compound (8) could efficiently promote receptor internalization, thus protecting the cell against HIV-1 infection.
CXCR4 inhibitors
Bicyclams are the first discovered low-molecular-weight compounds with a specific interaction with CXCR4 [28]. The most potent bicyclam was AMD3100 with an IC50 of 1–10 ng ml−1. It was withdrawn from clinical trials, however, because of its poor oral absorption and toxicity. Pettersson et al. [29] designed a combinatorial library of non-cyclam polynitrogenated compounds that preserve the main structural features of AMD3100: at least two nitrogen atoms on each side of the p-phenylene moiety with similar distances as the nitrogen atoms presented in cyclam. The library was then screened using the Program for Rational Analysis of Libraries in silico [30] through sequential criteria, including 2D (physicochemical, and topological based on information theory) and 3D (potential energy, surface, shape and volume) descriptors. 19 compounds synthesized were tested for anti-HIV activity and cytotoxicity. The most active compound (9) has an EC 50 value of 0.008 mg ml−1 and a CC 50 of >25 mg ml−1 Molecular docking of AMD3100 and compound 9 to the binding sites in CXCR4 shows that compound 9 interacts with CXCR4 in a similar way to AMD3100. Two main electrostatic interactions were identified between two positively charged nitrogen atoms in compound 9 and the negatively charged Asp262 and Glu288 residues of CXCR4, whereas in the case of AMD3100, three acidic residues of CXCR4 – Asp171, Asp262 and Glu288 – served as the main electrostatic interaction points for positively charged AMD3100 bicyclam rings binding. Finally, time-of-drug-addition experiments on the four most active compounds developed in this study confirmed that these compounds were selective ligands for CXCR4.
Lapidot and Borkow designed and synthesized a set of novel peptidomimetic substances, aminoglycoside-arginine conjugates (AACs) [31] and poly-arginine aminoglycoside conjugates (pAACs) [32], which showed considerable anti-HIV activities. AACs and pAACs are proposed to be able to interfere with both CD4–gp120 binding and gp120–CXCR4 binding. To investigate the inhibiting mechanism of these inhibitors, they performed molecular docking on the two most potent compounds, NeoR6 (AAC) and Neo-r9 (pAAC) [33] (Fig. 2). Homology models of CXCR4 and unliganded HIV-1IIIB gp120 were used as receptors. The binding sites were recognized through a geometric-electrostatic docking full scan by MolFit [34]. Flexible ligand docking was then carried out using AutoDock and the binding complexes were imported to the Discover3 module in Insight II [35] for final refinement. Binding free energies were calculated by the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) method implemented in Amber9. The results indicate that the gp120 binding site on CXCR4 is a more probable target for these two compounds. Their interference with the CD4–gp120 binding might also contribute to the inhibiting ability, as suggested by the docking and binding free-energy calculation results. On the basis of the findings in this study, Berchanski and Lapidot [36] designed novel poly-arginine–neomycin–poly-arginine conjugates (PA–Neo–PAs), which they predict can block gp120–CXCR4 binding, as well as gp120–CD4 binding like Neo-r9 and NeoR6. The same molecular modeling strategy was adopted to explore the potential interactions of PA–Neo–PAs and CXCR4/gp120. As expected, PA–Neo–PAs bind satisfactorily with both the receptors, although the complexes with CXCR4 were more energetically favorable. The locations of two negatively charged patches on CXCR4 were observed to be more favorable binding sites for the highly positively charged ligands. This might contribute mainly to the preference of AACs and/or pAACs and PA–Neo–PAs to bind CXCR4.
FIGURE 2.
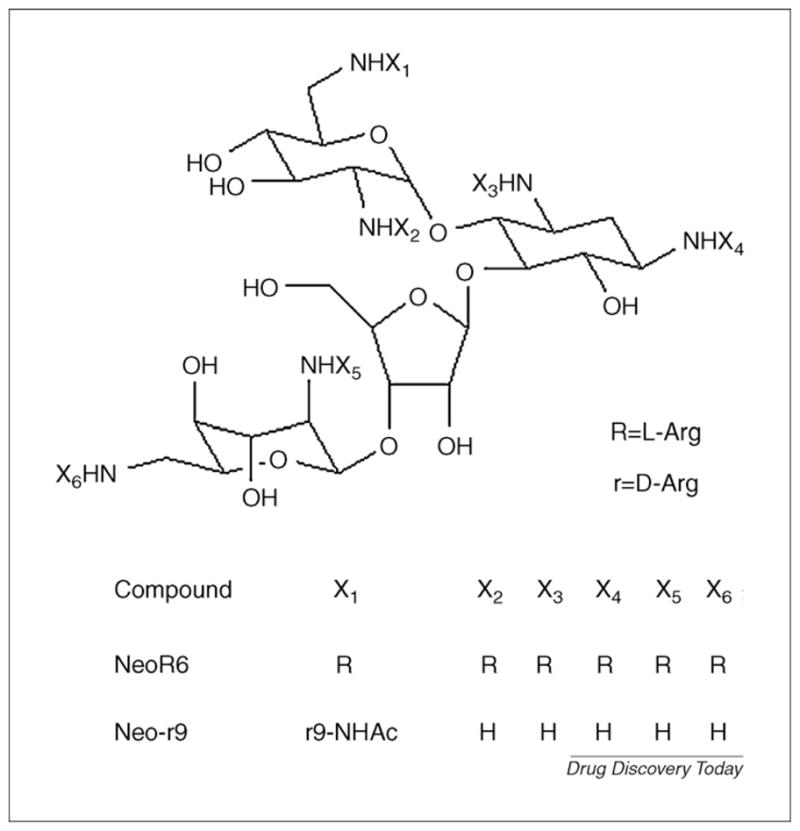
Structures of NeoR6 and Neo-r9.
Fusion inhibitors
During viral entry, HIV-1 gp41 plays a key part in the virus–cell fusion process. After gp120 binds to chemokine co-receptors, gp41 adopts a transient conformation known as ‘prehairpin intermediate’ in which a highly conserved therapeutic target, named the N-helix trimer, is exposed. In each of the grooves on the surface of the N-helix trimer, there is a hydrophobic pocket that accommodates conserved hydrophobic residues in the gp41 C-terminal heptad repeat regions (C-helix) to form a stable six-helical bundle. The formation of the six-helical bundle is a crucial step in the fusion process and has gained a lot of interest for developing fusion inhibitors.
T-20 (Enfuvirtide, approved by the FDA in 2003) is a synthetic peptide of 36 amino acids that is based on the sequence of the C-helix of gp41 and has been used to treat HIV/AIDS patients who have failed to respond to RTIs and PIs. T-20 has been proved to be able to interact with the gp41 N-helix and block the six-helical bundle formation, thereby inhibiting membrane fusion [9]. Five-helix is another fusion inhibitor [37], which consists of five helical sequences. Three of the sequences form a coiled core structure equivalent to the gp41 N-helix trimer. The other two sequences are equivalent to the gp41 C-helix and bind against the coiled core. Five-helix is able to bind with one of the gp41 C-helices to form a stable six-helical structure, thus interfering with the formation of the gp41 six-helical bundle. The crystal structure of gp41 ectodomain core region has been resolved by Chan et al. [9] (PDB code: 1AIK). Unfortunately, the crystal structure of the gp41–inhibitor complex or the complete gp41 protein has yet to become available for drug design. Our laboratory has explored the interactions between gp41 and its inhibitors [38–40], aiming to reveal the mechanism and key factors for drug binding.
Green and Tidor [41] described their computational study on electrostatic interactions between 5-helix and a C-terminal helix at the binding interface. Structure of 5-helix was built according to the crystal structure of gp41 core, which consists of a six-helical bundle of three 36-residue N-terminal sequences and three 34-residue C-terminal sequences. A C34 helix was bound rigidly to 5-helix and a continuum electrostatic method was used to analyze the electrostatic contributions to binding affinities. A following ‘electrostatic affinity optimization’ produced ‘the most favorable electrostatic binding free energy’ by mutating the residues of 5-helix. Binding free energy calculations of the 5-helix mutants showed improvements in binding of up to 100-fold for each single mutant and 500-fold for triple mutants. The authors pointed out that all the mutations should be feasible and could probably be extended to other systems of related structure.
There has lately been considerable interest in developing effective small molecular inhibitors of gp41. Jiang et al. discovered two pyrrole derivatives, NB-2 and NB-64, which can inhibit HIV-1 entry by interfering with gp41 six-helical bundle formation at concentrations lower than 10 μg/ml [42]. Molecular docking and 3D-QSAR approaches have been employed to explore the mechanism and binding mode of pyrrole derivatives including NB-2 and NB-64 into the gp41 hydrophobic pocket [43]. After integrating experimental data with molecular modeling studies on these compounds, Liu et al. [43] designed and synthesized a series of N-carboxyphenylpyrrole derivatives. Biological assays showed that some of these compounds have improved activities on inhibiti ng six-helical bundle formation and/or p24 production.
Reverse transcription inhibitors
HIV RT is a multifunctional enzyme involved in several essential activities for viral replication [44]. These activities include DNA-and RNA-dependent DNA polymerase, ribonuclease H (RNase H), strand transfer and strand displacement activities. Wild-type HIV-1 RT is an asymmetric heterodimer of a p66 subunit (consisting of a N-terminal polymerase domain, a C-terminal RNase H domain and a connection domain) and a p51 subunit (in which the RNase H domain is missing). RT has been the major target of current antiviral therapies against AIDS. NRTIs have been widely used in HAART, combined with PIs and/or NNRTIs. The high error rates characteristic of HIV-1 RT, however, are a presumptive source of the viral hypermutability that contributes mainly to the emergence of resistant variants, although the significant toxicity associated with current anti-HIV drugs also results in treatment failure. These factors together urge pharmacologists to develop more potent and less toxic RT inhibitors (RTIs) against the native RT and its drug-resistant variants. The X-ray structures of RT/RT mutants in complex with different ligands provide precious information for the computer-aided design of novel RTIs.
Da Silva et al. [45] published a computational study on several NRTIs (ddI, d4T, ddC and 3TC) and a novel proposed compound (10). Crystal structure of HIV-1 RT in complex with efavirenz (SUSTIVA®, NNRTI) [46] (PDB code: 1FK9), was used to build the receptor structure. GOLD was used to dock the ligands to the receptor. Docking results indicate that compound 10 occupies the similar region as the four NRTIs. All the ligands keep a good hydrophobic contact with the binding site. Interestingly, the chain extension at the hydroxyl group in compound 10 enables a new H-bond interaction between the hydroxyl group and Trp229 of the receptor. GoldScores rank the four inhibitors in the same order as their bioactivities, and compound 10 gets the highest score among all the ligands. MD simulation on the docking complex of RT compound 10 suggests a high binding affinity. Compound 10 obeys Lipinski’s rule of five, and its potential toxicity and metabolic properties were predicted by DEREK and METEOR programs. The authors believe that compound 10 could be a good potential HIV-1 RT inhibitor.
NNRTIs are a group of structurally dissimilar hydrophobic compounds that bind to a hydrophobic pocket on the RT adjacent to the substrate-binding site, thus leading to a noncompetitive inhibition of the enzyme. The past few years have seen encouraging achievements in developing RTIs (especially NNRTIs) with CADD approaches. Starting with X-ray structures of seven complexes of wild-type RT with diverse NNRTIs, Barreca et al. [47] developed a 3D pharmacophore model for NNRTIs. The seven training NNRTIs – efavirenz, MKC442 (emivirine), HBY097, MSC204, UC781, 739W94 and TMC120 – were selected on their abilities to form hydrogen bonds with the backbone of Lys101 and/or Lys103 of RT. The pharmacophore model was constructed using the common features hypothesis generation approach (HipHop) implemented in the program Catalyst 9.0 [48]. Common features among the inhibitors were identified to generate ‘qualitative hypothesis without the use of activity data. The corresponding pharmacophore models represent the essential 3D arrangement of functional groups common to these inhibitors. The final model attained in this study contains five features (hydrogen bond donors, hydrogen bond acceptors and hydrophobic regions). On the basis of pharmacophore features, three compounds were first designed and docked to the receptor by AutoDock. The docked positions were then mapped back to the pharmacophore model to analyze their potential functionalities. Taking together the information from pharmacophore model and docking results, another five compounds were designed. The eight compounds were synthesized and tested for activities and toxicities. All the compounds were proved to be active as inhibitors of HIV-1 RT, and one of them (11) exhibited robust anti-HIV activity against the wild-type and drug-escaped mutants with minimal cytotoxicity.
K103N and Y181C mutant RTs are the two most frequently observed RT mutants in patients failing therapy [49]. Nevirapine (Viramune ®), the first generation of NNRTI that has been approved by the FDA, has failed in interacting with the K103N and Y181C mutants. To develop novel Nevirapine analogs insensitive to the K103N and Y181C mutants, Saparpakorn et al. [50] designed a combinatorial library of 363 Nevirapine analogs and used molecular docking for virtual screening. The receptor structures were extracted from the X-ray structures of K103N and Y181C HIV-1 RT mutants in complex with Nevirapine (PDB code: 1FKP and 1JLB). Three docking methods (FlexX, GOLD and Surflex) were first tested by docking Nevirapine back to the receptors. GOLD exhibited a good ability to reproduce the X-ray bound conformation with RMSD less than 0.5 Å for both mutants and, thus, was selected to dock the designed compounds into the mutants. 124 hits with a higher GoldScore than Nevirapine were retrieved to the SILVER program for post-selection. SILVER selected 25 compounds that have H-bond interaction with Asn103 of K103N mutant or Cys181 of Y181C mutant and more than 80% of their surface buried upon binding. These compounds were observed to keep the specific interaction of Nevirapine with the receptors besides their particular interaction with Asn103 or Cys181. Quantum chemical calculation of the interaction energies of these compounds with Asn103 or Cys181 confirmed the existence of H-bond interactions.
Zhang et al. [51] reported their study on 25 Efavirenz analogs intending to discover novel inhibitors against both wild-type and K103N mutant RTs. The25 NNRTIs were first docked tothe receptors by using AutoDock 3.0.3. The conformations with the lowest binding free energies were then selected as binding conformations for structural alignment and 3D-QSAR analyses. CoMFA and CoMSIA were used to construct the 3D-QSAR models. With the information from molecular docking and 3D-QSAR models, a 3D pharmacophore model was established with Catalyst 4.6. A test of the pharmacophore model was carried out by screening a set of 500 compounds from the SPECS database mixed with 50 known inhibitors. As a result, only the 50 inhibitors were filtered out. An application of the pharmacophore model on virtual screening in the SPECS database yielded 50 hits. The preliminary bioassay on 12 compounds of them shows that two of them have good inhibitory activity against wild-type RT (IC50 < 10 μM).
Tetrahydroimidazo-[4,5,1-jk][1, 4]-benzodiazepinone (TIBO) derivatives (12) are a set of NNRTIs developed by Pauwels et al [52]. Several crystal structures of TIBO/RT complexes have been solved. A TIBO compound, Tivirapine, is already in the clinical trial stage. Aiming to find potent compounds for lead optimization, Sapre et al. [53] used flexible docking simulation for the virtual screening of the PubChem database. In this study, molecular docking was carried out using Moldock [54] with Grid scoring function (MolDock Grid). To validate the docking protocol, flexible docking was first performed on 9-Cl-TIBO to the crystal structure of RT extracted from the 9-Cl-TIBO/HIV-RT complex (PDB code: 1REV). The attained model exhibited an excellent alignment with the crystal coordinates (RMSD = 0.269 Å). The following docking simulations on 53 TIBO-derivate NNRTIs achieved a good correlation (r2 = 0.849, q2 = 0.843) between the biological activity and binding affinity of the inhibitors. After steps of prefilters, a docking screening on the PubChem database finally yielded 20 compounds that might have enhanced binding affinities. In a later study, Sapre et al. [55] improved the docking protocol by using incorporated templates, an enhanced pose clustering technique and a simplex evolution algorithm (MolDock SE) along with MolDock Grid. The more efficient docking protocol retrieved 25 novel TIBO-like compounds and six novel scaffolds from the PubChem database.
Research has shown that the activities of RT are strongly related to its dimer formation process [56]. A highly conserved cluster six tryptophans (tryptophan cluster) on the RT p66 subunit is important for RT dimerization [57] and might serve as a promising target for drug development. Recently, a structure-based ligand design implemented by Grohmann et al. [58] successfully discov ered a small molecule (MAS0) that strongly reduced the association of p66 and p51. Notably, the molecule also inhibited the activities of both the polymerase and the RNase H domain. The research started with computational and mutational studies on the contribution of individual residues to the HIV-1 RT heterodimer stability. MD simulation was then performed on the crystal structure of p66 subunit [59] (PDB code: 1RTH) by using the NAMD 2.5 [60] software package and CHAMM27 force field [61] to explore the receptor flexibility. On the basis of information of key residues for dimerization, six conformations of the receptor were selected from the MD trajectory. The software GRID21 [62] was used analyzing the conformations and constructing pharmacophore models. A virtual screening using the pharmacophore models as queries recognized potent hits, which were then docked into the p66 connection subdomain. This approach finally yielded ten compounds and after bioassays identified the promising inhibitor MAS0.
Integrase inhibitors
The integration of a DNA copy of HIV-1 genome into the host chromosome is achieved through a series of DNA cutting and joining reactions regulated by HIV-1 integrase [63,64]. In the step, known as ‘3′-processing reaction’, integrase removes two nucleotides from each 3′ end of the linear double-stranded viral DNA synthesized by reverse transcription from the viral RNA genome. The second step is termed ‘strand transfer’, in which the integrase protein joins the previously processed 3′ ends to 5′ ends of strands of target DNA at the site of integration. The end of HIV-1 integration, termed ‘disintegration’, involves host DNA repair synthesis. In this step, integrase might catalyze the excision of viral DNA. Integrase consists of three distinct structural domains: the zinc binding N-terminal; the catalytic core and the DNA-binding C-terminal. X-ray structure of the core domain [65], core plus C-terminal domain [66] and core plus N-terminal domain [67] have been resolved, respectively. The X-ray structure of the full-length enzyme remains elusive. An X-ray structure of a dimer of core domain in complex with an inhibitor, 5CITEP (PDB code: 1QS4), was also reported [16].
S-1360 [68] and L-870810 [69], the first two integrase inhibitors that entered clinical trials, belong to the class of β-diketo acids (DKAs), which have gained wide interest as integrase strand transfer inhibitors. S-1360 has failed, however, because of its metabolic instability. Another integrase strand transfer inhibitor, Raltegravir [70] (MK-0518), was approved by the FDA in October, 2007. GS-9137, an integrase inhibitor currently in late-stage clinical trials, demonstrated excellent antiviral activity in former clinical studies [71]. Besides strand transfer inhibitors, several compounds have been reported to inhibit the 3′-processing reaction [72]. Interest ingly, many compounds showed activity to inhibit both strand transfer and the 3′-processing reaction.
Given the large number of known inhibitors and lack of information on binding sites, pharmacophore model building followed by virtual screening has been frequently and successfully used in discovering novel integrase inhibitors [73]. This strategy has made more encouraging achievements in the past few years. Barreca et al [74] selected 33 strand-transfer-selective DKA derivate inhibitors to build a ‘quantitative predictive’ pharmacophore model for virtual screening. The model was built using a HypoGen [75] algorithm implemented in Catalyst with 17 of the inhibitors for the training set and 16 for the test set. The lead used in this study (13) has a strand transfer inhibitory activity of 0.03 μM. All the novel compounds attained could inhibit HIV-1 replication at micromolar concentration in in vitro assays. The most active two compounds (14, 15) have a strand transfer activity of 0.004 μM and 0.01 μM, respectively.
Dayam et al. [76] used S-1360 and three analogs to build a four-featured pharmacophore model with the HipHop module in Catalyst 4.8. Virtual screening yielded 1700 hits out of 150,000 small molecules. All 1700 compounds were docked into the binding area of 5CITEP in the IN-5CITEP complex crystal structure (using GOLD 1.2). According to the docking score, Lipinski’s rule of five and structural novelty, 110 compounds were selected for integrase assays, anti-HIV assays and toxicity determination. The results showed that the most potent compounds had a salicylic acid group connected to a rhodanine ring. A 2D substructure database search using salicylic acid group or rhodanine ring as the query structure was then carried out. Among all the compounds reported in this study, 11 compounds inhibited 3′-processing or strand transfer activity of integrase with IC50 < 25 μM. In the recent work of Dayam et al. [77], quinolone 3-carboxylic acids (including GS-9137) were used for pharmacophore model design. The best compound (16) identified by database screening showed inhibitory activity of 14 μM for 3′-processing and 5 μM for strand transfer. A substructure database search for compound 16 analogs discovered two compounds with higher activities than compound 16.
A pharmacophore model based on 30 compounds that inhibit the 3′-processing step with IC50 < 1 μM was developed by Mugnaini et al. [78]. Virtual screening on the ASINEX database (more than 200,000 compounds) was performed through sequential filters: electron–ion interaction potential, Lipinski’s rule of five, number of rotatable bonds <10 and, finally, pharmacophore model screening. The hits were then docked into the DNA-binding region in the IN core domain, based on which 12 compounds were selected for in vitro assays. One of the 12 compounds (compound 17) has a completely new scaffold and considerable anti-integrase activity (IC50 = 164 μM). Therefore, 29 analogs of compound 17 were selected and tested for activity. The most potent (18) of all the tested compounds has an IC50 value of 12 μM.
Chalcones and analogs have multiple biology activities, such as anticancer, antiviral, antiprotozoal and insecticidal properties. Some chalcones have been found to inhibit HIV-1 integrase strand transfer and 3′-processing processes [79]. Their utility is limited, however, by cytotoxicity and non-specificity. Starting with two of the chalcone leads, Deng et al. [80] designed a pharmacophore model to discover non-chalcone-based integrase inhibitors. To explore the rational binding conformation of the ligands, the two leads (chalcone 1 and chalcone 2) were first docked to the binding region of 5CIETP using the eHiTS docking program [81] The best pharmacophore model derived from the favorable docked orientation identified 407 compounds through database screening. 71 compounds were tested in this study. The most potent compound (19) could inhibit strand transfer with IC 50 = 0.6 μM and 3′-processing, IC50 = 1.9 μM.
It has been demonstrated that some DKAs as strand transfer inhibitors bind to integrase after the integrase has formed a complex with substrate DNA [82]. A lack of structure information on the active site after integrase binds with its DNA substrate hampers the structure-based drug design. Zhu et al. [83] docked dinucleotides to the NMR structure of the dimeric C-terminal domain of HIV integrase and identified two possible DNA-binding sites. Wang et al. [84] constructed integrase tetramer with available crystal structures of integrase domains. A 27 bp segment of viral DNA was then docked into the tetramer model including different number of metal ions. This study reveals the important roles of metal ions in integrase–DNA binding.
Using the 3D structure of Tn5 bacterial transposase/DNA complex as a template, Chen et al. [85] modeled the HIV-1 integrase/DNA complex with the structure of catalytic core and C-terminal domain. First, they aligned the integrase structure onto the template and modeled the missing loop region, Gly140-Gln148, in the IN core domain. MD simulation was then performed on the model and simulated annealing was applied to determine the significance of predicted loop conformation. After a minimizing step, the HIV RT DNA was built into the model by superimposing the nucleotide-heavy atoms onto the corresponding atoms of the transposase DNA. In the finally validated complex model, a potential hydrophobic binding pocket was observed at the active site, which could adopt different classes of IN inhibitors. Docking of L-870810 and two analogs to this integrase/DNA complex enabled the researchers to observe specific interactions between the ligands and the binding site. A series of novel compounds were then designed and synthesized [86], one of which (compound 20) showed higher anti-HIV activity and lower cytotoxicity than L-870810 in cell-based assays.
Ferro et al. [87] integrated two IN-Mg-DNA ternary complexes to build a new model of the IN-Mg-DNA complex and used it for docking the DKA inhibitors they had discovered previously (compound 13 and analogs) into the active site. Inspired by the molecular modeling results, the authors further designed a series of fluorine analogs and tested their biological activities. Seven compounds were more active than the lead compound 13.
Peptides derived from the interfacial helices of integrase dimer have been reported to block integrase dimerization [88]. The helix-forming tendency of the peptide inhibitors, as well as their binding affinity with integrase, is essential for their inhibitory activity. Binding affinities of these peptides with integrase were evaluated by docking and binding free energy estimation. Some of the designed peptides that showed improved helicity and binding abilities with intergrase might inhibit integrase dimerization and activity.
Protease inhibitors
In a later stage of the HIV life cycle, HIV protease (PR) hydrolyzes precursor polyproteins into functional proteins that are essential for viral assembly and subsequent activity. The functional structure of HIV-1 protease is a homodimer containing an active site created in the cleft between the monomers as part of a four-stranded β turn [89]. The active-site region is capped by two identical β-hairpin loops (the flaps, residues 45–55 in each monomer), which experience big conformational changes upon substrate binding [90]. A structural water molecule that forms hydrogen bonds with the enzyme flaps was observed in the X-ray crystal structure of the protease dimer. Hundreds of structures of protease in complex with its inhibitors have been resolved by X-ray crystallography. All protease inhibitors that are currently licensed for the treatment of HIV infections (namely saquinavir, ritonavir, indinavir, nelfinavir, amprenavir and lopinavir) mimic the substrate and block the active site. Another strategy is to develop compounds that bind to the subunit interface and thus block the dimerization [91].
Late in the last century, great achievements were made in developing HIV PR inhibitors (PRIs) with computer-aided approaches, which represent the most successful examples of the application of CADD. In the past few years, CADD research on PRIs has been more focused on QSAR studies of different classes of PRIs. New inhibitors discovered using QSAR models, however, are not so commonly seen in literature. Very recently, Jorissen et al [35] reported a successful application of ‘additive models’ to guide PRI design. Additive models are models that consider binding free energy contributions of substituents of a compound as independent and additive [92]. The first model was built with 61 compounds, which had been previously synthesized and tested for activities. Estimation of the affinity contributions of the various substituents led to the synthesis of 39 new compounds, which –together with the original 61 molecules – were used to build a second additive model. Six more molecules were then synthesized and a third model of the best estimating ability was constructed. Several of the newly designed compounds bind to the protease target with affinities of an order of magnitude of 10 pM. QSAR models with standard global molecular descriptors were also built for comparison but showed inferior predictions in this study.
Cyclic urea derivatives (21) are a class of non-peptide PT inhibitors that have long been proved and studied. The carbonyl oxygen on the cyclic urea ring mimics the structural water and forms a H-bond interaction with the enzyme flaps. Frecer et al. [93] analyzed several crystal structures of PR–PRIs using Cerius 2 and identified 11 different descriptors of the structural requirements for active ligands. The descriptors were used to generate a combinatorial library of non-symmetrically substituted cyclic ureas. The designed cyclic urea analogs, as well as the known inhibitors, were docked to the PR receptor (retrieved from the crystal structure of the PR–XV-638 complex to explore the plausible binding conformations and to build a QSAR scoring function capable of predicting the inhibitory activities of the analogs). The scoring function was validated and applied to screen the combinatorial library. The most potent nine hits have predicted activities of 0.5–2.2 pM. AMDET properties of the compounds were evaluated using the QikProp program of Schrödinger [94].
Using the same methodology, Frecer et al. [95] recently designed a series of peptidomimetic potential PR inhibitors containing –PheΨPro– core and a variety of flanking residues. Molecular modeling studies on the designed compounds that have high predicted inhibitory potencies indicate that two of the compounds were able to form H-bond interactions with a PR backbone and another compound can bind to PR, driven mainly by solvation effect. It has been proposed that these three compounds are active against drug-resistant PR mutants owing to their specific interactions with the receptor. AMDET property predictions also suggest these three compounds are potent lead candidates for further drug development.
Durdagi et al. [96] performed a computational study on binding interactions between a series of fullerene derivate PR inhibitors with the binding site. All the inhibitors were first docked into the receptor (structure retrieved from PR–haloperidol complex PDB code: 1AID) using FlexX in SYBYL. The selected docking complexes, as well as the non-bound receptor, were imported to GRO MACS 3.3.1 [98] for MD simulation with the GMX force field Binding affinities of the complexes were then estimated using FlexX and the results showed good correlation with the experimental data The structures from MD simulation revealed notable conformational changes of the PR flaps and the binding pocket from non bound to fullerene-bound status. 3D-QSAR (CoMFA and CoMSIA) models were then constructed for the inhibitors and employed for LeapFrog de novo design of fullerene analogs as PR inhibitors. Some of the designed compounds showed high predicted potency. It is worth noting that experiments are necessary to examine the feasibility and reliability of this de novo design.
Other inhibitors
RNA as a target designing the novel anti-HIV inhibitors has, until now, proceeded largely without incorporating direct input from structure-based design methodology, partly because of a lack of structural data and complications arising from substrate flexibility Some scientists, however, obtained some process on this field. Davis et al. [99] propose a paradigm to explain the physical mechanism for ligand-induced refolding of transactivation response element from HIV-1. They test this hypothesis by using NMR and computational methods to model the interaction of a series of novel inhibitors of the in vitro RNA-binding activities for a peptide derived from Tat Comparison of the interactions of two of these ligands with the RNA and structure–activity relationships observed within the compound series confirm the importance of the two specific electrostatic interactions in the stabilization of the Tat-bound RNA conformation. Their work illustrates how the use of medicinal chemistry and structural analysis can provide a rational basis for the prediction of ligand-induced conformational change, a necessary step towards the application of structure-based methods in the design of novel RNA. By using rational design, synthesis of 1′-acetoxychavicol acetate derivatives and biological evaluation of inhibitory activities, Liu et al. [100] revealed new salient pharmacophore features potential lead drug targeting the stem-loop IIB of Rev-responsible element against the HIV virus. Nef is an attractive target for drug discovery against HIV-1, but the lack of a 3D structure makes Nef difficult in CADD. Emert-Sedlak et al. [101] developed a high throughput screening assay for inhibitors of Nef function by coupling it to one of its host cell binding partners, the Src-family kinase Hck. Using this method, a novel diphenylfuropyrimidine-4-amino propanol (22) was demonstrated as a strong inhibitor of Nef-dependent Hck activation. This compound also displayed remarkable antiretroviral activity, blocking Nef-dependent HIV replication in cell culture. Its analogs were synthesized and have shown similar Nef-dependent anti-HIV activity, identifying the diphenylfuropyr imidine substructure as a new lead for developing antiretroviral drugs.
Drug resistance in HIV-1 PIs and RT
Genotypic and phenotypic mutations have been observed in more than 50% of residues of HIV PR, and more than 20 residues are associated with resistance to clinically available PIs [102]. With the aid of computational approaches, researchers are able to reveal the structural features and dynamical mechanism involved in the drug resistance of different HIV PR mutants. By virtue of MD simulation, Wartha et al. [103] explored the specific resistance of the D30N PR mutant and the N88S PR mutant to nelfinavir (NLF). A substrate and another PI, amprenavir, were used as comparison. The starting structures of wild-type PR, the two mutants and protease bound with drug/substrate were generated by modifying several X-ray structures of protease–drug and protease–substrate complexes. MD simulations were carried out in AMBER 7.0 under standard NPT conditions and data were collected during the 1 ns productive simulation for each system. A significant decrease in the van der Waals interaction energy between the D30N mutant and NLF was observed, which was related to the steric clashes caused by this active-site mutation. The consequent loss of hydrogen bond between D30 and NLF also contributes to the decrease of drug susceptibility. The non-active-site mutation N88S, by contrast, enhances the hydrogen bond between the 88 and 30 residues and results in a weakened binding of NLF to the 30 residue. Another successful implement of MD simulation on investigating drug resistance is reported by Ode et al. [104] on M36I mutation of HIV-1 PR. The authors discovered that this non-active-site mutation dramatically affects the conformation of the ligand-binding cavity, mainly through the interactions with residues L33 and V77.
A series of researches reported by Hannongbua [48,105,106] have revealed their findings on the resistance of PR G48V and G48V/L90M mutants against saquinavir (SQV), a selective PI approved by the FDA. The protonation state of catalytic aspartic acids Asp25 and Asp25 was first identified by performing the DFT calculations and the QM/MM ONIOM method [107,108]. Further quantum calculations and MD simulations followed by free energy calculations have discovered that the G48V mutation introduces steric conflict with SQV. The steric conflict results in a conformational change of the protein and dramatically weakens the hydrogen bond formed between the backbone carbonyl of residue 48 and SQV. The L90M mutation, although not located at the active site, causes repositioning throughout the entire protein structure. This mutation leads not only to a loss in the enzyme–inhibitor binding affinity but also, interestingly, to an increase in the enzyme stability.
Because of the high genetic variability of HIV, the rapid emergence of drug-resistant mutants has been a severe problem in clinical therapy. In recent years, computational strategies have shown particular advantages in investigating the drug-resistant mechanism and in designing specific or wide-range drugs. In 2005, Das et al. [109] presented a thorough review on the successful uses of computer-aided methods in developing NNRTIs that are effective against drug-resistant viral variants. K103N mutant RT is the most common HIV RT mutation that causes a high level of drug resistance [110]. Combining molecular docking with 3D-QSAR (CoMFA and CoMSIA), Juan [111] analyzed a set of 53 NNRTIs binding to the K103N mutant RT. The models generated in this study revealed the hydrophobic properties and flexibility at the active site of the mutant RT, as well as the corresponding features in the active inhibitors.
The prediction of energetics and kinetics
MD simulations have long been employed to give insights into the potencies and manners of ligands binding to a pharmaceutical target. Although molecular docking has been proved efficient in studying the ligand–protein binding process, especially in virtual screening for novel drugs, it suffers from two major deficiencies: unreliable scoring functions and the neglect of protein flexibility. With the advancement of computer performance and calculation techniques, it is now practical to combine molecular docking with MD simulations and free energy calculations to improve the enrichment and accuracy [112]. MD simulation treats the receptor structure in a flexible manner and collects multiple conformations as targets in docking. The docking results are then submitted to MD simulations for relaxing the complexes and a more accurate evaluation of the binding affinities. Among the numerous MD-based techniques for estimating binding free energy, free energy perturbation [113] and thermodynamic integration (TI) [113] have long been recognized as the most rigorous methods. The linear interaction energy (LIE) method [114] and the MM/PB-SA method [115] represent the alternative approximate approaches that have been widely used to meet practical demands, however. In a newly published study, Okimoto et al. [116] carried out approximately 6000 MD simulations for the top-ranked 1000 compounds docked to four target proteins (trypsin, HIV PR, acetylcholine esterase and cyclin-dependent kinase 2, or CDK2), in about approximately a week. The binding affinities were evaluated using the MM/PB-SA method, except for CDK2, where MM-PBSA had failed to improve the docking results. Alternatively, an approach based on the linear response and MM/PB-SA methods (the LR-MM/PB-SA approach [117]) performed effectively in the case of CDK2. In general, this strategy improved the enrichment performance of molecular dockings 1.6–4.0-fold.
Fragment-based drug design offers efficient access to the molecular diversity of drug agents [118]. This method identifies small drug-like fragments for a target protein and then evolves or links them to create molecules with higher affinities. Computational methods significantly improve the efficiency and reduce the cost of experimental fragment-based drug discovery. It is inevitably limited, however, by the issues concerning protein flexibility, solvent effects and the entropy loss upon assembling fragments. A site identification by ligand competitive saturation (SILCS) method was reported recently by Guvench et al. [119]; it addresses the fragment-based strategy with all-atom explicit-solvent MD. Essentially, the SILCS method performs multiple MD simulations of an aqueous system containing the target protein and various small molecules and computes the probability maps (FragMaps) for different fragment types binding around the protein. The resulting 3D free-energy-based FragMaps characterize the binding pocket and can be used as docking grids for high-throughput virtual screening. At the meantime, Clark et al. [120] developed a fragment-based method for evaluating binding free energies of whole molecules from those of their component fragments. Systematic sampling is first carried out in the six translational and rotational dimensions for rigid fragments, and molecular mechanics force field energy between the fragment and a protein for each pose is calculated. The fragments are then assembled and the binding free energies of the assembled molecules to the protein are integrated. The systematic sampling enables that estimation of binding affinities of many molecule poses with little computation and without a prior determination of the binding pose. Because of the approximations adopted in this method, however, it fails to give absolute free energy.
When the protein–ligand binding pose is known or attained by docking, the absolute (standard) binding free energy can be calculated with a variety of approaches. Recent years have seen encouraging improvements and achievements in free energy computing techniques. Binding free energy calculations can be categorized into two general classes: the pathway approaches (PMF, or the potential of mean force methods, FEB and TI) and the endpoint approaches (MM/PB-SA and LIE). Deng and Roux [121] have provided us with an overview of the theory, methods and recent applications of the pathway approaches. The filling potential method [122] is an umbrella potential sampling method that enables the ligand to drift from the bound state to the unbound state. The weighted histogram analysis method (WHAM) [123] then combines several trajectories of different umbrella potentials, and the PMF along the dissociation path can be obtained. A modified filling potential method named the smooth reaction path generation method [124] has been revealed recently, with TI used in the place of the time-consuming WHAM. In 2007, Almlöf et al. [125] further developed the LIE method by modifying the scaling factor for estimating the electrostatic component of solvation free energy. The electrostatic term was combined with an empirical non-polar term to predict the total solvation free energy. This derived model successfully reproduced the experimental hydration free energies for more than one hundred molecules. It should also improve the accuracy of LIE method for calculating binding free energies.
Future perspectives
This review gives a brief summary of the recent five-year achievements in discovering anti-HIV agents with the assistance of computer-aided approaches. Readers are referred to the original articles for detailed information. In addition to NRTIs, NtRTIs, NNRTIs and PIs, compounds that target viral entry and virus–cell fusion have great potential for the treatment of HIV infections. Studies of the viral proteins Tat, Rev and Nef might further identify a group of new drug targets. The latest technological advances (e.g. protein crystallography, X-ray crystallography, computer resource, cheminformatics and bioinformatics), the growing number of chemical and biological databases, and an explosion in programs and softwares are together opening a new chapter in anti-HIV drug design.
Acknowledgments
The authors thank Chinese Natural Science Foundation project (Nos. 30670497 and 30970784), National Key Basic Research Program of China (2009CB930200), Beijing Natural Science Foundation (Nos. 5072002 and 7082006), Research Fund for the Doctorate Program of Higher Education of China (X0015001200801), Chinese Academy of Sciences (CAS) ‘Hundred Talents Program’ (07165111ZX) and China-Finland Nanotechnology (No. 2008DFA01510) for financial support.
Biography
DR. XING-JIE LIANG
Dr. Xing-Jie Liang got Ph.D. at National Key Laboratory of Biomacromolecules, Institute of Biophysics at CAS. He finished his postdoc at Center for Cancer Research, NCI, NIH, and worked as a Research Fellow at Surgical Neurology Branch, NINDS. He worked on Molecular imaging at School of Medicine, Howard University before he became deputy director of CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety, National Center for Nanoscience and Technology of China. He is a founder member of International Society of Nanomedicine, member of American Association for Cancer Research, and member of American Society of Cell Biology. He is current editorial board member of ‘Acta Biophysica Sinica’ and ‘Current Nanoscience’. Developing drug delivery strategies for prevention/treatment of AIDS and cancers are current program ongoing in Dr. Liang’s lab based on understanding of basic physiochemical and biological processes of nanomedicine.

References
- 1.Girard MP, et al. A review of vaccine research and development: the human immunodeficiency virus (HIV) Vaccine. 2006;24:4062–4081. doi: 10.1016/j.vaccine.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 2.Barbaro G, et al. Highly active antiretroviral therapy: current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr Pharm Des. 2005;11:1805–1843. doi: 10.2174/1381612053764869. [DOI] [PubMed] [Google Scholar]
- 3.Louie M, Markowitz M. Goals and milestones during treatment of HIV-1 infection with antiretroviral therapy: a pathogenesis-based perspective. Antiviral Res. 2002;55:15–25. doi: 10.1016/s0166-3542(02)00022-0. [DOI] [PubMed] [Google Scholar]
- 4.Greene WC. The brightening future of HIV therapeutics. Nat Immunol. 2004;5:867–871. doi: 10.1038/ni0904-867. [DOI] [PubMed] [Google Scholar]
- 5.Hazuda D, et al. Emerging pharmacology: inhibitors of human immunodeficiency virus integration. Annu Rev Pharmacol Toxicol. 2009;49:377–394. doi: 10.1146/annurev.pharmtox.011008.145553. [DOI] [PubMed] [Google Scholar]
- 6.DiMasi JA, et al. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22:151–185. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 7.Kwong PD, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speck RF, et al. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J Virol. 1997;71:7136–7139. doi: 10.1128/jvi.71.9.7136-7139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan DC, et al. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 10.Kong R, et al. Prediction of the binding mode between BMS-378806 and HIV-1 gp120 by docking and molecular dynamics simulation. Biochim Biophys Acta. 2006;1764:766–772. doi: 10.1016/j.bbapap.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Lifson JD, Engleman EG. Role of Cd4 in normal immunity and HIV infection. Immunol Rev. 1989;109:93–117. doi: 10.1111/j.1600-065x.1989.tb00021.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang JH, et al. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci U S A. 2001;98:10799–10804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myszka DG, et al. Energetics of the HIV gp120-CD4 binding reaction. Proc Natl Acad Sci U S A. 2000;97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neffe AT, Meyer B. A peptidomimetic HIV-entry inhibitor directed against the CD4 binding site of the viral glycoprotein gp120. Angew Chem Int Ed Engl. 2004;43:2937–2940. doi: 10.1002/anie.200353271. [DOI] [PubMed] [Google Scholar]
- 15.Neffe AT, et al. Synthesis and optimization of peptidomimetics as HIV entry inhibitors against the receptor protein CD4 using STD NMR and ligand docking. Org Biomol Chem. 2006;4:3259–3267. doi: 10.1039/b605380g. [DOI] [PubMed] [Google Scholar]
- 16.Goldgur Y, et al. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc Natl Acad Sci U S A. 1999;96:13040–13043. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neffe AT, et al. Rational optimization of the binding affinity of CD4 targeting peptidomimetics with potential anti HIV activity. J Med Chem. 2007;50:3482–3488. doi: 10.1021/jm070206b. [DOI] [PubMed] [Google Scholar]
- 18.Schlyer S, Horuk R. I want a new drug: G-protein-coupled receptors in drug development. Drug Discov Today. 2006;11:481–493. doi: 10.1016/j.drudis.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Roy K, Leonard JT. QSAR analyses of 3-(4-benzylpiperidin-1-yl)-N-phenylpropylamine derivatives as potent CCR5 antagonists. J Chem Inf Model. 2005;45:1352–1368. doi: 10.1021/ci050205x. [DOI] [PubMed] [Google Scholar]
- 20.Aher YD, et al. 3D-QSAR studies of substituted 1-(3,3-diphenylpropyl)-piperidinyl amides and ureas as CCR5 receptor antagonists. J Mol Model. 2007;13:519–529. doi: 10.1007/s00894-007-0173-z. [DOI] [PubMed] [Google Scholar]
- 21.Dessalew N. QSAR study on piperidinecarboxamides as antiretroviral agents: an insight into the structural basis for HIV coreceptor antagonist activity. QSAR Comb Sci. 2008;27:901–912. [Google Scholar]
- 22.Afantitis A, et al. Investigation of substituent effect of 1-(3,3-diphenylpropyl)-piperidinyl phenylacetamides on CCR5 binding affinity using QSAR and virtual screening techniques. J Comput Aided Mol Des. 2006;20:83–95. doi: 10.1007/s10822-006-9038-2. [DOI] [PubMed] [Google Scholar]
- 23.Leonard JT, Roy K. Comparative QSAR modeling of CCR5 receptor binding affinity of substituted 1-(3,3-diphenylpropyl)-piperidinyl amides and ureas. Bioorg Med Chem Lett. 2006;16:4467–4474. doi: 10.1016/j.bmcl.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 24.Zhuo Y, et al. Three-dimensional QSAR analyses of 1,3,4-trisubstituted pyrrolidine-based CCR5 receptor inhibitors. Eur J Med Chem. 2008;43:2724–2734. doi: 10.1016/j.ejmech.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 25.Finke PE, et al. Antagonists of the human CCR5 receptor as anti-HIV-1 agents. Part 4: synthesis and structure–activity relationships for 1-[N(methyl)-N-(phenylsulfonyl)amino]-2-(phenyl)-4-(4-(N-(alkyl)-N-(benzyloxycarbonyl)amino)piperidin-1-yl)butanes. Bioorg Med Chem Lett. 2001;11:2475–2479. doi: 10.1016/s0960-894x(01)00492-9. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y, et al. Molecular docking and 3D QSAR studies on 1-amino-2-phenyl-4-(piperidin-1-yl)-butanes based on the structural modeling of human CCR5 receptor. Bioorg Med Chem. 2004;12:6193–6208. doi: 10.1016/j.bmc.2004.08.045. [DOI] [PubMed] [Google Scholar]
- 27.Kellenberger E, et al. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J Med Chem. 2007;50:1294–1303. doi: 10.1021/jm061389p. [DOI] [PubMed] [Google Scholar]
- 28.Este JA, et al. Activity of different bicyclam derivatives against human immunodeficiency virus depends on their interaction with the CXCR4 chemokine receptor. Mol Pharmacol. 1999;55:67–73. doi: 10.1124/mol.55.1.67. [DOI] [PubMed] [Google Scholar]
- 29.Pettersson S, et al. Discovery of novel non-cyclam polynitrogenated CXCR4 coreceptor inhibitors. ChemMedChem. 2008;3:1549–1557. doi: 10.1002/cmdc.200800145. [DOI] [PubMed] [Google Scholar]
- 30.Pascual R, et al. Design and analysis of a combinatorial library of HEPT analogues: comparison of selection methodologies and inspection of the actually covered chemical space. J Chem Inf Comput Sci. 2003;43:199–207. doi: 10.1021/ci0255681. [DOI] [PubMed] [Google Scholar]
- 31.Lapidot A, Borkow G. Multi-targeting the entrance door to block HIV-1 by aminoglycoside-arginine conjugates (AACs) Biopolymers. 2005;80:536. doi: 10.2174/1568005053174645. [DOI] [PubMed] [Google Scholar]
- 32.Hegde R, et al. Structure–function relationship of novel X4 HIV-1 entry inhibitors – L- and D-arginine peptide-aminoglycoside conjugates. FEBS J. 2007;274:6523–6536. doi: 10.1111/j.1742-4658.2007.06169.x. [DOI] [PubMed] [Google Scholar]
- 33.Berchanski A, Lapidot A. Prediction of HIV-1 entry inhibitors neomycin-arginine conjugates interaction with the CD4-gp120 binding site by molecular modeling and multistep docking procedure. Biochim Biophys Acta. 2007;1768:2107–2119. doi: 10.1016/j.bbamem.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 34.Katchalski-Katzir E, et al. Molecular-surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci U S A. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorissen RN, et al. Additivity in the analysis and design of HIV protease inhibitors. J Med Chem. 2009;52:737–754. doi: 10.1021/jm8009525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berchanski A, Lapidot A. Computer-based design of novel HIV-1 entry inhibitors: neomycin conjugated to arginine peptides at two specific sites. J Mol Model. 2009;15:281–294. doi: 10.1007/s00894-008-0401-1. [DOI] [PubMed] [Google Scholar]
- 37.Root MJ, et al. Protein design of an HIV-1 entry inhibitor. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 38.Tan JJ, et al. Investigating interactions between HIV-1 gp41 and inhibitors by molecular dynamics simulation and MM-PBSA/GBSA calculations. J Mol Struct THEOCHEM. 2006;766:77–82. [Google Scholar]
- 39.Tan JJ, et al. Studies on binding free energies and the binding mode by docking and MM-PBSA in gp41-ligand complex. Mol Simul. 2005;31:1051–1056. [Google Scholar]
- 40.Tan JJ, et al. Gp41 hydrophobic pocket entry inhibitor linear interaction energy MD simulation. J Mol Struct THEOCHEM. 2004;682:9–15. [Google Scholar]
- 41.Green DF, Tidor B. Design of improved protein inhibitors of HIV-1 cell entry: optimization of electrostatic interactions at the binding interface. Proteins: Struct Func Bioinf. 2005;60:644–657. doi: 10.1002/prot.20540. [DOI] [PubMed] [Google Scholar]
- 42.Jiang SB, et al. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Ch. 2004;48:4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu K, et al. Design, synthesis, and biological evaluation of N-carboxyphenylpyrrole derivatives as potent HIV fusion inhibitors targeting gp41. J Med Chem. 2008;51:7843–7854. doi: 10.1021/jm800869t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goff SP. Retroviral reverse-transcriptase: synthesis, structure, and function. J Acquir Immune Defic Syndr Hum Retrovirol. 1990;3:817–831. [PubMed] [Google Scholar]
- 45.Da Silva CHTP, et al. Molecular dynamics, docking, density functional, and admet studies of HIV-1 reverse transcriptase inhibitors. J Theor Comput Chem. 2006;5:579–586. [Google Scholar]
- 46.Ren J, et al. Binding of the second generation non-nucleoside inhibitor S-1153 to HIV-1 reverse transcriptase involves extensive main chain hydrogen bonding. J Biol Chem. 2000;275:14316–14320. doi: 10.1074/jbc.275.19.14316. [DOI] [PubMed] [Google Scholar]
- 47.Barreca ML, et al. Computational strategies in discovering novel non-nucleoside inhibitors of HIV-1 RT. J Med Chem. 2005;48:3433–3437. doi: 10.1021/jm049279a. [DOI] [PubMed] [Google Scholar]
- 48.Wittayanarakul K, et al. Insights into saquinavir resistance in the G48V HIV-1 protease: quantum calculations and molecular dynamic simulations. Biophys J. 2005;88:867–879. doi: 10.1529/biophysj.104.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Menendez-Arias L. Targeting HIV: antiretroviral therapy and development of drug resistance. Trends Pharmacol Sci. 2002;23:381–388. doi: 10.1016/s0165-6147(02)02054-0. [DOI] [PubMed] [Google Scholar]
- 50.Saparpakorn P, et al. Design of nevirapine derivatives insensitive to the K103N and Y181CHIV-1 reverse transcriptase mutants. SAR QSAR Environ Res. 2006;17:183–194. doi: 10.1080/10659360600636147. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, et al. Towards discovering dual functional inhibitors against both wild type and K103N mutant HIV-1 reverse transcriptases: molecular docking and QSAR studies on 4,1-benzoxazepinone analogues. J Comput Aided Mol Des. 2006;20:281–293. doi: 10.1007/s10822-006-9050-6. [DOI] [PubMed] [Google Scholar]
- 52.Pauwels R, et al. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature. 1990;343:470–474. doi: 10.1038/343470a0. [DOI] [PubMed] [Google Scholar]
- 53.Sapre NS, et al. Molecular docking studies on tetrahydroimidazo-[4,5,1-jk][1,4]-benzodiazepinone (TIBO) derivatives as HIV-1NNRT inhibitors. J Comput Aided Mol Des. 2008;22:69–80. doi: 10.1007/s10822-007-9161-8. [DOI] [PubMed] [Google Scholar]
- 54.Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 55.Sapre NS, et al. Data mining using template-based molecular docking on tetrahydroimidazo-[4,5,1-jk][1,4]-benzodiazepinone (TIBO) derivatives as HIV-1RT inhibitors. J Mol Model. 2008;14:1009–1021. doi: 10.1007/s00894-008-0335-7. [DOI] [PubMed] [Google Scholar]
- 56.Restle T, et al. Rnase-H activity of HIV reverse transcriptases is confined exclusively to the dimeric forms. FEBS Lett. 1992;300:97–100. doi: 10.1016/0014-5793(92)80172-d. [DOI] [PubMed] [Google Scholar]
- 57.Mulky A, et al. Identification of amino acid residues in the human immunodeficiency virus type-1 reverse transcriptase tryptophan-repeat motif that are requiredfor subunitinteractionusing infectiousvirions. J Mol Biol. 2005;349:673–684. doi: 10.1016/j.jmb.2005.03.057. [DOI] [PubMed] [Google Scholar]
- 58.Grohmann D, et al. Small molecule inhibitors targeting HIV-1 reverse transcriptase dimerization. ChemBioChem. 2008;9:916–922. doi: 10.1002/cbic.200700669. [DOI] [PubMed] [Google Scholar]
- 59.Ren J, et al. High-resolution structures of HIV-1 RT from 4 RT-inhibitor complexes. Nat Struct Biol. 1995;2:293–302. doi: 10.1038/nsb0495-293. [DOI] [PubMed] [Google Scholar]
- 60.Kale L, et al. NAMD2: greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151:283–312. [Google Scholar]
- 61.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 62.Boobbyer DNA, et al. New hydrogen-bond potentials for use in determining energetically favorable binding sites on molecules of known structure. J Med Chem. 1989;32:1083–1094. doi: 10.1021/jm00125a025. [DOI] [PubMed] [Google Scholar]
- 63.Esposito D, Craigie R. HIV integrase structure and function. Adv Virus Res. 1999;52:319–333. doi: 10.1016/s0065-3527(08)60304-8. [DOI] [PubMed] [Google Scholar]
- 64.Fenollar-Ferrer C, et al. HIV-1 integrase-DNA interactions investigated by molecular modelling. Comput Math Methods Med. 2008;9:231–243. [Google Scholar]
- 65.Goldgur Y, et al. Three new structures of the core domain of HIV-1 integrase: an active site that binds magnesium. Proc Natl Acad Sci U S A. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen JCH, et al. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci U S A. 2000;97:8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang JY, et al. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosemond MJC, et al. Enzymology of a carbonyl reduction clearance pathway for the HIV integrase inhibitor, S-1360: role of human liver cytosolic aldo–keto reductases. Chem Biol Interact. 2004;147:129–139. doi: 10.1016/j.cbi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Johnson AA, et al. HIV-1 integrase inhibitors: a decade of research and two drugs in clinical trial. Curr Top Med Chem. 2004;4:1059–1077. doi: 10.2174/1568026043388394. [DOI] [PubMed] [Google Scholar]
- 70.Markowitz M, et al. Antiretroviral activity, pharmacokinetics, and tolerability of MK-0518, a novel inhibitor of HIV-1 integrase, dosed as monotherapy for 10 days in treatment-naive HIV-1-infected individuals. J Acquir Immune Defic Syndr. 2006;43:509–515. doi: 10.1097/QAI.0b013e31802b4956. [DOI] [PubMed] [Google Scholar]
- 71.DeJesus E, et al. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr. 2006;43:1–5. doi: 10.1097/01.qai.0000233308.82860.2f. [DOI] [PubMed] [Google Scholar]
- 72.Deprez E, et al. Mechanism of HIV-1 integrase inhibition by styrylquinoline derivatives in vitro. Mol Pharmacol. 2004;65:85–98. doi: 10.1124/mol.65.1.85. [DOI] [PubMed] [Google Scholar]
- 73.Hong H, et al. Discovery of HIV-1 integrase inhibitors by pharmacophore searching. J Med Chem. 1997;40:930–936. doi: 10.1021/jm960754h. [DOI] [PubMed] [Google Scholar]
- 74.Barreca ML, et al. Pharmacophore-based design of HIV-1 integrase strand-transfer inhibitors. J Med Chem. 2005;48:7084–7088. doi: 10.1021/jm050549e. [DOI] [PubMed] [Google Scholar]
- 75.Li H, et al. Pharmacophore Perception, Development, and Use in Drug Design. International University; 2000. HypoGen: An Automated System for Generating 3D Predictive Pharmacophore Models. [Google Scholar]
- 76.Dayam R, et al. beta-Diketo acid pharmacophore hypotesis. 1 Discovery of a novel class of HIV-1 integrase inhibitors. J Med Chem. 2005;48:111–120. doi: 10.1021/jm0496077. [DOI] [PubMed] [Google Scholar]
- 77.Dayam R, et al. Quinolone 3-carboxylic acid pharmacophore: design of second generation HIV-1 integrase inhibitors. J Med Chem. 2008;51:1136–1144. doi: 10.1021/jm070609b. [DOI] [PubMed] [Google Scholar]
- 78.Mugnaini C, et al. Toward novel HIV-1 integrase binding inhibitors: molecular modeling, synthesis, and biological studies. Bioorg Med Chem Lett. 2007;17:5370–5373. doi: 10.1016/j.bmcl.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 79.Nicklaus MC, et al. HIV-1 integrase pharmacophore: discovery of inhibitors through three-dimensional database searching. J Med Chem. 1997;40:920–929. doi: 10.1021/jm960596u. [DOI] [PubMed] [Google Scholar]
- 80.Deng J, et al. Discovery of structurally diverse HIV-1 integrase inhibitors based on a chalcone pharmacophore. Bioorg Med Chem. 2007;15:4985–5002. doi: 10.1016/j.bmc.2007.04.041. [DOI] [PubMed] [Google Scholar]
- 81.Zsoldos Z, et al. eHITS: an innovative approach to the docking and scoring function problems. Curr Protein Pept Sci. 2006;7:421–435. doi: 10.2174/138920306778559412. [DOI] [PubMed] [Google Scholar]
- 82.Espeseth AS, et al. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc Natl Acad Sci U S A. 2000;97:11244–11249. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu HM, et al. Docking dinucleotides to HIV-1 integrase carboxyl-terminal domain to find possible DNA binding sites. Bioorg Med Chem Lett. 2005;15:475–477. doi: 10.1016/j.bmcl.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 84.Wang LD, et al. Constructing HIV-1 integrase tetramer and exploring influences of metal ions on forming integrase-DNA complex. Biochem Biophys Res Commun. 2005;337:313–319. doi: 10.1016/j.bbrc.2005.08.274. [DOI] [PubMed] [Google Scholar]
- 85.Chen X, et al. Modeling, analysis, and validation of a novel HIV integrase structure provide insights into the binding modes of potent integrase inhibitors. J Mol Biol. 2008;380:504–519. doi: 10.1016/j.jmb.2008.04.054. [DOI] [PubMed] [Google Scholar]
- 86.Jin H, et al. Design, synthesis, and SAR studies of novel and highly active tri-cyclic HIV integrase inhibitors. Bioorg Med Chem Lett. 2006;16:3989–3992. doi: 10.1016/j.bmcl.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 87.Ferro S, et al. Docking studies on a new human immuodeficiency virus integrase-Mg–DNA complex: phenyl ring exploration and synthesis of 1H-benzylindole derivatives through fluorine substitutions. J Med Chem. 2009;52:569–573. doi: 10.1021/jm8009266. [DOI] [PubMed] [Google Scholar]
- 88.Maroun RG, et al. Peptide inhibitors of HIV-1 integrase dissociate the enzyme oligomers. Biochemistry. 2001;40:13840–13848. doi: 10.1021/bi011328n. [DOI] [PubMed] [Google Scholar]
- 89.Lapatto R, et al. X-ray-analysis of HIV-1 proteinase at 2.7 a resolution confirms structural homology among retroviral enzymes. Nature. 1989;342:299–302. doi: 10.1038/342299a0. [DOI] [PubMed] [Google Scholar]
- 90.Levy Y, Caflisch A. Flexibility of monomeric and dimeric HIV-1 protease. J Phys Chem B. 2003;107:3068–3079. [Google Scholar]
- 91.Boggetto N, Reboud-Ravaux M. Dimerization inhibitors of HIV-1 protease. Biol Chem. 2002;383:1321–1324. doi: 10.1515/BC.2002.150. [DOI] [PubMed] [Google Scholar]
- 92.Free SM, Wilson JW. A mathematical contribution to structure–activity studies. J Med Chem. 1964;7:395–399. doi: 10.1021/jm00334a001. [DOI] [PubMed] [Google Scholar]
- 93.Frecer V, et al. Combinatorial design of nonsymmetrical cyclic urea inhibitors of aspartic protease of HIV-1. Bioorg Med Chem. 2005;13:5492–5501. doi: 10.1016/j.bmc.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 94.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 95.Frecer V, et al. Design of peptidomimetic inhibitors of aspartic protease of HIV-1 containing –Phe Psi Pro– core and displaying favourable ADME-related properties. J Mol Graph Model. 2008;27:376–387. doi: 10.1016/j.jmgm.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 96.Durdagi S, et al. Computational design of novel fullerene analogues as potential HIV-1 PR inhibitors: analysis of the binding interactions between fullerene inhibitors and HIV-1 PR residues using 3D QSAR, molecular docking and molecular dynamics simulations. Bioorg Med Chem. 2008;16:9957–9974. doi: 10.1016/j.bmc.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 97.Rutenber E, et al. Structure of a nonpeptide inhibitor complexed with HIV-1 protease – developing a cycle of structure-based drug design. J Biol Chem. 1993;268:15343–15346. [PubMed] [Google Scholar]
- 98.Van der Spoel D, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 99.Davis B, et al. Rational design of inhibitors of HIV-1 TAR RNA through the stabilisation of electrostatic “hot spots”. J Mol Biol. 2004;336:343–356. doi: 10.1016/j.jmb.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 100.Liu Y, et al. Structure–activity relationships of 1′-acetoxychavicol acetate homologues as new nuclear export signal inhibitors. Pharmazie. 2007;62:659–662. [PubMed] [Google Scholar]
- 101.Emert-Sedlak L, et al. Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem Biol. 2009;4:939–947. doi: 10.1021/cb900195c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shafer RW, et al. Identification of biased amino acid substitution patterns in human immunodeficiency virus type 1 isolates from patients treated with protease inhibitors. J Virol. 1999;73:6197–6202. doi: 10.1128/jvi.73.7.6197-6202.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wartha F, et al. Molecular dynamics simulations of HIV-1 protease suggest different mechanisms contributing to drug resistance. J Chem Theory Comput. 2005;1:315–324. doi: 10.1021/ct049869o. [DOI] [PubMed] [Google Scholar]
- 104.Ode H, et al. Computational characterization of structural role of the non-active site mutation M36I of human immunodeficiency virus type 1 protease. J Mol Biol. 2007;370:598–607. doi: 10.1016/j.jmb.2007.04.081. [DOI] [PubMed] [Google Scholar]
- 105.Aruksakunwong O, et al. Structural and dynamical properties of different protonated states of mutant HIV-1 protease complexed with the saquinavir inhibitor studied by molecular dynamics simulations. J Mol Graph Model. 2006;25:324–332. doi: 10.1016/j.jmgm.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 106.Saen-oon S, et al. Insight into analysis of interactions of saquinavir with HIV-1 protease in comparison between the wild-type and G48V and G48V/L90M mutants based on QM and QM/MM calculations. J Mol Graph Model. 2007;26:720–727. doi: 10.1016/j.jmgm.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 107.Friesner RA, Beachy MD. Quantum mechanical calculations on biological systems. Curr Opin Struct Biol. 1998;8:257–262. doi: 10.1016/s0959-440x(98)80048-1. [DOI] [PubMed] [Google Scholar]
- 108.Morokuma K. New challenges in quantum chemistry: quests for accurate calculations for large molecular systems. Philos Transact A: Math Phys Eng Sci. 2002;360:1149–1164. doi: 10.1098/rsta.2002.0993. [DOI] [PubMed] [Google Scholar]
- 109.Das K, et al. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 110.Bacheler L, et al. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J Virol. 2001;75:4999–5008. doi: 10.1128/JVI.75.11.4999-5008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Juan AAS. 3D-QSAR models on clinically relevant K103N mutant HIV-1 reverse transcriptase obtained from two strategic considerations. Bioorg Med Chem Lett. 2008;18:1181–1194. doi: 10.1016/j.bmcl.2007.11.134. [DOI] [PubMed] [Google Scholar]
- 112.Amaro RE, et al. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J Comput Aided Mol Des. 2008;22:693–705. doi: 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kollman P. Free-energy calculations: applications to chemical and biochemical phenomena. Chem Rev. 1993;93:2395–2417. [Google Scholar]
- 114.Aqvist J, et al. Ligand binding affinities from MD simulations. Acc Chem Res. 2002;35:358–365. doi: 10.1021/ar010014p. [DOI] [PubMed] [Google Scholar]
- 115.Kollman PA, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 116.Okimoto N, et al. High-performance drug discovery: computational screening by combining docking and molecular dynamics simulations. PLOS Comput Biol. 2009;5:e1000528. doi: 10.1371/journal.pcbi.1000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou ZG, Madura JD. Relative free energy of binding and binding mode calculations of HIV-1 RT inhibitors based on dock-MM-PB/GS. Proteins: Struct Func Bioinf. 2004;57:493–503. doi: 10.1002/prot.20223. [DOI] [PubMed] [Google Scholar]
- 118.Tickle I, et al. High-throughput protein crystallography and drug discovery. Chem Soc Rev. 2004;33:558–565. doi: 10.1039/b314510g. [DOI] [PubMed] [Google Scholar]
- 119.Guvench O, MacKerell AD. Computational fragment-based binding site identification by ligand competitive saturation. PLOS Comput Biol. 2009;5:e1000435. doi: 10.1371/journal.pcbi.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Clark M, et al. fragment-based computation of binding free energies by systematic sampling. J Chem Inf Model. 2009;49:1901–1913. doi: 10.1021/ci900132r. [DOI] [PubMed] [Google Scholar]
- 121.Deng Y, Roux B. Computations of standard binding free energies with molecular dynamics simulations. J Phys Chem B. 2009;113:2234–2246. doi: 10.1021/jp807701h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fukunishi Y, et al. The filling potential method: a method for estimating the free energy surface for protein-ligand docking. J Phys Chem B. 2003;107:13201–13210. [Google Scholar]
- 123.Kumar S, et al. The weighted histogram analysis method for free-energy calculations on biomolecules. 1 The method. J Comput Chem. 1992;13:1011–1021. [Google Scholar]
- 124.Fukunishi Y, et al. Protein-ligand binding free energy calculation by the smooth reaction path generation (SRPG) method. J Chem Inf Model. 2009;49:1944–1951. doi: 10.1021/ci9002156. [DOI] [PubMed] [Google Scholar]
- 125.Almlöf M, et al. Improving the accuracy of the linear interaction energy method for solvation free energies. J Chem Theory Comput. 2007;3:2162–2175. doi: 10.1021/ct700106b. [DOI] [PubMed] [Google Scholar]