

Abstract
Genome sequences of human Rhinoviruses (HRV) have primarily been from stocks collected in the 1960’s, with genomes and phylogeny of modern HRVs remaining undefined. Here, two modern isolates (hrv-A101 and hrv-A101-v1) collected ~8 years apart were sequenced in their entirety. Incorporation into our full-genome HRV alignment with subsequent phylogenetic network inference indicated that these represent a unique HRV-A, localized within an early diverging clade. They appear to have resulted from recombination of the hrv-65 and hrv-78 lineages. These results support our contention that there are unrecognized distinct HRV-A strains, and that recombination is evident in currently circulating strains.
Human Rhinoviruses (HRV) are positive-sense, single-stranded RNA viruses, of the family Picornaviridae, genus Enterovirus, with three species denoted HRV-A, -B and -C. The members of this viral family share a common genome structure that consists of a 5′ untranslated region (5′UTR), one long open reading frame, and a 3′ untranslated region (3′UTR). After translation, HRV-derived proteases cleave the product into 11 proteins, including four that form the capsid and the remaining 7 that are involved in translation, replication, and virus-host interactions [17]. HRV infection is frequently the etiologic agent of upper respiratory tract infections, and besides the morbidity associated with the “common cold”, HRV infection can lead to sinusitis, otitis media, and exacerbations of asthma, chronic obstructive lung disease, and cystic fibrosis [2, 3, 6, 12, 15]. What is not apparent from epidemiologic studies to date are the genomic features that are responsible for these more complex phenotypes (such as asthma exacerbations) amongst the >100 known HRV strains. Tapparel et al., Kistler et al., and our group recently undertook full genome sequencing and analysis of recognized HRV-A and -B strains [8, 11, 16], the majority of which were from the American Type Culture Collection, a repository for the canonical reference set. (HRV-C is a recently recognized species that has yet to be propagated in cells, with seven full genome sequences submitted to GenBank between November 2007 and August 2009.) By construction of a structure-based alignment of this reference set and the HRV-C strains, we established a framework for subsequent analysis of newly identified strains [8, 11, 16]. Virtually all of these HRV-A and -B were from samples collected primarily in the 1960’s, with the few more recent field samples being found to fit into the phylogeny as minor variants of a known reference strain [11]. Certain features of the phylogeny, our identification of previously unrecognized recombination events amongst HRVs, and the likely high error rate of the HRV polymerase, have all suggested that other distinct strains of HRV-A and -B are likely to be in current circulation within the population. Indeed, several recent studies of clinical samples report partial sequence that suggests previously unrecognized HRV strains [1, 7, 9, 14]. In this study, we have identified one such novel strain from two recently obtained respiratory samples, and herein report the complete genome sequences and subsequent analyses.
The HRV samples were both collected using nasal lavage from two individuals with upper respiratory tract infections. The samples, denoted hrv-A101 and hrv-A101-v1, were collected at two different points in time and geographic locations: one in 2000 in Madison, Wisconsin, and the other in 2008 in Baltimore, Maryland. Sample hrv-A101 was selected because it was identified as a potential new HRV-A strain by its 5′UTR sequence as described previously [9]. The samples were stored at −80°C until RNA preparation and extraction. Full HRV genome sequence was obtained using the modified sequence-independent single-primer amplification method as we have recently described in detail [11]. Briefly, after RNAse and DNAse treatments to reduce exogenous contaminants, viral RNA was extracted and cDNA synthesized using random hexamers. The cDNA was PCR amplified, purified, and then cloned and transformed using the Topo TA Cloning kit (Invitrogen). Several hundred individual clones were picked, expanded, and sequenced using an Applied Biosystems ABI 3700 instrument. HRV genomes were assembled from these clones with the Cap3 program (http://pbil.univ-lyon1.fr/cap3.php), and regions of low coverage or missing sequence were addressed by specific PCRs and sequencing. The average coverage for both genomes was >33-fold.
Using the statistical profiles generated from our reference set alignment, hrv-A101 and hrv-A101-v1 sequences were aligned using the program HMMER 2.3.2 (http://hmmer.janelia.org/) [11]. To generate the phylogenetic network, the programs PAUP (http://paup.csit.fsu.edu/paupfaq/faq.html) and Modeltest, were used to select the best model of nucleotide substitution based on fitting 56 different models to the data and comparing the fits via Likelihood ratio tests and Akaike Information Criterion [13]. Using the best-fit model (GTR + γ + I), a network was inferred with Neighbor-Net calculated splits and constructed with EqualAngle in the SplitsTree4 v4.10 program [4, 5]. The sequences were analyzed for recombination with all known full-length HRV sequences using the suite of programs within the Recombination Detection Program 3 (RDP3) [10]. The default settings were used except for changing the sequence type to ‘Linear’, setting the P-value = 0.001, requiring that more than 2 programs predict the event, and using 5 programs (RDP, GENECONV, MaxChi, 3Seq, and Chimaera) to initially detect recombination events before refinement of the results with Bootscan and SiScan. The program MEGA (http://www.megasoftware.net/) was used to calculate pairwise p-distances (used to calculate percentage identity).
Neighbor-Net analysis of the full-length genome nucleotide alignment resulted in the phylogenetic network shown in Fig. 1. Because the reference data set showed evidence of recombination among HRV strains [11], a phylogenetic network rather than a bifurcating tree was generated to infer the evolutionary relationships. Unlike a single bifurcating tree, a network can illustrate conflicting phylogenetic signals that are associated with more complex models of evolution such as recombination [4, 5]. There are 106 taxa represented in this network including 76 HRV-A, 25 HRV-B, and 5 HRV-C strains, and statistical support for their associations are shown as percentages of 1000 bootstrap replicates (the reference strains correspond to those found in Palmenberg et al. [11]). The two new sequences localize within the HRV-A species; they form a monophyletic cluster within a clade containing hrv-71, -51, and -65 among others (indicated in Fig. 1 as ‘clade 1’). The bootstrap support values on the outermost branches, ranging from 93-100%, indicate the high reliability of the relationships. The separate branching of the hrv-A101 lineage and the supporting bootstrap values are consistent with these isolates representing a novel and distinct HRV. Furthermore, the nucleotide difference between hrv-A101 and its closest strain was 76%, a value that is lower than virtually all other HRV-A analyzed in the same manner (Fig. 2A). We note that based on its position in the network, hrv-A101 most likely binds to cells via the ICAM-1 receptor [11]. Taken together, these data indicate that hrv-A101 represents a distinct lineage localized within the HRV-A species.
Fig. 1.
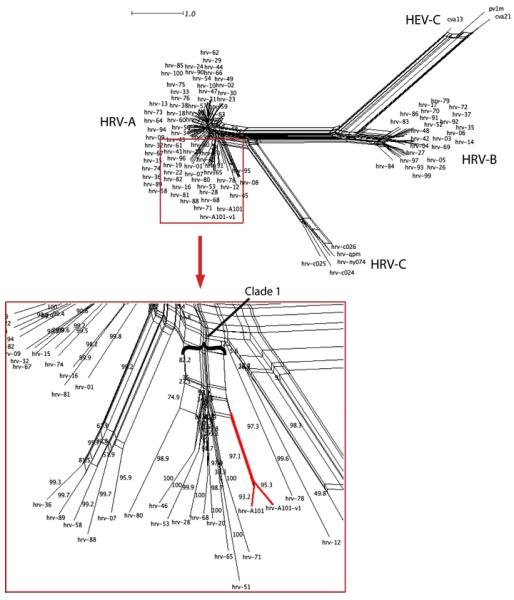
Neighbor-Net analysis of 106 full nucleotide genomes of HRV-A, HRV-B, and HRV-C strains. The network, shown in the upper right, demonstrates the expected clustering of the three Enterovirus species formed by their respective strains. The red box (bottom left) is a magnified view of a portion of the HRV-A species showing the detailed localization of hrv-A101 and hrv-A101-v1 (highlighted in red) as a monophyletic cluster within clade 1. The complex shapes further in the network (parallelograms) represent regions of parallel evolution resulting from either recombination or selection.
Fig. 2.
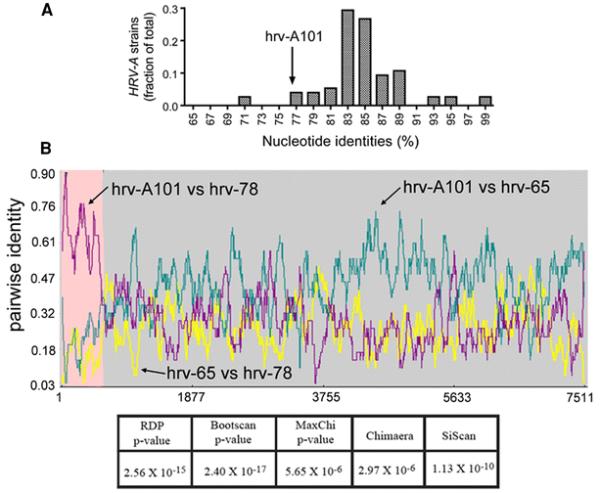
Identity comparisons and evidence for recombination of hrv-A101. In A), a plot of the frequency of the percentage nucleotide identity between each HRV-A strain and its closest sister strain is shown. hrv-A101 (arrow) had its highest identity (76.6%) with reference strain hrv-53. This value is lower than the majority of identities found between all other HRV-A analyzed in a similar manner, supporting the inferred phylogenetic relationship of hrv-A101. In B), the genome-wide pairwise identities between hrv-A101 and hrv-78 and hrv-65 are shown. Recombination between ancestral hrv-65 (major parent) and hrv-78 (minor parent) was predicted to occur at nucleotide ~506, representing recombination of the majority of the 5′UTR of hrv-65. The prediction was highly statistically significant as indicated by the P-values from the five tests.
Recombination analysis detected that hrv-A101 was formed by a recombination event between two HRV-A strains in the hrv-65 and hrv-78 lineages (Fig. 2B). The event was detected by all five of the RDP3 programs, with p-values < 1E-5. The majority of the 5′UTR of hrv-A101 was donated by hrv-78 lineage, while the coding region and 3′UTR of the viral genome were donated by hrv-65. Fig. 2B also illustrates that despite the strong evidence for recombination, hrv-A101 shows substantial genomic variation from the apparent parental strains. Thus, the three genome sequences shown here, which provide evidence of a past recombination event, represent closely related members of their respective lineages (hrv-65, hrv-78, and hrv-A101). Similar evidence for recombination was found for hrv-A101-v1 (data not shown).
hrv-A101 and hrv-A101-v1 lie very close together in the network, with evolutionary relationships and identities indicative of these being from the same lineage with a small degree of sequence variation. At the nucleotide level, there is a 92.3% identity between the two, and a 97.0% identity when comparing the amino acids. Fig. 3 shows the distribution of these variations between hrv-A101 and hrv-A101-v1 by genomic feature. For the open reading frame, the prevalence of variations ranged from ~6% to ~11% based on region. The 3′UTR was without variation, while the 5′UTR had an ~3% difference between the two. When examining only the nonsynonymous variations, several observations were noted. First, VP4 was the only region without differences in amino acid sequence. Although relatively small, VP4 is similar in total amino acids to 2B, 3A, and 3B, each of which do exhibit nonsynonymous variations. Thus the lack of variation in VP4 may not necessarily be attributed to its small size. When considering the nonsynonymous variations, the relatively large 2C region also appears to be minimally variant (0.31%). Sequences from additional isolates will be necessary, though, to ascertain whether VP4 and 2C amino acid sequence is maintained in other hrv-A101 clinical samples. Further stratification of the nonsynonymous variations as conservative and nonconservative (no change, or change, respectively, in polarity or charge) revealed that the few variations observed in 2C and 3A were conservative in nature. In contrast, the moderately variable 3Dpol, VP1, VP2, and VP3, were skewed towards a higher percentage of nonconservative variations compared to other regions. Finally, in VP2 we note six instances of in-frame insertion/deletions of multiples of 3 nucleotides. The net effect, though, was no difference in the total number of amino acids in this region between the two strains.
Fig. 3.
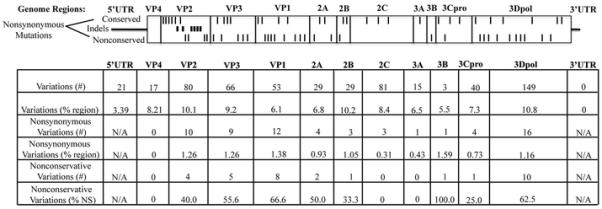
Sequence variation between hrv-A101 and hrv-A101-v1 stratified by genome location. The nonsynonymous mutations are shown in approximate scale to their localization within the HRV proteins (ticks: top, conservative substitution; bottom, nonconservative substitution; middle, insertion/deletion). The variations are further shown in absolute numbers or as a percentage of the annotated gene segment as indicated.
Using local nucleotide sequence alignment tools (NCBI Blast), we found partial sequences in GenBank consistent with hrv-A101-like isolates from patient samples (data not shown). Comparisons of portions of the hrv-A101 sequence (primarily 5′UTR, VP4/2) against all HRV-like (>90% identity) entries of greater than 100 nucleotides yielded 33 sequences. These were isolates collected in China, Germany, Spain, Australia, Belgium, and 3 states in the United States (Georgia, Connecticut, and Tennessee). The isolates were found in patients with a range of illnesses including upper respiratory infections, asthma exacerbations, and bronchitis [1, 7, 14].
In summary, we have identified and sequenced the complete genomes of two closely related HRVs from human respiratory samples. Unlike what we have found from full genome sequencing of other isolates [11], which were variants of recognized HRVs, hrv-A101 represents a distinct and novel strain within the HRV-A species. The more recent isolate, hrv-A101-v1, is a closely related variant with respect to hrv-A101. Both are localized within a small clade of HRV-A, shared by 8 other recognized strains. They evolved from a distant recombination event involving 2 reference hrv lineages and represent a subset of modern, globally circulating hrv strains. As sequencing capabilities continue to improve in terms of throughput and costs, we urge complete genome sequencing of clinical isolates so that the evolutionary relationships, mutation frequencies, recombination potential, and structure/function predictions for HRVs can be ascertained. With this approach, specific motifs or features of the HRVs determined from genome-wide data can then be correlated with in vitro and clinical phenotypes.
Acknowledgment
Funded by National Institutes of Health grant HL071609.
Footnotes
Contributor Information
Jennifer A. Rathe, Departments of Medicine and Physiology, University of Maryland School of Medicine, 20 Penn Street, HSF-II, Room S-114, Baltimore, MD 21201 USA
Xinyue Liu, Institute for Genome Sciences, University of Maryland School of Medicine, BioPark II, 801 West Baltimore Street, Room 623, Baltimore, MD 21201 USA.
Luke J. Tallon, Institute for Genome Sciences, University of Maryland School of Medicine, BioPark II, 801 West Baltimore Street, Room 623, Baltimore, MD 21201 USA
James E. Gern, Departments of Pediatrics and Medicine, University of Wisconsin, Madison, K4/198 CSC, 600 Highland Avenue, Madison, WI 53792 USA
Stephen B. Liggett, Departments of Medicine and Physiology, University of Maryland School of Medicine, 20 Penn Street, HSF-II, Room S-114, Baltimore, MD 21201 USA
References
- 1.Briese T, Renwick N, Venter M, Jarman RG, Ghosh D, Kondgen S, Shrestha SK, Hoegh AM, Casas I, Adjogoua EV, Akoua-Koffi C, Myint KS, Williams DT, Chidlow G, van den Berg R, Calvo C, Koch O, Palacios G, Kapoor V, Villari J, Dominguez SR, Holmes KV, Harnett G, Smith D, Mackenzie JS, Ellerbrok H, Schweiger B, Schonning K, Chadha MS, Leendertz FH, Mishra AC, Gibbons RV, Holmes EC, Lipkin WI. Global distribution of novel rhinovirus genotype. Emerg Infect Dis. 2008;14:944–947. doi: 10.3201/eid1406.080271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Camargo CA, Jr., Ginde AA, Clark S, Cartwright CP, Falsey AR, Niewoehner DE. Viral pathogens in acute exacerbations of chronic obstructive pulmonary disease. Intern Emerg Med. 2008;3:355–359. doi: 10.1007/s11739-008-0197-0. [DOI] [PubMed] [Google Scholar]
- 3.Chonmaitree T, Howie VM, Truant AL. Presence of respiratory viruses in middle ear fluids and nasal wash specimens from children with acute otitis media. Pediatrics. 1986;77:698–702. [Google Scholar]
- 4.Huson DH. SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998;14:68–73. doi: 10.1093/bioinformatics/14.1.68. [DOI] [PubMed] [Google Scholar]
- 5.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 6.Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, Symington P, O’Toole S, Myint SH, Tyrrell DA, et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ (Clinical research ed. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khetsuriani N, Lu X, Teague WG, Kazerouni N, Anderson LJ, Erdman DD. Novel human rhinoviruses and exacerbation of asthma in children. Emerg Infect Dis. 2008;14:1793–1796. doi: 10.3201/eid1411.080386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kistler AL, Webster DR, Rouskin S, Magrini V, Credle JJ, Schnurr DP, Boushey HA, Mardis ER, Li H, DeRisi JL. Genome-wide diversity and selective pressure in the human rhinovirus. Virology journal. 2007;4:40. doi: 10.1186/1743-422X-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WM, Kiesner C, Pappas T, Lee I, Grindle K, Jartti T, Jakiela B, Lemanske RF, Jr., Shult PA, Gern JE. A diverse group of previously unrecognized human rhinoviruses are common causes of respiratory illnesses in infants. PLoS ONE. 2007;2:e966. doi: 10.1371/journal.pone.0000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–262. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- 11.Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, Fraser-Liggett CM, Liggett SB. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science. 2009;324:55–59. doi: 10.1126/science.1165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitkaranta A, Arruda E, Malmberg H, Hayden FG. Detection of rhinovirus in sinus brushings of patients with acute community-acquired sinusitis by reverse transcription-PCR. Journal of clinical microbiology. 1997;35:1791–1793. doi: 10.1128/jcm.35.7.1791-1793.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 14.Renwick N, Schweiger B, Kapoor V, Liu Z, Villari J, Bullmann R, Miething R, Briese T, Lipkin WI. A recently identified rhinovirus genotype is associated with severe respiratory-tract infection in children in Germany. The Journal of infectious diseases. 2007;196:1754–1760. doi: 10.1086/524312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth AR, Smyth RL, Tong CY, Hart CA, Heaf DP. Effect of respiratory virus infections including rhinovirus on clinical status in cystic fibrosis. Arch Dis Child. 1995;73:117–120. doi: 10.1136/adc.73.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tapparel C, Junier T, Gerlach D, Cordey S, Van Belle S, Perrin L, Zdobnov EM, Kaiser L. New complete genome sequences of human rhinoviruses shed light on their phylogeny and genomic features. BMC genomics. 2007;8:224. doi: 10.1186/1471-2164-8-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitton JL, Cornell CT, Feuer R. Host and virus determinants of picornavirus pathogenesis and tropism. Nature reviews. 2005;3:765–776. doi: 10.1038/nrmicro1284. [DOI] [PubMed] [Google Scholar]