

Abstract
Background
Ras-extracellular signal-regulated kinase (Ras-ERK) signaling is central to the molecular machinery underlying cognitive functions. In the striatum, ERK1/2 kinases are co-activated by glutamate and dopamine D1/5 receptors, but the mechanisms providing such signaling integration are still unknown. The Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1), a neuronal specific activator of Ras-ERK signaling, is a likely candidate for coupling these neurotransmitter signals to ERK kinases in the striatonigral medium spiny neurons (MSN) and for modulating behavioral responses to drug abuse such as cocaine.
Methods
We used genetically modified mouse mutants for Ras-GRF1 as a source of primary MSN cultures and organotypic slices, to perform both immunoblot and immunofluorescence studies in response to glutamate and dopamine receptor agonists. Mice were also subjected to behavioral and immunohistochemical investigations upon treatment with cocaine.
Results
Phosphorylation of ERK1/2 in response to glutamate, dopamine D1 agonist, or both stimuli simultaneously is impaired in Ras-GRF1– deficient striatal cells and organotypic slices of the striatonigral MSN compartment. Consistently, behavioral responses to cocaine are also affected in mice deficient for Ras-GRF1 or overexpressing it. Both locomotor sensitization and conditioned place preference are significantly attenuated in Ras-GRF1– deficient mice, whereas a robust facilitation is observed in overexpressing transgenic animals. Finally, we found corresponding changes in ERK1/2 activation and in accumulation of FosB/ΔFosB, a well-characterized marker for long-term responses to cocaine, in MSN from these animals.
Conclusions
These results strongly implicate Ras-GRF1 in the integration of the two main neurotransmitter inputs to the striatum and in the maladaptive modulation of striatal networks in response to cocaine.
Keywords: Cocaine responses, dopamine, ERK1/2 signaling, glutamate, Ras-GRF1, striatum
In the adult central nervous system behavioral plasticity is governed by various intracellular signaling pathways, including the Ras-ERK cascade (1–3). Whereas the input level of this signaling module is provided by the Ras subfamily of small GTPase, the amplification/stability core component consists of the Raf-MEK-ERK protein kinase cascade (4–8). Thus, neurotransmitter-dependent ERK activation in the brain leads to a sequence of cellular events culminating in the phosphorylation of key nuclear factors, chromatin remodeling, and gene expression (9–13).
In recent years, the Ras-ERK signaling cascade has been implicated in responses to most drugs of abuse, supporting the notion that drug experience plasticity might share common mechanisms with learning and memory processes (14–19). More specifically, psychostimulants rapidly activate the main isoform ERK2 in a number of structures, including the dorsal striatum, nucleus accumbens (NAc), amygdala, and the deep layers of the prefrontal and cingulate cortex (20–25). The functional relevance of ERK2 activation for behavioral plasticity in the striatum has been demonstrated in a number of studies through the use of either specific inhibitors of MEK such as SL327 and U0126 or genetically modified mice lacking the negative regulatory isoform ERK1 (21,26–34). In this context ERK2 activation seems to be critical in the striatonigral compartment although, upon certain behavioral manipulations or genetic enhancement of ERK signaling, some contribution of the striatopallidal pathway has also been observed (16,21,24,33–43). Importantly, in the striatonigral pathway, D1R-dependent downstream signals also involve the cAMP, protein kinase A (PKA), and dopamine- and cAMP-regulated phosphoprotein (DARPP-32) pathway, which contributes to maintain ERK1/2 in their active, phosphorylated forms via the concerted inhibitory action of striatal-enriched tyrosine phosphatase (STEP) and protein phosphatase 1 (PP1) (24,44).
Thus, ERK-dependent signaling might represent an integration point between glutamatergic inputs (mainly amino-3-hydroxy-5-methylisoxazole propionate receptor [AMPAR]/N-methyl d-aspartate receptor [NMDAR] dependent) from cortical and limbic regions and dopaminergic inputs (mainly D1-like receptor-dependent) from mesolimbic nuclei in the striatonigral pathway, and its concomitant activation by these two distinct inputs might create a “permissive” state for behaviorally salient plasticity and neuronal adaptations within the medium spiny neuron (MSN) network (19). This model, however, requires the action of “signaling integrators,” located in close proximity to the plasma membrane, to simultaneously “sense” both stimuli and to provide a tight regulation of the downstream ERK cascade at the input level.
In this study, we report the identification of a candidate for such an integrative role in the striatum, Ras-GRF1. This central nervous system-specific guanine-exchange factor, together with its close homologue Ras-GRF2, a distinct Ras-GEF isoform abundantly expressed in cortical regions and in the hippocampal formation, is localized at the synapse where it is able to activate Ras- and Rac-dependent signaling in neurons, both in response to calcium influx and metabotropic receptor activation (45–63). Here we show that Ras-GRF1 is crucially involved in both D1 and glutamate receptor-dependent ERK activation in the MSN striatonigral network and that its expression levels significantly influence long-term behavioral responses to cocaine.
Methods and Materials
See Supplement 1 for details.
Results
Ras-GRF1 Act as Signaling Integrator for Glutamate and Dopamine to Activate ERK2 in the Striatonigral MSN Network
A large body of evidence indicates that cell signaling mechanisms downstream of glutamate and dopamine in the striatum play a central role in the onset of addiction to psychostimulants (14,18,19,64). However, crucial signaling modulators linking neurotransmitter function to Ras and ERK signaling in the striatum still need to be identified. With Ras-GRF1 mutant mice, we explored the possibility that this signaling component might couple glutamate and dopamine D1/5 receptors to Ras in the striatonigral compartment, thus activating the downstream ERK pathway (59,65).
We first determined expression levels of both p140Ras-GRF1 and p135Ras-GRF2, a distinct isoform with partially overlapping functions, at different developmental stages in striatum, cortex, and hippocampus of wild-type (WT) animals. As shown in Figure 1A, Ras-GRF1 is virtually absent at birth, although its expression progressively increases until adulthood (postnatal day P30). The Ras-GRF2, by contrast, appears earlier during postnatal development and reaches an expression plateau as early as P14 (Figure 1B). In addition, analysis of brains prepared from 2-month-old animals showed no change in expression of Ras-GRF2 or of ERK1/2 mitogen-activated protein kinases (MAPK) in Ras-GRF1 null mice (Figure 1C).
Figure 1.
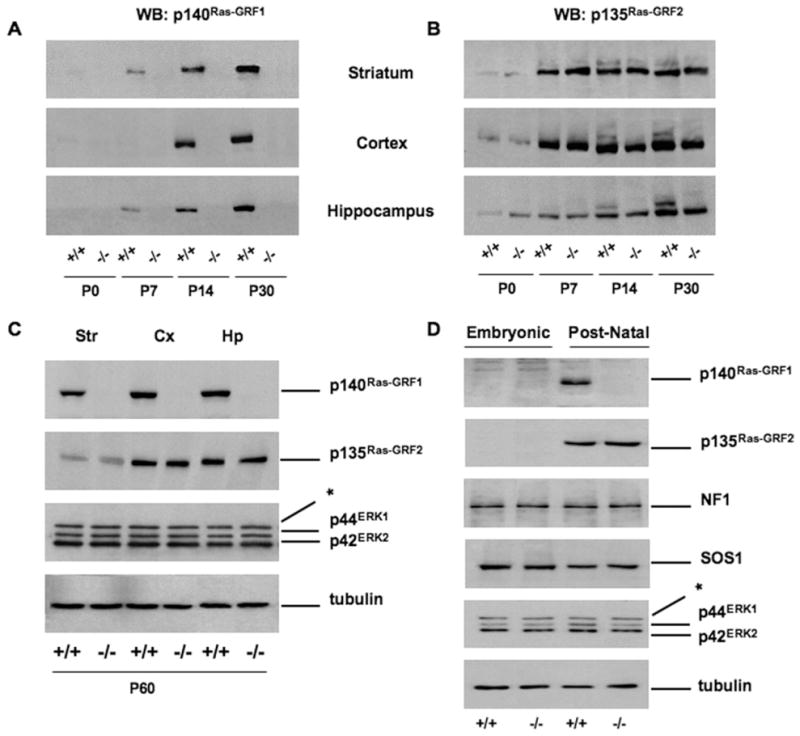
Expression profile of Ras-GRF1 and Ras-GRF2 in brain extracts and primary striatal cultures of wild-type (WT) and Ras-GRF1 knockout (KO) mice. (A) Brain extracts from WT (+/+) and Ras-GRF1 KO (−/−) animals killed at four different time points were prepared for Western blot (WB) analysis and probed with an antibody against p140Ras-GRF1. (B) Same extracts as in (A), probed with p135Ras-GRF2 antibodies. Expression of both Ras-GRF1 and Ras-GRF2 gradually increases over time: postnatal day P0, P7, P14, and P30. Equal amounts of proteins (30 μg) for each time point were loaded. Ras-GRF1 KO and WT mice showed equivalent levels of Ras-GRF2 immunoreactivity in striatum (Str), cortex (Cx), and hippocampus (Hp) at each time point. (C) Representative immunoblots from Str, Cx, and Hp of adult (P60) WT and Ras-GRF1 KO mice. Both Ras-GRF1 and Ras-GRF2 seemed to be highly expressed in Cx and Hp and to a lesser extent in the Str of WT mice. No obvious alterations in the Ras-GRF2 levels could be seen in extracts from Ras-GRF1 KO mice. The p44ERK1 and p42ERK2 kinases were also equally expressed in mice of the different genotypes. (D) Protein extracts were prepared from primary embryonic (E16) striatal cells or postnatal (P1) striatal cultures and analyzed by WB. The p140Ras-GRF1 was only expressed in WT postnatal cells, whereas equal levels of p135Ras-GRF2 were found in postnatal WT and KO cells. Expression of neurofibromin (NF1), SOS-1, p44ERK1, and p42ERK2 are neither developmentally regulated nor affected by the Ras-GRF1 mutation. Tubulin was used as loading control. *Nonspecific band. ERK, extracellular signal-regulated kinase.
To directly investigate the possibility that Ras-GRF1 might be involved in glutamate and/or dopamine activation of ERK signaling in the striatum, we prepared primary neuronal cultures from either embryonic or newborn striata. Whereas WT embryonic cells do not express Ras-GRF1, postnatal neurons kept in culture for 8 days do express p140Ras-GRF1 (Figure 1D). As expected, Ras-GRF1– deficient cells do not express the corresponding p140 protein at any stage of development. Expression of Ras-GRF2 protein seems to be unaffected by the Ras-GRF1 mutation and was found in both WT and Ras-GRF1 knockout (KO) postnatal cells but not in embryonic striatal neurons. We confirmed, as a control, that neurofibromin, SOS-1, ERK1, and ERK2—all core components of the Ras-ERK pathway—are equally expressed in embryonic and postnatal striatal cells and do not seem to be affected by the lack of Ras-GRF1 (Figure 1D).
We then investigated whether activation of ERK1/2 in post-natal MSN after stimulation with glutamate might be influenced by the lack of Ras-GRF1. After 10-min exposure to 100 μmol/L glutamate, ERK1/2 phosphorylation was 63% less in Ras-GRF1–deficient cells in comparison with controls (Figures 2A and 2D).
Figure 2.

Absence of Ras-GRF1 in striatal neurons reduces activation of ERK1/2 in response to glutamate (GLU), SKF 38393 (SKF), and forskolin (Forsk). (A,B,C) WT (upper panels) or Ras-GRF1 KO (KO) (lower panels) postnatal neurons were infected at 8 days after plating with a Semliki-Forest virus expressing green fluorescent protein (SFV/GFP, green) and 8 hours later stimulated for 10 min with either 100 μmol/L GLU (A), 100 μmol/L SKF a D1 receptor agonist (B), or 20 μmol/L Forsk, a direct activator of adenylyl cyclase (C). After stimulation, neurons were fixed and labeled for ERK1/2 activation (p-ERK, red). (D,E,F) Quantification of active-ERK intensity in SFV/GFP positive neurons (merge). The histograms show the mean ± SEM of p-ERK fluorescence intensities normalized to nonstimulated neurons (control [Ctr]). Loss of Ras-GRF1 caused a significant reduction of p-ERK1/2 in response to GLU (D), SKF (E), and Forsk (F). Cells were counted from 5 independent experiments. Two-way analysis of variance, Scheffé test post hoc comparison, WT versus KO, treated only: *p < .01; **p < .001. Abbreviations as in Figure 1.
Previous experimental evidence supports the idea that ERK might be activated by D1-like receptors via a PKA-dependent mechanism in the striatonigral pathway (24). This observation, together with the evidence that PKA might phosphorylate and activate Ras-GRF1 in cortical preparations (57), has prompted us to investigate the possibility that this signaling mediator might be involved in both dopaminergic and PKA-dependent activation of the ERK cascade in the striatum. The Ras-GRF1–deficient striatal cells challenged with the D1-R agonist SKF 38393 failed to show a significant activation of ERK1/2 at 10 min, with 77% less phosphorylation of ERK than in control cells (Figures 2B and 2E). Moreover, Ras-GRF1–deficient cells only partially responded (50% less than controls) to forskolin, a direct activator of adenylyl cyclase (Figures 2C and 2F).
One of the major limitations of using dissociated neuronal cultures for studying cell signaling is the loss of the functional cytoarchitecture of the brain structure of interest. Therefore, to provide definitive evidence for a role of Ras-GRF1 in the integration between glutamate and dopamine signals in a more physiological setting, we prepared organotypic cultures from postnatal mice (P4). We first determined the most effective concentration of both glutamate and SKF 38393 to elicit ERK2 phosphorylation, after a stimulation of 15 min (Figure S1A in Supplement 2). Fifty micromolar of either glutamate or SKF 38393 resulted in a maximal activation of ERK2, without further increase when 100 μmol/L was applied. Importantly, only costimulation of glutamate and SKF 38393 at 50 μmol/L caused a clear additive effect, with an approximately 100% increase of phosphorylated ERK2 (p-ERK2) in comparison with that observed upon single stimulation. Subsequently, we compare the effect on ERK2 of single glutamate, SKF 38393 stimulation, or costimulation at 50 μmol/L (for 15 min). As indicated in Figures 3A and 3B, either single or double stimulation failed to induce p-ERK2 in Ras-GRF1 KO organotypic preparations. In a separate experiment, we also investigated whether forskolin-mediated activation of ERK2 was impeded in the KO slices (Figures 3C and 3D). Indeed, we found, similarly to the results in dissociated cultures (Figure 2F), a significant albeit not complete inhibition of p-ERK2 induction in response to adenylyl cyclase activation. Importantly, we also stimulated slices with brain-derived growth factor (BDNF), a neurotrophin able to activate ERK2 in a calcium-, cAMP-independent manner (53) and confirmed that the Ras-GRF1 mutation did not globally weaken the ability of these slices to respond to extracellular stimuli (Figures 3E and 3F).
Figure 3.

Striatal organotypic slices from Ras-GRF1 KO mice show a marked decrease of p-ERK2 induction in response to glutamate (GLU), SKF 38393, and forskolin (Forsk) but not to brain-derived growth factor (BDNF). (A,B) Organotypic striatal slices (P4) from WT (+/+) and Ras-GRF1 KO (−/−) mice after 3 days in vitro were stimulated for 15 min either with 50 μmol/L GLU, 50 μmol/L SKF, or both (C,D) with 10 μmol/L Forsk and (E,F) with 100 ng/mL BDNF. The histograms show the mean ± SEM of p-ERK2 induction expressed as the ratio normalized to nonstimulated slices (control [CTR]). (B) Absence of Ras-GRF1 caused loss of p-ERK1/2 in response to GLU, SKF, or both (two-way analysis of variance [ANOVA], Scheffé test post hoc comparison GLU, SKF, and GLU+ SKF, *p < .01 genotype effect). (D) Forskolin-mediated activation of ERK2 is partially reduced in Ras-GRF1 slices (two-way ANOVA, Scheffé test post hoc comparison, *p < .01 genotype effect). (F) p-ERK2 is normally induced in Ras-GRF1 KO striatal slices upon BDNF in comparison with WT slices. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; other abbreviations as in Figure 1.
An important open question is the potential crosstalk, in MSN striatonigral circuitry, between glutamate receptor and dopamine D1/5 receptors. Early evidence indicated that cocaine-mediated induction of ERK in vivo in the striatum is not only dependent on dopamine D1 receptors but also on NMDAR (21). However, to elucidate this point in detail it is necessary to use an in vitro system, such as our organotypic slices. We surprisingly found, as indicated in Figure S1B in Supplement 2, a reciprocal dependence for both glutamate and SKF 38393 on NMDAR and D1/5 R subclasses of receptors. In fact, whereas glutamate- and SKF 38393-dependent ERK2 activation is totally abolished by a pretreatment with the corresponding antagonist (APV and SCH 23390, respectively), the same is true when either glutamate is applied in the presence of the dopaminergic antagonist or when SKF 38393 is used in combination with APV. This result clearly indicates that a receptor crosstalk is necessary in striatonigral MSN to activate ERK2, possibly via Ras-GRF1.
Bidirectional Alterations in Ras-GRF1 Expression Do Not Affect Locomotor Activity, Coordination, or Spatial Learning but Influence Procedural Memory Formation
We previously demonstrated that protein levels rather than simply activity of certain core components of the ERK pathway, including ERK1, might critically affect signaling output (32–34). To demonstrate whether increased protein levels of Ras-GRF1 might affect ERK signaling, we first overexpressed p140 (approximately 10-fold) in embryonic striatal cells, which normally do not express it (see Figure 1). Results indicate that Ras-GRF1 might elevate basal ERK phosphorylation while leaving intact glutamate- and SKF 38393-dependent ERK activation (Figure S2 in Supplement 3).
In addition, to further address the role of Ras-GRF1 levels in a more physiological condition in vivo, we took advantage of a mouse model showing a mild overexpression of Ras-GRF1 in the mouse brain. In this transgenic mouse model the silencing element in the Ras-GRF1 locus that causes genomic imprinting and maternal allele inactivation has been removed (66). As shown in Figure 4, biallelic expression in the Ras-GRF1tm2Pds (hereinafter Ras-GRF1 OE) transgenic mice resulted in a moderate increase in p140 protein (threefold WT levels) in striatum, cortex, and hippocampus. Importantly, this genomic modification did not affect the level of p135Ras-GRF2 protein, providing additional evidence that gene expression from the two loci is essentially independent.
Figure 4.

Expression profile of Ras-GRF1 and Ras-GRF2 in Ras-GRF1 overexpressing (OE) mice during postnatal development. (A) Time course analysis of p140Ras-GRF1 expression in striatal, cortical, and hippocampal lysates from WT (+/+) and Ras-GRF1 overexpressing (OE) transgenic mice. At P0 Ras-GRF1 is low in both WT and transgenic animals, whereas its expression becomes pronounced at P30. Note the increased levels of Ras-GRF1 in transgenic animals when compared with WT at this stage. (B) In the same extracts as in (A), p135 Ras-GRF2 gradually increases over time (0, 7, 14, 30 days) without any significant difference between WT and Ras-GRF1 OE transgenic mice. Equal amounts of proteins (30 μg) for each time point were loaded. (C) Western blot analysis of Str, Cx, and Hp dissected from adult (P60) WT and Ras-GRF1 OE mice confirmed the overexpression of Ras-GRF1 in transgenic mice in comparison with WT control subjects. Moreover, p135Ras-GRF2 is similarly expressed in each structure of both genotypes. p44ERK1 and p42ERK2 kinases were also found to be equally expressed. Tubulin was used as loading control. *Nonspecific band. (D) Densitometry measurements of Ras-GRF1 and Ras-GRF2 from same brain extracts used in (C) demonstrate that p140Ras-GRF1 was approximately threefold increased in Ras-GRF1 OE transgenic mice. Mean ± SEM from 3 animals/condition is indicated. One-way ANOVA, genotype effect, *p < .01 OE vs. +/+ mice. Other abbreviations as in Figures 1 and 3.
To study the behavioral consequences of Ras-GRF1 expression, we analyzed both mouse lines, one lacking (Ras-GRF1 KO) and the other mildly overexpressing (Ras-GRF1 OE) Ras-GRF1. Initial data indicate that rotarod performance and spatial learning in the Morris water maze test are essentially unaffected by any of the Ras-GRF1 expression changes (Figure S3 in Supplement 4 and Figure S4 in Supplement 5). These data were obtained by analyzing two distinct, independently generated lines of Ras-GRF1–deficient animals and the Ras-GRF1 OE strain. On the contrary, some forms of procedural memory formation, as assayed in passive avoidance learning, are either impaired in Ras-GRF1 loss of function or facilitated in the mildly overexpressing mouse lines (Figure S5 in Supplement 6).
Ras-GRF1 Levels Affect Striatal ERK1/2 Activation in Response to Acute Cocaine Treatment
To investigate the potential role of Ras-GRF1 in mediating the response to cocaine, we first measured locomotor activity upon acute injection in both Ras-GRF1 KO and Ras-GRF1 OE mice (Figure 5). Before treatment, habituation to the test environment was carried out over 3 days, one trial/day. No differences in the initial horizontal activity or in habituation were found, in any of the genotype groups (Figures 5A and 5B). On day 4, each animal group received saline or cocaine, and locomotor activity was recorded for 15 min. All cocaine-treated animals manifested a significant enhancement in horizontal locomotion in comparison with saline-treated groups. However, neither Ras-GRF1 KO (Figure 5C) nor Ras-GRF1 OE (Figure 5D) mice showed alterations in comparison with their WT control subjects.
Figure 5.

Effects of an acute cocaine (coc) administration on behavioral responses and ERK1/2 activation in Ras-GRF1 mutant mice. (A,B) Spontaneous locomotor activity was measured during 3 consecutive days for 15 min: (A) Ras-GRF1 KO (n = 15) and WT control mice (n = 15); (B) Ras-GRF1 OE (n = 14) and WT littermates (n = 14). All mice displayed similar locomotor responses and pronounced habituation to the test environment. Data are expressed as mean of beam breaks ± SEM. Two-way ANOVA for repeated measures: Ras-GRF1, time effect: [F(2,28) = 106.29, #p < .0001]; Ras-GRF1 OE, time effect: [F(2,26) = 83.74, #p < .0001]. (C,D) On day 4, animals received either a coc injection (15 mg/kg i.p.) or a saline (sal) injection, and locomotor responses were measured for 15 min: (C) (WT coc, n = 10; KO coc, n = 10; WT sal, n = 5; KO sal, n = 5); (D) (WT coc, n = 10; OE coc, n = 10; WT sal, n = 4; OE sal, n = 4). Cocaine-induced locomotor activity was significantly different in comparison with sal administration for both mouse strains, with no difference between genotypes. Two-way ANOVA, Scheffé test post hoc comparison, coc effect WT [F(1,25) = 19.125, p < .0001] and coc effect Ras-GRF1 KO mice [F(1,25) = 60.806, p < .0001]; coc effect WT [F(1,21) = 20.64, p < .0001] and coc effect RasGRF1 OE mice [F(1,23) = 13.26, p < .001]. (E) Photomicrographs showing p-ERK immunoreactivity in dorsal striatum (CPu), nucleus accumbens (NAc), and cingulate cortex (Cig Cx) of sal- and coc-treated animals perfused immediately after locomotor activity on day 4 (C,D). (F,G,H) Quantification of the p-ERK1/2 positive cells from CPu (F), NAc (G), and Cig Cx (H), expressed as mean ± SEM. WT samples from both Ras-GRF1 KO and OE strains were pooled, because they did not show any statistical difference. p-ERK activation was found to be significantly reduced both in CPu and in the NAc of Ras-GRF1 KO coc-treated animals but not in the Cig Cx. Two-way ANOVA, genotype effect (WT vs. KO, treated only, CPu and NAc, *p < .01). Conversely, in Ras-GRF1 OE mice an enhanced ERK activation was observed in the same structures. Genotype effect (WT vs. OE, treated only, CPu and NAc, **p < .001). Other abbreviations as in Figures 1–4.
To evaluate possible changes of MAPK activation, we examined the levels of p-ERK1/2 by immunohistochemical techniques in dorsal striatum, NAc, and cingulate cortex, structures in which ERK phosphorylation has previously been found to be upregulated in response to acute cocaine administration (Figure 5E). Fifteen minutes after single cocaine administration ERK activation was detectable in all three brain areas but with remarkable differences between genotypes. The Ras-GRF1 KO animals showed significant attenuation in both dorsal striatum and NAc (core and shell), whereas Ras-GRF1 OE mice showed a clear increase of p-ERK immunoreactivity in comparison with their WT littermates (Figures 5F and 5G). This last observation indicates that changes in Ras-GRF1 levels affect the initial activation phase of the ERK pathway in response to acute treatment with cocaine in the striatum. In the cingulate cortex, however, p-ERK induction was found essentially comparable in all genotypes, indicating that Ras-GRF1 might not be critically involved in cocaine responses in this cerebral structure (Figure 5H). We consistently found that glutamate-dependent ERK activation in Ras-GRF1 KO cortical preparations is normal, further supporting the idea that removal of Ras-GRF1 from the cortex is not sufficient to impair ERK signaling (Figure S6 in Supplement 7).
Ras-GRF1 Expression Regulates Long-Term Responses to Cocaine
The observed Ras-GRF1–dependent changes in p-ERK levels in response to acute cocaine treatment led us to investigate whether repeated drug administration might cause significant behavioral changes. Behavioral sensitization to cocaine in rodents has been proposed as a model of an initial stage of cocaine addiction in humans, possibly an important step in the development of severe cocaine dependence (67). Not only does this form of behavioral plasticity require ERK activation in the striatum but its onset can be significantly potentiated upon stimulus-dependent enhancement of ERK2 activity, as observed in ERK1-deficient mice (33,34).
As shown in Figure 6, Ras-GRF1 KO and OE mice were chronically treated with cocaine for 9 days. All drug-treated groups showed significant increase in locomotor activity over time in comparison with the saline-treated group. Interestingly, cocaine-related locomotor activity in Ras-GRF1 KO animals was significantly less than in WT mice (Figure 6A). By contrast, Ras-GRF1 OE mice showed a significant increase in locomotion from day 5 onward, in comparison with the corresponding WT control group (Figure 6B). After 9 days of cocaine treatment, conditioned responses to the sensitization context were measured by injecting saline (day 13). No differences were found among cocaine-treated groups. In addition, we also tested the strength of the acquired sensitization response. Animals left for 10 days in the home cage and then rechallenged with a single dose of cocaine manifested a robust locomotor response to the drug, identical to that observed after the last day of chronic treatment, with the Ras-GRF1 KO and Ras-GRF1 OE animals showing attenuated and enhanced responses, respectively, in comparison with control groups.
Figure 6.

Ras-GRF1 bidirectionally regulates coc sensitization and FosB/ΔFosB accumulation. (A,B) Repeated coc injections (15 mg/kg i.p.) promoted a progressive induction of locomotor sensitization as indicated: days 1–3: spontaneous locomotor activity; days 4–12: once daily injection of coc; day 13: single sal injection to monitor conditioned responses; day 23: additional challenge with coc/sal after 10 days of withdrawal. (A) Ras-GRF1 KO and WT coc-treated mice, n = 15 each group; Ras-GRF1 KO and WT sal, n = 10 each group. Ras-GRF1 KO mice clearly manifested a decrease in the locomotor sensitization induced by chronic coc when compared with their littermate control subjects. Two-way ANOVA for repeated measures, genotype effect [F(1,28) = 11.857, *p < .01]. Scheffé test post hoc comparison, Ras-GRF1 mutants vs. WT from days 5–12, *p < .01. When re-exposed to coc 10 days later, WT mice continued to show a much greater locomotor response than Ras-GRF1 mutants, (day 23, Scheffé test post hoc comparison, *p < .05). (B) Ras-GRF1 OE (n = 14) and WT (n = 12) coc-injected mice; Ras-GRF1 OE (n = 10) and WT (n = 10) sal-injected mice. Ras-GRF1 OE animals showed enhanced locomotor sensitization to coc in comparison with their control subjects. Two-way ANOVA for repeated measures, genotype effect [F(1,24) = 12.466, *p < .01]. Ten days after the end of the chronic coc treatment, the sensitization to coc effects on locomotor activity were still greater in Ras-GRF1 OE mice than in the WT group (day 23, Scheffé test post hoc comparison, *p < .01) (C) Photomicrographs showing immunohistochemical labeling of FosB/ΔFosB in CPu and NAc of sal- and chronically coc-treated animals perfused 15 min after the last injection on day 12 (A,B). (D,E) Quantification of the FosB/ΔFosB positive cells from CPu (D) and NAc (E), expressed as mean ± SEM (WT sal = 5; WT coc = 12; KO sal = 5; KO coc = 12; OE sal = 5; OE coc = 11). WT samples from both Ras-GRF1 KO and OE strains were pooled, because they did not show any statistical difference. According to the observed behavioral responses, significant changes in the number of FosB/ΔFosB positive cells were found in both Ras-GRF1 KO and OE mice. Genotype effect (WT vs. KO, WT vs. OE, treated only *p < .01). Other abbreviations as in Figures 1–5.
Long-term treatments with drugs of abuse cause significant changes in gene expression in relevant brain regions, such as the striatum and the NAc (68). To confirm that key molecular markers for cellular adaptations to cocaine are affected by the Ras-GRF1 manipulations, we prepared sections for immunohistochemistry analysis from a distinct set of WT, Ras-GRF1 KO, and Ras-GRF1 OE mice treated with either saline or chronic cocaine, killed 15 min after the last injection, on day 12. In this experimental condition, as a marker for long-term cellular responses to cocaine, the chronic form of FosB, ΔFosB, is detected (69). Quantitative analyses in the dorsal striatum and in the NAc (Figures 6C–6E) indicate that FosB/ΔFosB immunoreactive cells are consistently more abundant in Ras-GRF1 OE mice and remarkably less abundant in Ras-GRF1 KO striatum and NAc, in comparison with the WT, cocaine-treated animals.
We also measured the rewarding effect of cocaine with the conditioned place preference (CPP) paradigm. In this classical Pavlovian conditioning procedure, the drug serves as an unconditioned stimulus that is repeatedly paired with one side of the testing chamber (conditioned stimulus). On the day of testing, an increase in time spent in the drug-paired context relative to a control value is taken as evidence that the unconditioned stimulus was rewarding (33,70). In CPP analysis, Ras-GRF1–deficient mice spent significantly less time than control subjects in the drug-paired compartment, indicating the presence of a clear impairment in the perception of reward from cocaine in these mice (Figure 7A). In contrast, Ras-GRF1 OE mutants showed a marked enhancement in reward sensitivity to the same doses of cocaine (Figure 7B). The analysis of both Ras-GRF1 lines indicates that the level of expression of this molecule is important to the formation of the CPP response as well as locomotor sensitization to cocaine.
Figure 7.

Loss and increase of Ras-GRF1 cause opposite effects in conditioned place preference (CPP). Conditioned place preference is expressed as difference between postconditioning and preconditioning time spent in the drug-paired compartment of the training apparatus. (A) Rewarding properties of cocaine (15 mg/kg i.p.) were measured in Ras-GRF1 KO (n = 12) and WT (n = 14) animals. Corresponding saline-treated groups were used as control (WT = 15; KO = 15). Ras-GRF1 KO mice clearly manifested a reduction in CPP responses to cocaine when compared with their littermate control subjects. Two-way ANOVA revealed a significant effect of treatment [F(1,53) = 90.643, p < .0001], genotype [F(1,53) = 13.915, p < .0001], and interaction between these two factors [F(1,53) = 18.383, p < .0001]. Subsequent one-way ANOVA indicated that Ras-GRF1– deficient mice spent significantly less time than control subjects in the drug-paired compartment [F(1,28) = 79.968, ***p < .0001]. (B) Ras-GRF1 OE (n = 19) and WT (n = 16) animals underwent the same CPP protocol as in (A), including the saline-treated groups (WT = 12; OE = 14). Ras-GRF1 OE animals showed enhanced CPP responses to cocaine in comparison with the control subjects. Two-way ANOVA revealed a significant effect of treatment [F(1,57) = 245.202, p < .0001], genotype [F(1,57) = 12.038, p < .01], and interaction between these two factors [F(1,57) = 29.644, p < .0001]. One-way ANOVA for genotype effects indicated a significant difference between cocaine-treated Ras-GRF1 OE mutants and relative control subjects [F(1,33) = 32.893, ***p < .0001]. Other abbreviations as in Figures 1 and 3.
Discussion
Molecular mechanisms underlying striatal physiology are central to a variety of motor and cognitive processes, including procedural learning, reward, habit formation, and drug addiction (17,68,71).
With the present work we have focused on a signal transduction pathway, the ERK cascade, which is activated in both dorsal and ventral striatum by most of the drugs of abuse upon single administration (22). From the signaling point of view striatal networks are peculiar, in comparison with other brain structures commonly activated by drugs, in the sense that their activity largely relies on a complex interplay between glutamate- and dopamine-dependent inputs (16). Several studies clearly indicate that cocaine-dependent ERK activation in the striatum, specifically in the striatonigral MSN compartment, requires both calcium influx from AMPA/NMDA receptors and cAMP/PKA activity harboring from D1/5 receptor activation, making cocaine an excellent pharmacological tool to investigate the interaction between these two receptor systems in vivo (18,19,42). Our in vitro data extended previous observations on glutamate and dopamine cell signaling by firmly establishing a unique role for Ras-GRF1 in the control of the striatal ERK cascade. In particular, two important conclusions can be drawn from our in vitro results. First, we showed for the first time that an apparently linear (additive) signal integration between glutamate and dopamine converges on ERK1/2 in the striatonigral MSN pathway and that integration is totally lost in the absence of Ras-GRF1. Second, a direct crosstalk seems to exist between NMDA receptors and D1/5 receptors even when a single stimulus is applied, because both neurotransmitter systems require, to induce ERK1/2 activation, the presence not only of their direct receptors (i.e., NMDAR for glutamate, D1/5 R for SKF 38393) but also the reciprocal one. This observation might suggest that Ras-GRF1, being down-stream to both receptor systems, must be simultaneously activated to transduce an integrated signal downstream to ERK1/2.
These observations about the integration of glutamate and dopamine on the ERK pathway might be unique to the striatum. For instance, it has been shown in hippocampal preparations that a nonlinear (synergistic) integration of glutamate and dopamine signals exist and that only dopaminergic activity requires NMDA receptors for stimulating ERK1/2 but not the opposite (72). This is probably also linked to the limited ability of dopamine to activate ERK in cortical areas (see also our data in Figure 6B in Supplement 7). Certainly, in cortical areas, the role of Ras-GRF1 seems to be limited too. Our expression data in WT and Ras-GRF1 KO animals in combination with in situ hybridization data recently made available by the Allen Institute for Brain Science (http://www.brain-map.org) provide a likely explanation of the compensatory effect between Ras-GRF1 and Ras-GRF2 in cortical areas. Both homologues are highly expressed in cortex, although Ras-GRF2 seems to be largely confined to the external layers. Similarly, in the hippocampus, Ras-GRF2 is almost exclusively expressed in the dentate gyrus, whereas Ras-GRF1 can also be found in the CA3-CA1 region. Remarkably, no RNA signal can be detected in the striatum for Ras-GRF2, and a very low-level of p135 immunoreactivity can be found by WB analysis. The Ras-GRF1 instead is significantly expressed in this structure, at somewhat lower levels than in cortical region. We propose that the phenotypes we found in striatum-dependent signaling and behavior in the Ras-GRF1 KO animals are likely to be due to a lack of compensatory effect exerted by Ras-GRF2. Importantly, in agreement with a recent microarray analysis, we failed to observe changes in Ras-GRF2 levels in either our Ras-GRF1 KO or OE animals, confirming the idea that the two loci are independently regulated (73).
An unusual feature of the MSN network is their NMDAR composition. A common developmental pattern in most brain regions is the decline with maturation of the initial high NR2B levels found early in development and a concomitant increase in NR2A expression levels in the forebrain (74–76). One exception is the striatum, where NR2B expression remains high in MSN through adulthood (77–80). Considering that Ras-GRF1 (but not Ras-GRF2) preferentially binds to the NR2B subunit (56) and that the deactivation of NR2B-containing receptors is much slower (81), it is conceivable to assume a stronger activation and a more prominent role in striatal signaling of Ras-GRF1 in comparison with cortical or hippocampal signaling. Obviously, this observation does not exclude the possibility that other calcium-sensitive Ras-GEFs might also contribute in the striatum to ERK activation, including members of Ras-GRP/CalDAG GEF family that are striatal enriched (82–86).
An important aspect that will require further investigation is the link among D1-like receptors, adenylyl cyclase-dependent signaling, and Ras-GRF1. Our in vitro findings, although indicating that most of D1R-dependent ERK1/2 activation requires Ras-GRF1, show only a limited dependence from p140 of forskolin-mediated activation. So far, all models linking cAMP to ERK in the striatum have suggested a major role of the PKA-dependent phosphorylation of DARPP-32 on Thr-34 that would causes an inhibition of the PP-1 and STEP phosphatase cascade, normally inhibiting MEK1/2 activity (24,87). Considering that PKA has previously been shown to phosphorylate p140 in culture cells on Ser916 (45,51,57,88), our data support the idea that the cAMP pathway mediates ERK activation via a direct mechanism (Ras-GRF1–dependent) and an indirect one (DARPP-32–dependent). Thus, we believe that Ras-GRF1 and DARPP-32 cooperate in striatonigral MSN networks in sustaining ERK activity in addition to the general role played by DARPP-32 in regulating striatal cell signaling (44,89).
At the behavioral level, bidirectional changes in Ras-GRF1 levels, as observed in two independent lines of KO and one OE strain, do not seem to significantly impact on locomotor activity and hippocampal-dependent learning in the Morris water maze but do affect performance in procedural learning tasks such as the passive avoidance. This largely agrees with our original report indicating that both hippocampal long-term potentiation and spatial memory in the Ras-GRF1 KO animals are intact, whereas fear conditioning and procedural learning are impaired (59), although it is somewhat in disagreement with an earlier independent investigation (61).
Consistently, with a bidirectional change in procedural memory formation and altered glutamate and dopamine signaling in the striatum—both largely dependent on Ras-GRF1 gene dosage—we found corresponding changes in cocaine-dependent behavior. There are two reasons that cocaine was used in our Ras-GRF1 mouse models: to confirm, with an in vivo approach, our hypothesis that p140 acts in the striatonigral compartment as an integrator for glutamate and dopamine signaling; and to demonstrate that gene dosage of this signaling intermediate is important for early phases of drug addiction. Despite the extensive work on the role of ERK signaling in cocaine-dependent responses, the large majority of studies have been confined to simply demonstrate a permissive role of this pathway in the process by using pharmacological blockade of MEK1/2 kinase. Two notable exceptions are the studies on ERK1- and MSK1-deficient mice in which ERK-dependent complex behavioral changes in response to cocaine were observed (13,34). The effect of ERK1 ablation is particularly relevant in this context, because it essentially produces opposite changes to those found by inhibiting both ERK1 and ERK2 kinases simultaneously, demonstrating that ERK1 might act in response to appropriate stimuli as a partial agonist to ERK2 in the MEK-dependent activation process. The enhancement of both locomotor sensitization and CPP responses in the ERK1 KO animals is remarkably similar to what we observed in the Ras-GRF1 OE mice. In our opinion this finding conclusively demonstrates that Ras-GRF1-controlled ERK activation in the striatum plays both permissive and instructive roles downstream to dopamine and glutamate, thus affecting global cellular adaptations occurring in response to drugs of abuse.
Supplementary Material
Acknowledgments
This work was supported by the Italian Ministry for the University and Research (MIUR) (RB, TP, and MG) and by the Mariani Foundation for Neurological Research (RB). We would like to thank Steven Clapcote for the GENA53 mice and Marzia Indrigo for excellent technical assistance.
Footnotes
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 2.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Davis S, Laroche S. Mitogen-activated protein kinase/extracellular regulated kinase signalling and memory stabilization: A review. Genes Brain Behav. 2006;5(suppl 2):61–72. doi: 10.1111/j.1601-183X.2006.00230.x. [DOI] [PubMed] [Google Scholar]
- 4.Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatiotemporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- 5.Malumbres M, Barbacid M. RAS oncogenes: The first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 6.Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- 7.Shaul YD, Seger R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–176. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valjent E, Caboche J, Vanhoutte P. Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: A molecular substrate for learning and memory? Mol Neurobiol. 2001;23:83–99. doi: 10.1385/MN:23:2-3:083. [DOI] [PubMed] [Google Scholar]
- 10.Adams JP, Sweatt JD. Molecular psychology: Roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. doi: 10.1146/annurev.pharmtox.42.082701.145401. [DOI] [PubMed] [Google Scholar]
- 11.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 12.Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 13.Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, et al. Parsing molecular and behavioral effects of cocaine in mitogen-and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 15.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- 16.Fasano S, Brambilla R. Cellular mechanisms of striatum-dependent behavioral plasticity and drug addiction. Curr Mol Med. 2002;2:649–665. doi: 10.2174/1566524023362005. [DOI] [PubMed] [Google Scholar]
- 17.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: The role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 18.Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Girault JA, Valjent E, Caboche J, Herve D. ERK2: A logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Pierce RC, Pierce-Bancroft AF, Prasad BM. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/mitogen-activated protein kinase signal transduction cascade. J Neurosci. 1999;19:8685–8695. doi: 10.1523/JNEUROSCI.19-19-08685.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valjent E, Pages C, Herve D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- 23.Radwanska K, Caboche J, Kaczmarek L. Extracellular signal-regulated kinases (ERKs) modulate cocaine-induced gene expression in the mouse amygdala. Eur J Neurosci. 2005;22:939–948. doi: 10.1111/j.1460-9568.2005.04286.x. [DOI] [PubMed] [Google Scholar]
- 24.Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen PC, Lao CL, Chen JC. Dual alteration of limbic dopamine D1 receptor-mediated signalling and the Akt/GSK3 pathway in dopamine D3 receptor mutants during the development of methamphetamine sensitization. J Neurochem. 2007;100:225–241. doi: 10.1111/j.1471-4159.2006.04203.x. [DOI] [PubMed] [Google Scholar]
- 26.Gerdjikov TV, Ross GM, Beninger RJ. Place preference induced by nucleus accumbens amphetamine is impaired by antagonists of ERK or p38 MAP kinases in rats. Behav Neurosci. 2004;118:740–750. doi: 10.1037/0735-7044.118.4.740. [DOI] [PubMed] [Google Scholar]
- 27.Mizoguchi H, Yamada K, Mizuno M, Mizuno T, Nitta A, Noda Y, et al. Regulations of methamphetamine reward by extracellular signal-regulated kinase 1/2/ets-like gene-1 signaling pathway via the activation of dopamine receptors. Mol Pharmacol. 2004;65:1293–1301. doi: 10.1124/mol.65.5.1293. [DOI] [PubMed] [Google Scholar]
- 28.Valjent E, Corbille AG, Bertran-Gonzalez J, Herve D, Girault JA. Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci U S A. 2006;103:2932–2937. doi: 10.1073/pnas.0511030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valjent E, Corvol JC, Trzaskos JM, Girault JA, Herve D. Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006;7:20. doi: 10.1186/1471-2202-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci. 2005;8:212–219. doi: 10.1038/nn1383. [DOI] [PubMed] [Google Scholar]
- 32.Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Porrazzo A, Orban PC, et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 34.Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE. Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology. 2006;31:2660–2668. doi: 10.1038/sj.npp.1301014. [DOI] [PubMed] [Google Scholar]
- 35.Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 36.Girault JA, Greengard P. The neurobiology of dopamine signaling. Arch Neurol. 2004;61:641–644. doi: 10.1001/archneur.61.5.641. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Lou D, Jiao H, Zhang D, Wang X, Xia Y, et al. Cocaine-induced intracellular signaling and gene expression are oppositely regulated by the dopamine D1 and D3 receptors. J Neurosci. 2004;24:3344–3354. doi: 10.1523/JNEUROSCI.0060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao L, Tang Q, Samdani S, Liu Z, Wang JQ. Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur J Neurosci. 2004;19:1207–1216. doi: 10.1111/j.1460-9568.2004.03223.x. [DOI] [PubMed] [Google Scholar]
- 39.Yang L, Mao L, Tang Q, Samdani S, Liu Z, Wang JQ. A novel Ca2+-independent signaling pathway to extracellular signal-regulated protein kinase by coactivation of NMDA receptors and metabotropic glutamate receptor 5 in neurons. J Neurosci. 2004;24:10846–10857. doi: 10.1523/JNEUROSCI.2496-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ. The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci. 2005;25:2741–2752. doi: 10.1523/JNEUROSCI.4360-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voulalas PJ, Holtzclaw L, Wolstenholme J, Russell JT, Hyman SE. Metabotropic glutamate receptors and dopamine receptors cooperate to enhance extracellular signal-regulated kinase phosphorylation in striatal neurons. J Neurosci. 2005;25:3763–3773. doi: 10.1523/JNEUROSCI.4574-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Herve D, Valjent E, et al. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferguson SM, Norton CS, Watson SJ, Akil H, Robinson TE. Amphetamine-evoked c-Fos mRNA expression in the caudate-putamen: The effects of DA and NMDA receptor antagonists vary as a function of neuronal phenotype and environmental context. J Neurochem. 2003;86:33–44. doi: 10.1046/j.1471-4159.2003.01815.x. [DOI] [PubMed] [Google Scholar]
- 44.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: An integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 45.Mattingly RR, Macara IG. Phosphorylation-dependent activation of the Ras-GRF/CDC25Mm exchange factor by muscarinic receptors and G-protein beta gamma subunits. Nature. 1996;382:268–272. doi: 10.1038/382268a0. [DOI] [PubMed] [Google Scholar]
- 46.Zippel R, Orecchia S, Sturani E, Martegani E. The brain specific Ras exchange factor CDC25Mm: Modulation of its activity through Gi-protein-mediated signals. Oncogene. 1996;12:2697–2703. [PubMed] [Google Scholar]
- 47.Zippel R, Gnesutta N, Matus-Leibovitch N, Mancinelli E, Saya D, Vogel Z, et al. Ras-GRF, the activator of Ras, is expressed preferentially in mature neurons of the central nervous system. Mol Brain Res. 1997;48:140–144. doi: 10.1016/s0169-328x(97)00120-4. [DOI] [PubMed] [Google Scholar]
- 48.Sturani E, Abbondio A, Branduardi P, Ferrari C, Zippel R, Martegani E, et al. The Ras guanine nucleotide exchange factor is present at the synaptic junction. Exp Cell Res. 1997;235:117–123. doi: 10.1006/excr.1997.3660. [DOI] [PubMed] [Google Scholar]
- 49.Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–526. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- 50.Buchsbaum R, Telliez JB, Gooneskera S, Feig LA. The N-terminal pleckstrin, coiled-coil, and IQ domains of the exchange factor Ras-GRF act cooperatively to facilitate activation by calcium. Mol Cell Biol. 1996;16:4888–4896. doi: 10.1128/mcb.16.9.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mattingly RR, Saini V, Macara IG. Activation of the Ras-GRF/CDC25Mm exchange factor by lysophosphatidic acid. Cell Signal. 1999;11:603–610. doi: 10.1016/s0898-6568(99)00034-0. [DOI] [PubMed] [Google Scholar]
- 52.Innocenti M, Zippel R, Brambilla R, Sturani E. CDC25(Mm)/Ras-GRF1 regulates both Ras and Rac signaling pathways. FEBS Lett. 1999;460:357–362. doi: 10.1016/s0014-5793(99)01374-5. [DOI] [PubMed] [Google Scholar]
- 53.Orban PC, Chapman PF, Brambilla R. Is the Ras-MAPK signalling pathway necessary for long-term memory formation? Trends Neurosci. 1999;22:38–44. doi: 10.1016/s0166-2236(98)01306-x. [DOI] [PubMed] [Google Scholar]
- 54.Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 55.Tian X, Feig LA. Age-dependent participation of Ras-GRF proteins in coupling calcium-permeable AMPA glutamate receptors to Ras/Erk signaling in cortical neurons. J Biol Chem. 2006;281:7578–7582. doi: 10.1074/jbc.M512060200. [DOI] [PubMed] [Google Scholar]
- 56.Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, et al. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- 57.Yang H, Cooley D, Legakis JE, Ge Q, Andrade R, Mattingly RR. Phosphorylation of the Ras-GRF1 exchange factor at Ser916/898 reveals activation of Ras signaling in the cerebral cortex. J Biol Chem. 2003;278:13278–13285. doi: 10.1074/jbc.M209805200. [DOI] [PubMed] [Google Scholar]
- 58.Tian X, Gotoh T, Tsuji K, Lo EH, Huang S, Feig LA. Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J. 2004;23:1567–1575. doi: 10.1038/sj.emboj.7600151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, et al. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 60.Tonini R, Franceschetti S, Parolaro D, Sala M, Mancinelli E, Tininini S, et al. Involvement of CDC25Mm/Ras-GRF1-dependent signaling in the control of neuronal excitability. Mol Cell Neurosci. 2001;18:691–701. doi: 10.1006/mcne.2001.1050. [DOI] [PubMed] [Google Scholar]
- 61.Giese KP, Friedman E, Telliez JB, Fedorov NB, Wines M, Feig LA, et al. Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine-nucleotide releasing factor 1 (Ras-GRF1) Neuropharmacology. 2001;41:791–800. doi: 10.1016/s0028-3908(01)00096-x. [DOI] [PubMed] [Google Scholar]
- 62.Li S, Tian X, Hartley DM, Feig LA. The environment versus genetics in controlling the contribution of MAP kinases to synaptic plasticity. Curr Biol. 2006;16:2303–2313. doi: 10.1016/j.cub.2006.10.028. [DOI] [PubMed] [Google Scholar]
- 63.Li S, Tian X, Hartley DM, Feig LA. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26:1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelley AE. Memory and addiction: Shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 65.Clapcott SJ, Peters J, Orban PC, Brambilla R, Graham CF. Two ENU-induced mutations in Rasgrf1 and early mouse growth retardation. Mamm Genome. 2003;14:495–505. doi: 10.1007/s00335-002-2258-4. [DOI] [PubMed] [Google Scholar]
- 66.Yoon B, Herman H, Hu B, Park YJ, Lindroth A, Bell A, et al. Rasgrf1 imprinting is regulated by a CTCF-dependent methylation-sensitive enhancer blocker. Mol Cell Biol. 2005;25:11184–11190. doi: 10.1128/MCB.25.24.11184-11190.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson TE, Berridge KC. The neural basis of drug craving: An incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 68.Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med. 2004;55:113–132. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- 69.Kelz MB, Nestler EJ. deltaFosB: A molecular switch underlying long-term neural plasticity. Curr Opin Neurol. 2000;13:715–720. doi: 10.1097/00019052-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 70.Maldonado R, Saiardi A, Valverde O, Samad TA, Roques BP, Borrelli E. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature. 1997;388:586–589. doi: 10.1038/41567. [DOI] [PubMed] [Google Scholar]
- 71.Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: From actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- 72.Kaphzan H, Doron G, Rosenblum K. Co-application of NMDA and dopamine-induced rapid translation of RSK2 in the mature hippocampus. J Neurochem. 2007;103:388–399. doi: 10.1111/j.1471-4159.2007.04774.x. [DOI] [PubMed] [Google Scholar]
- 73.Fernandez-Medarde A, Porteros A, de las Rivas J, Nunez A, Fuster JJ, Santos E. Laser microdissection and microarray analysis of the hippocampus of Ras-GRF1 knockout mice reveals gene expression changes affecting signal transduction pathways related to memory and learning. Neuroscience. 2007;146:272–285. doi: 10.1016/j.neuroscience.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 74.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, et al. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 75.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 76.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 77.Li L, Fan M, Icton CD, Chen N, Leavitt BR, Hayden MR, et al. Role of NR2B-type NMDA receptors in selective neurodegeneration in Huntington disease. Neurobiol Aging. 2003;24:1113–1121. doi: 10.1016/j.neurobiolaging.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 78.Ghasemzadeh MB, Sharma S, Surmeier DJ, Eberwine JH, Chesselet MF. Multiplicity of glutamate receptor subunits in single striatal neurons: An RNA amplification study. Mol Pharmacol. 1996;49:852–859. [PubMed] [Google Scholar]
- 79.Standaert DG, Friberg IK, Landwehrmeyer GB, Young AB, Penney JB., Jr Expression of NMDA glutamate receptor subunit mRNAs in neurochemically identified projection and interneurons in the striatum of the rat. Brain Res Mol Brain Res. 1999;64:11–23. doi: 10.1016/s0169-328x(98)00293-9. [DOI] [PubMed] [Google Scholar]
- 80.Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB, Jr, Young AB. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–5307. doi: 10.1523/JNEUROSCI.15-07-05297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen N, Luo T, Raymond LA. Subtype-dependence of NMDA receptor channel open probability. J Neurosci. 1999;19:6844–6854. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:1082–1086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- 83.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- 84.Kawasaki H, Springett GM, Toki S, Canales JJ, Harlan P, Blumenstiel JP, et al. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc Natl Acad Sci U S A. 1998;95:13278–13283. doi: 10.1073/pnas.95.22.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 86.Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- 87.Fernandez E, Schiappa R, Girault JA, Le Novere N. DARPP-32 is a robust integrator of dopamine and glutamate signals. PLoS Comput Biol. 2006;2:e176. doi: 10.1371/journal.pcbi.0020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Norum JH, Methi T, Mattingly RR, Levy FO. Endogenous expression and protein kinase A-dependent phosphorylation of the guanine nucleotide exchange factor Ras-GRF1 in human embryonic kidney 293 cells. FEBS J. 2005;272:2304–2316. doi: 10.1111/j.1742-4658.2005.04658.x. [DOI] [PubMed] [Google Scholar]
- 89.Nairn AC, Svenningsson P, Nishi A, Fisone G, Girault JA, Greengard P. The role of DARPP-32 in the actions of drugs of abuse. Neuropharmacology. 2004;47(suppl 1):14–23. doi: 10.1016/j.neuropharm.2004.05.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.