

Abstract
Facioscapulohumeral dystrophy (FSHD) is a muscular hereditary disease with a prevalence of 1 in 20,000 caused by a partial deletion of a subtelomeric repeat array on chromosome 4q. However, very little is known about the pathogenesis as well as the molecular and biochemical changes linked to the progressive muscle degeneration observed in these patients. Several studies have investigated possible pathophysiological pathways in FSHD myoblasts and mature muscle cells but some of these reports were apparently in contradiction. The discrepancy between these studies may be explained by differences between the sources of myoblasts. Therefore, we decided to thoroughly analyze affected and unaffected muscles from patients with FSHD in terms of vulnerability to oxidative stress, differentiation capacity and morphological abnormalities. We have established a panel of primary myoblast cell cultures from patients affected with FSHD and matched healthy individuals. Our results show that primary myoblasts are more susceptible to an induced oxidative stress than control myoblasts. Moreover, we demonstrate that both types of FSHD primary myoblasts differentiate into multi-nucleated myotubes, which present morphological abnormalities. Whereas control myoblasts fuse to form branched myotubes with aligned nuclei, FSHD myoblasts fuse to form either thin and branched myotubes with aligned nuclei or large myotubes with random nuclei distribution. In conclusion, we postulate that these abnormalities could be responsible for muscle weakness in patients with FSHD and provide an important marker for FSHD myoblasts.
Keywords: Facioscapulohumeral dystrophy (FSHD), muscle differentiation, myoblasts, oxidative stress, cellular model
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is the third most frequent form of inherited muscle disease, following Duchenne Muscular Dystrophy (DMD) and Myotonic Dystrophy. Clinically, the disease is characterized by a progressive weakness and atrophy of facial, shoulder and upper arm muscles. There is a wide variability in the clinical spectrum of the disease, ranging from very mild muscle weakness, with patients who are almost unaware of being affected, to severe symptoms with patients who are wheelchair-dependent. The major form of FSHD maps to the subtelomeric region of the long arm of chromosome 4 (4q35) where a partial deletion of a repeat array (D4Z4) is associated with the disorder [1, 2]. In the general population, the D4Z4 repeat array is polymorphic and may contain between 11 and 150 units, while the majority of FSHD patients carry only 1 to 11 repeat units [3, 4]. There is an inverse correlation between the residual repeat size and the severity and the age of onset of the disease [5, 6]. However, not every study agrees with an inverse correlation between D4Z4 copy number and disease severity [7].
Several putative mechanisms have been proposed to explain the origin of FSHD. Gabellini et al.[8] suggested that FSHD might be caused by reduced affinity between transcriptional repressors and the D4Z4 region, leading to an inappropriate activation of FRG1, FRG2 and ANT1, which are proximal to the 4q35 region. Indeed, transgenic mice overexpressing FRG1 in skeletal muscle develop a muscular dystrophy [9]. However, patient with FSHD show a very limited, if any, overexpression of FRG1 as compared to healthy individuals [10, 11].
Another interesting feature of the D4Z4 repeated element is the presence of an open reading frame (ORF) containing a double homeobox sequence, DUX4[12], which is preceded by a putative promoter element that displays high transcriptional activity in transient expression studies. Some of us contributed to the demonstration that DUX4 is transcribed both in cells transfected with D4Z4 elements and endogenously in FSHD myoblasts [13, 14]. These data are suggestive for a pathogenic transcriptional role of the D4Z4 element in FSHD [12, 15].
Moreover, some of us have shown that the D4Z4 array contains a strong transcriptional activator which may up-regulate transcription of neighboring genes [16]. We have also demonstrated that a nuclear matrix attachment site (S/MAR) is located in the vicinity of the D4Z4 repeat and separates it from the transcriptional enhancer [17]. This S/MAR is prominent in normal human myoblasts and non-muscular human cells, and much weaker in muscle cells derived from patients with FSHD, suggesting that the D4Z4 repeat array and upstream genes are located in two distinct loops in non-muscular cells and healthy human myoblasts, whereas it is in a single loop in FSHD myoblasts. This S/MAR may also function as an insulator, thus blocking the D4Z4 enhancer in normal, but not in FSHD cells [16].
Contraction of the D4Z4 repeat alone is not sufficient to cause the disease. Indeed, similar repeat arrays are present also on chromosome 10 and on two equally common alleles of chromosome 4, but only contractions associated with the 4qA allele variant are associated with the disease [10]. Another polymorphic region proximal to D4Z4 [18] directly coincides with the S/MAR we previously described, suggesting that changes in the chromatin organization of the region may play a key role in the disease [16, 17].
Little is known also about the molecular mechanisms that induce the progressive muscle degeneration observed in FSHD, and many groups have observed apparently contradictory gene expression patterns, particularly in the 4q35 region, by using different types of cells from healthy individuals and patients with FSHD [19].
Moreover, in patients affected with FSHD, it is quite common to observe the co-existence of affected and apparently healthy muscles. In previous studies, myoblasts, which were obtained from muscle typically affected in FSHD, manifested an increased susceptibility to oxidative stress during proliferation [20]. On the contrary, in another study, cells expanded from unaffected FSHD muscles showed no morphological abnormalities and were proposed as a suitable tool for clinical trials of autologous cell transplantation [21]. The apparent discrepancy between these two studies may be explained by differences between the sources of myoblasts: from clinically affected muscles in the first study and from unaffected muscles in the second study.
Therefore, we decided to thoroughly analyze affected and unaffected muscles from patients with FSHD in terms of vulnerability to oxidative stress, differentiation capacity and morphological abnormalities. To this aim, we purified satellite cells from various affected and unaffected muscle of 14 patients with FSHD of both sexes and compared them with 14 control samples.
We found that satellite cell-derived myoblasts from both clinically unaffected and affected muscles of patients with FSHD are more susceptible to an induced oxidative stress than control myoblasts. Moreover, although myoblasts from patients with FSHD fully differentiated into multi-nucleated myotubes, they fused to form either thin and branched myotubes with aligned nuclei or large myotubes with random nuclei distribution. This defect could explain the muscle weakness observed in patients with FSHD and provides an important marker for FSHD myoblasts.
Materials and methods
Patients
We used fresh and frozen muscles obtained from 14 male and female patients with genetically confirmed diagnosis of FSHD (20–54 years; mean 37.86 ± 11.06) and 14 control patients (16–52 mean 32.78 ± 10.57) free of neuromuscular diseases. None of the patients was treated with steroids or immunosuppressive drugs. All patients and control patients have given informed written consent.
The patients with FSHD had a D4Z4 array length, which ranged between 4 (most severe cases) and 8 repeats [22] (Table 1). All patients were ranked between 1 and 4 for arms and between 1 and 5 for legs according to Brooke–Vignos functional scale [21].
Table 1.
Data of patients with FSHD. Recapitulative table of the data about each patient with FSHD: name of the FSHD cell line, age and sex of the patient (M:male; F:female), number of D4Z4 units, site of the muscle biopsy, and Brooke–Vignos scales defining the clinical status of upper and lower limb muscles, respectively. In the Brooke scale, high values define affected upper limb muscles and low values define non-affected upper limb muscles. In the Vignos scale, low values define non-affected lower limb muscles and high values define affected lower limb muscles.
| Name | Sex | Age (years) | D4Z4 copy number | Muscle | Brooke scale ARMS | Vignos scale LEGS |
|---|---|---|---|---|---|---|
| FSHD1 | M | 30 | 5 | Trapezius | 4 | 5 |
| FSHD2 | F | 54 | 5 | Piriformis | 3 | 4 |
| FSHD3 | F | 32 | 7 | Vastus lateralis | 1 | 1 |
| FSHD4 | M | 41 | 7 | Infraspinatus | 4 | 4 |
| FSHD5 | M | 53 | 6 | Vastus lateralis | 2 | 3 |
| FSHD6 | F | 23 | 8 | Vastus lateralis | 1 | 1 |
| FSHD7 | M | 53 | 9 | Femoral biceps | 2 | 2 |
| FSHD8 | M | 39 | 6 | Vastus lateralis | 2 | 1 |
| FSHD9 | M | 36 | 7 | Vastus lateralis | 2 | 1 |
| FSHD10 | F | 20 | 4 | Vastus lateralis | 1 | 1 |
| FSHD11 | M | 44 | 7 | Vastus lateralis | 3 | 3 |
| FSHD12 | F | 38 | 7 | Vastus lateralis | 1 | 1 |
| FSHD13 | F | 42 | 8 | Vastus lateralis | 4 | 3 |
| FSHD14 | M | 25 | 4 | Vastus lateralis | 1 | 1 |
Samples of skeletal muscle were obtained in accordance with the French and European legislations. Muscle tissues were obtained from the Banque de Tissus pour la Recherche de l’Association Française contre les Myopathies (AFM) or from patients followed in the Service de Physiologie Clinique of the Hôpital Lapeyronie (Montpellier, France). Muscle biopsies were performed on the trapezius muscle (in one patient during orthopaedic surgery), the piriformis (one patient), the femoral biceps (one patient) and the paraspinal lumbar (one patient during orthopaedic surgery). Quadriceps muscle samples from 10 patients with FSHD and 10 control patients were obtained by needle biopsies as the procedure routinely used in the Service de Physiologie Clinique [23, 24] (Table 1).
To assess whether the biopsied muscle was affected and to evaluate the severity of its involvement, we used clinical and histopathologic criteria.
Myoblast isolation
A total of 1 mm3 explants from each biopsy were scissor-minced and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Sigma, Saint Quentin Fallavier, France) supplemented with 20% foetal bovine serum (FBS, Hyclone Perbio, Brebière, France), 2% Ultroser G (Biosepra, Cercy Saint Christophe, France), 10 mM Hepes (Sigma), 50 μg/ml Gentamicin (Sigma). Three different procedures were used to isolate muscle satellite cells in collagen-coated Petri dishes (IWAKI, Starlab, Paris, France):
Non-adherent explants: explants were cultured in suspension in 60 mm collagen-coated Petri dishes for 8 days.
‘Adherent-explants: explants were placed in 60 mm collagen-coated Petri dishes with a minimal volume of medium for 6 hrs, then, the dish was slowly filled, leaving explants attached to the dish.
Explants in Matrigel: explants were trapped inside a thin layer of 3.6 mg/ml Matrigel (BD Matrigel Matrix from Beckton Dickinson Biosciences, Le Pont de Claix, France) in 60 mm collagen-coated Petri dishes with medium added on top of the layer. After 8 days of culture, migrant cells harvested in these three different conditions were counted. The total number of released cells was arbitrary fixed to 100% for matrigel. Cells were tested for their myogenicity by immunofluorescence with an anti-desmin antibody [25].
Myoblasts purification
Harvested cells were then purified with an immuno-magnetic sorting system (Miltenyi Biotec, Paris, France) using an anti-CD56/NCAM antibody [25]. Purified myoblasts were plated in collagen-coated Petri dishes and cultured in growth medium containing DMEM supplemented with 20% FBS at 37°C in humidified atmosphere with 5% CO2. At cell isolation, all human myoblasts were considered to be at 1 PD (population doubling). All experiments were performed between 2 PD and 10 PD, to avoid premature replicative senescence, which occurs after 15 PD and interferes with proliferation and differentiation processes.
Proliferation kinetics and analyses of myotube fusion
Myoblasts were seeded at 2 × 104 cells/dish onto 35 mm collagen-coated dishes and cultured in growth medium. Cells were counted using a cell counter (Coulter). The doubling time t was calculated as t = ((lny/x)(ln2)/96, where x is the number of cells counted at day 0 and y the number of cells counted at day 4.
Myogenic differentiation of confluent cells was induced after 5 days by changing to DMEM containing 2% FBS (differentiation medium). Cells were kept in differentiation medium for 4–6 days. At day 6 after induction of differentiation, cells were immunostained with anti-troponinT antibody coupled with DAPI (see Immunocytochemistry for details) to visualize myotubes and nuclei in the culture. The myogenic fusion index (MFI) was thus determined by dividing the number of nuclei in multi-nucleated myotubes by the total number of nuclei in a given microscopic field. Ten fields per culture were counted in three independent cultures (a total of 15,000 nuclei per cell line) using the MRI cell image analyzer software developed by Volker Baecker at the Montpellier RIO Imaging facility of CRBM [26].
The Deformed Myotube Index (DMI) was calculated as the proportion of myotubes with a deformed morphology characterized by an abnormal repartition of nuclei. Ten fields per culture were counted in three independent cultures.
Immunocytochemistry
Cell staining was performed as previously described [25]. The following antibodies and dilutions were used: anti-desmin 1/100 (mAb, SIGMA), anti-troponinT 1/100 (mAb, SIGMA), anti-myogenin 1/100 (pAb, Santa Cruz Biotechnology, Le Perray-en-Yvelines, France), anti-α-actinin 1/100 (mAb, SIGMA), anti-MyoD 1/100 (pAb, Santa Cruz Biotechnology), anti-MF20 1/100 (Developmental Studies Hybridoma Bank, University of Iowa, IA, USA), anti-p21 1/100 (pAb, Santa Cruz Biotechnology), anti-β-tubulin 1/100 (mAb, SIGMA), anti-α-tubulin 1/100 (mAb, SIGMA), Alexa-488 conjugated anti-mouse and Alexa-555 conjugated anti-rabbit 1/1000 (Invitrogen, Cergy Pontoise Cedex, France).
Phase contrast and fluorescence imaging
Image acquisitions and image analysis were performed on workstations of the Montpellier RIO Imaging facility at the CRBM (http://www.mri.cnrs.fr).
Western Blot analysis
Cells were lysed in hypertonic buffer containing 50 mM Tris pH7, 50 mM NaCl, 0.1% NP40, anti-proteases and 1 mM DTT. A total of 30 μg of each cell lysate were separated by SDS-polyacrylamide gel electrophoresis (NuPage system, Invitrogen), transferred to Nitrocellulose membranes (GE Healthcare, Orsay Cedex, France), blocked 1 hr at room temperature with 5% milk diluted in PBS and probed at 4°C overnight with anti-p21 antibodies (mAb, Sigma) in 0.5% bovine serum albumin (BSA)(PBS. Membranes were washed in PBS and incubated with a horseradish peroxidase-conjugated anti-mouse antibody (GE healthcare).
Oxidative stress experiments
Myoblasts were seeded at 2 χ 104 cells/dish in 35 mm collagen-coated dishes and cultured in growth medium. At day 2, cells were treated with increasing H2O2 concentrations (200, 400, 600, 800 and 10 mM) for 24 hrs. The LIVE/DEAD® Viability/Cytotoxicity Kit for mammalian cells (Invitrogen) was used according to the manufacturer’s instructions. Briefly, after exposure to the different H2O2 concentrations, myoblasts were stained with calcein (excitation 494 nm/emission 517 nm; to detect live cells) and ethidium homodimer-1 (excitation 528 nm/emission 617 nm; for dead cells) and were analyzed by flow cytometry performed with a FacsCalibur (Becton Dickinson) equipped with the CellQuest software. Experiments were performed twice on two independent cultures.
Statistical analysis
All data reported are presented as mean ±SEM. Pearson product-moment correlations were used to detect relationship between proportion of atrophied myotubes and MFI. All analyses were performed using Staview software. Statistical significance was set at P≤ 0.05.
Results
Clinical severity and muscle characterization in the FSHD patients
To assess vulnerability to oxidative stress, differentiation capacity and morphological abnormalities in myoblasts derived from muscles of control and patients with FSHD, we selected 14 healthy individuals and 14 patients affected with FSHD with different clinical severity and number of D4Z4 repeats on chromosome 4. Table 1 reports the clinical evaluation according to the Vignos and Brooke Scale [21] and genetic data of the patients studied.
Seven patients had clinical evidence of lower limb muscle involvement according to the Vignos Scale (grade >1) [21]. In the other patients, clinical manifestations of the disease were restricted to the scapular areas with, in general, a weak functional deficit according to the Brooke Scale (grade ≤2) [21]. Clinical investigation showed that the quadriceps (i.e. Vastus lateralis) was unaffected in 7 of the 10 patients who had a quadriceps biopsy, and did not present histo-pathological abnormalities. In the other three cases, the muscle (FSHD5, 11, 13) had clinical and histological signs of disease. A clear correlation between the clinical status of the FSHD muscles analyzed and the overall tissue organization, by histological examination, was observed: in contrast to clinically unaffected FSHD muscles, clinically affected FSHD muscles appear to have an increased variability in fibre shape and size (data not shown).
Optimization of myoblast purification
At the beginning of this study we used three different methods to isolate satellite cells: ‘non-adherent explants’ in collagen-coated Petri dishes, ‘adherent-explants’ in collagen-coated Petri dishes and explants grown in Matrigel. Migrating cells harvested in these three different conditions, were then counted and tested for myogenicity.
The number of cells that migrated from the explants was higher for the explants grown in Matrigel (Fig. 1A). Migrating cells were then analyzed for their myogenicity by immunofluorescence staining for desmin, a marker of muscle precursor cells [27]. Myogenicity of the culture was calculated by dividing the number of desmin-positive cells by the total number of cells (DAPI-stained nuclei) in a given microscopic field. As shown in (Fig. 1B), 35–40% of cells derived from explants in Matrigel and 25–30% of those from ‘adherent explants’ expressed desmin, whereas only 10% of the migrating cells from ‘non-adherent explants’ were positive for desmin. Therefore, the use of Matrigel greatly improves myoblast isolation from muscular biopsies by increasing both yield and myogenicity. For this reason, we decided to use this technique throughout the study.
Fig 1.
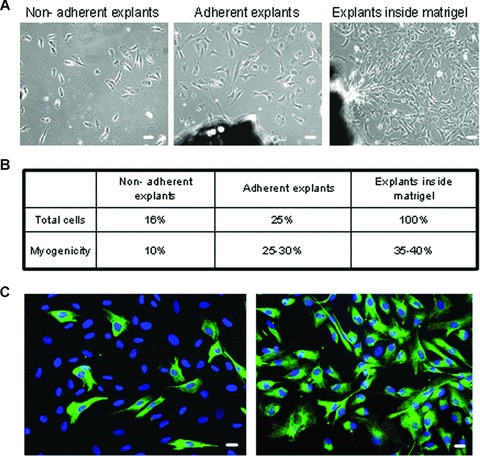
Optimization of FSHD myoblast purification. (A) Phase contrast images of cells migrating away from non-adherent explants (left) adherent explants (middle) and explants in Matrigel (right), 10× magnification. (B) After 8 days of culture, cells were harvested and counted. The total number of released cells was arbitrary fixed to 100% for matrigel. Myogenicity was defined as the percentage of cells expressing desmin. (C) Immunofluorescence showing desmin positive cells (green) before and after MACS purification performed with CD56 antigen. Nuclei were revealed after DNA staining with DAPI (blue). 20× magnification, Bar = 10μm.
Then, to avoid contamination by non-muscle cells, mainly fibroblasts, we purified our myogenic cells by using an immuno-magnetic sorting system coupled with an antibody against CD56/NCAM, a cell surface marker that is highly expressed in myoblasts [28] but not in fibroblasts. This technique enabled us to obtain highly purified myoblasts cultures, as shown by staining with the anti-desmin antibody, a specific marker of muscle cells (i.e., 90–95% of desmin-positive cells in both FSHD and control cell cultures) (Fig. 1C, right panel).
Myoblasts derived from patients with FSHD and controls have similar proliferation rates and gene expression patterns
The proliferation rate and morphology of control and FSHD myoblasts were analyzed in cells kept in growth medium. The growth capacities of control and FSHD cells were very similar. The mean doubling time for each FSHD and control cell line was calculated at an early stage of culture and was established to 42.6 hrs (±2.7 SEM) for controls and 43.3 hrs (±4.7 SEM) for FSHD. Notably, the proliferation rates of all FSHD cultures were comparable and did not correlate with the severity of the disease or patient age.
We did not detect any morphological differences in FSHD myoblasts from clinically affected and unaffected FSHD muscles and control muscles by phase contrast microscopy (Fig. 2A). We did not observe necrotic cells in any of the FSHD cultures in agreement with recently published data [29]. As myoblast structure directly influences the organization of the resulting myotube, we decided to stain myoblasts with phalloidin to analyze the arrangement of the actin microfilaments. Normal stress fibres were observed in both control and FSHD myoblast cell cultures (Fig. 2B).
Fig 2.
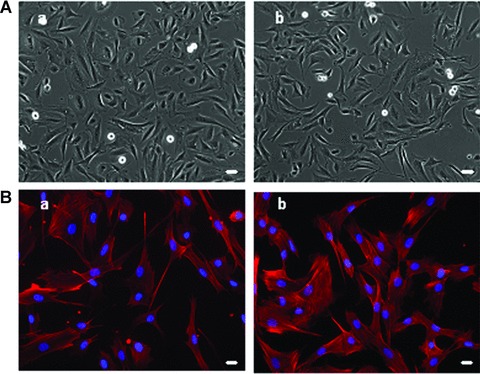
Comparative morphological analysis of control and FSHD myoblasts. (A) Phase contrast images of a representative control (a; CTL 6) and FSHD (b; FSHD13) myoblast culture showing that FSHD myoblasts have a normal morphology, 10× magnification. (B) Observation of actin microfilaments using phalloidin (red) and DAPI (blue) immunostaining on a representative control (a; CTL1) and a representative FSHD (b; FSHD3) myoblast cultures show normal cytoskeleton morphology, 20× magnification, bar = 10 μm. All these observations were done on the 14 CTL and FSHD myoblast cultures.
We then examined by immuno-fluorescence the expression and localization patterns of MyoD, a myoblast lineage-specific marker, and Ki67, a cell-proliferation marker. In growth conditions, both MyoD and Ki67 were localized in the nuclear compartment and similarly expressed in FSHD and control primary myoblasts (Fig. 3).
Fig 3.
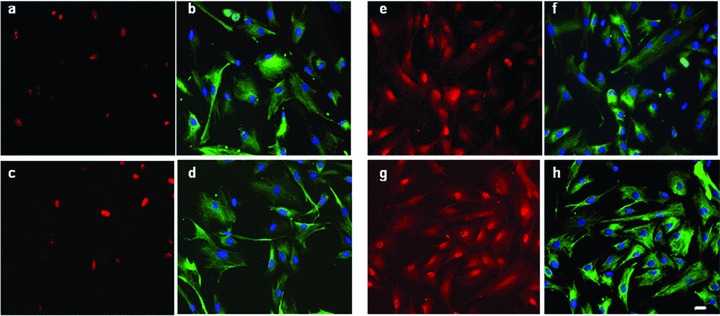
Characterization of FSHD and control cells proliferation. Representative images of a CTL (a, b, e, f; CTL1) and FSHD (c, d, g, h; FSHD13) showing co-immunofluorescence with anti-Ki67 (a, c; red) and anti-desmin (b,d; green), anti-MyoD (e, g; red) and anti-desmin (f, h; green) antibodies and DAPI nuclear staining (blue). MyoD and Ki67 are localized into the nucleus 20× magnification, bar = 10 μm. Experiments were performed on two controls and three FSHD cultures.
In conclusion, we did not detect differences in either the proliferation or the expression of MyoD in FSHD myoblasts.
FSHD myoblasts from affected and unaffected muscles are both susceptible to an induced oxidative stress
As a previous study demonstrated that myoblasts from affected FSHD muscles exhibit increased susceptibility to oxidative stress [20], we decided to examine whether FSHD myoblasts derived from clinically unaffected FSHD muscles were also susceptible to an induced oxidative stress. To this aim, we acutely exposed FSHD and control myoblasts to different concentrations of hydrogen peroxide (H2O2), a pro-apoptotic compound and determined the percentage of living and dead cells with the LIVE/DEAD® Viability/Cytotoxicity Kit (see Materials and Methods). We observed that for both clinically affected and unaffected FSHD myoblasts mortality started at concentration of 500 μM H2O2 and almost all cells were dead with 700 μM H2O2, whereas for control myoblasts mortality occurred between 700 and 800 μM H2O2 (Fig. 4A and B).
Fig 4.
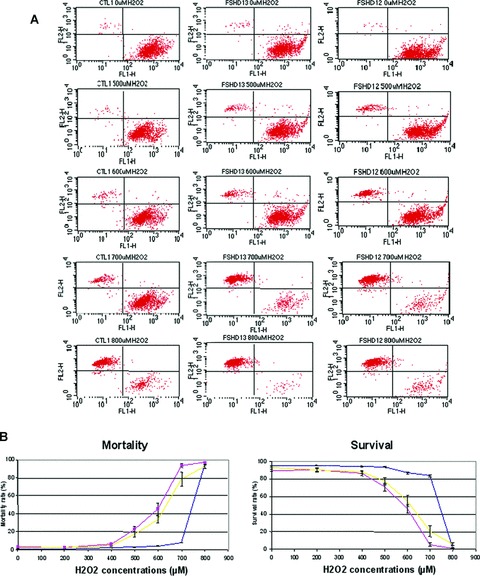
Comparative analysis of control and FSHD myoblasts response to H202-induced oxidative stress. (A) A representative flow cytometry analysis on myoblasts from one control (CTL1), one clinically affected FSHD (FSHD13) and one clinically unaffected FSHD (FSHD12). FACS experiments were conducted on FSHD and control myoblasts after 24 hrs exposure to increasing doses of H2O2. Live cells stained with calcein (green) were sorted with the FL1 channel (down-right window) and dead cells stained with ethidium homodimer-1 (red) were sorted with the FL2 channel (up-left window). (B) Flow cytometry analysis of myoblasts from all control (blue), unaffected FSHD (yellow) and affected FSHD (red). Experiments were performed twice on two independent cultures.
This result indicates that all FSHD myoblasts, independently from the clinical status of the muscle they were derived from, are more susceptible to an induced oxidative stress than control cells.
Differentiating FSHD myoblasts from affected and unaffected muscles have an abnormal morphology
Next we wanted to check whether myoblasts from clinically affected and unaffected FSHD muscles could differentiate normally into myotubes.
We then induced myoblast differentiation by shifting to differentiating medium (2% foetal calf serum). The expression and localization of p21 and of one-muscle differentiation markers (myogenin) were similar in FSHD and control cells (Fig. 5A). In addition, we examined the maturation degree of these myotubes by following the immuno-localization of the MF20 antibody (which recognizes all myosins) and an anti-α-actinin antibody, which stains Z lines and is used to check the sarcomeric organization (Fig. 5B).
Fig 5.
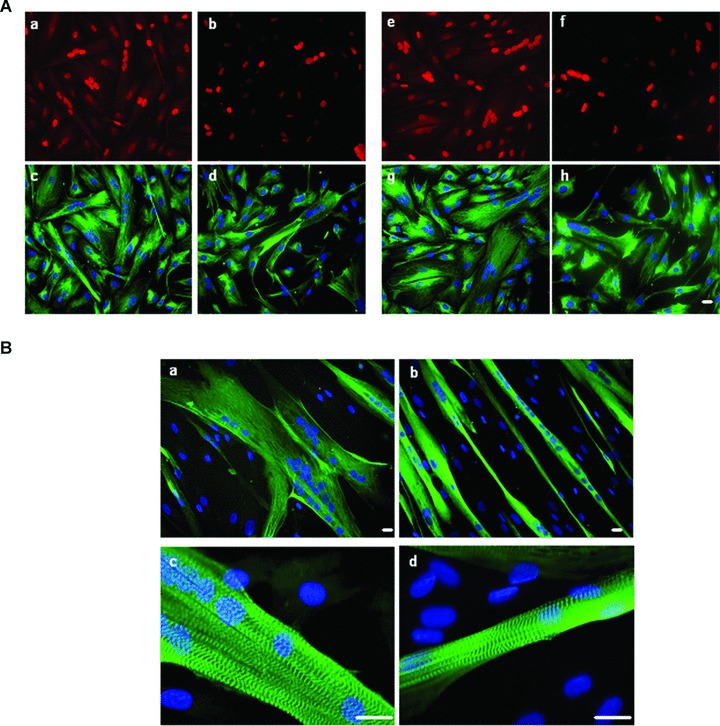
FSHD and control cells upon induction of differentiation. (A) A representative control (a, b, c, d; CTL7) and FSHD (e, f, g, h; FSHD4) culture at the onset of cell fusion: co-immunofluorescence with anti-myogenin (a,e; red) and anti-desmin (c, g; green), anti-p21 (b, f; red) and anti-desmin (d, h; green) antibodies and DAPI nuclear staining (blue), myogenin and P21 localize into the nucleus, 20× magnification. Experiments were performed on three controls and three FSHD cultures. (B) Terminal differentiation (day 6; see Materials and Methods) of a representative culture of CTL (a, c; CTL1) and FSHD (b, d; FSHD9) myotubes: immunofluorescence with either an anti-myosin (a, b; green) and DAPI nuclear staining (blue), 20× magnification, bar = 10 μm or with an anti-α-actinin (c, d; green) antibody and nuclear (blue) staining showing correct localization of these proteins in FSHD cells, 40× magnification, Bar × 10 μm. Experiments were performed on 11 controls and 12 FSHD cultures.
We did not detect defects in the maturation and sarcomeric organization of the myotubes derived from both affected and unaffected FSHD muscles.
To assess fusion competence, we calculated the MFI, which is the ratio between the nuclei present in myotubes versus the total number of nuclei in a given field, where a myotube is defined as a muscle cell containing at least three or more nuclei. We obtained MFI values between 37% and 70% in most of the control and FSHD cell cultures, except for FSHD1 and FSHD14. In FSHD1, the fusion index was much lower (18%) and in FSHD14 the fusion index was higher (85%) than in the other cell cultures (Fig. 6).
Fig 6.

Myogenic Fusion Index (MFI) of FSHD and control cultures. MFI values range from 37% to 70% for both FSHD and control cell cultures, except for FSHD1 and FSHD14 (respectively, 18% and 85%). To calculate MFI, 10 fields per culture were counted in three independent cultures for each cell line.
Although the biochemical features we analyzed were similar in both FSHD and control cells, we observed an abnormal morphology in all FSHD myoblasts undergoing differentiation. Whereas control myoblasts fuse to form branched myotubes with aligned nuclei (Fig. 7A), FSHD myoblasts appeared either thinner (Fig. 7B) or deformed (Fig. 7C) with randomly localized nuclei. Eight of the 14 FSHD cell cultures (i.e., FSHD 1, 4, 3, 2, 7, 5, 6, 9) had a significant increase in the proportion of myotubes containing 3 to 10 nuclei each compared to controls (Fig. 8A), but no myotubes with more than 50 nuclei were detected in these FSHD cell cultures. Moreover, in these cultures a large proportion of myotubes had a cell diameter <20 μm (Fig. 8B). In these cell cultures we did not find very large myotubes (>100 μm), which were observed in the other FSHD and control cell cultures. These data suggest that, in the FSHD 1, 4, 3, 2, 7, 5, 6 and 9 cultures, a large proportion of myotubes are atrophic. In addition, we observed a significant inverse correlation (r=−0.77; P < 0.02) between MFI and the proportion of atrophied myotubes in FSHD culture (Fig. 8C).
Fig 7.
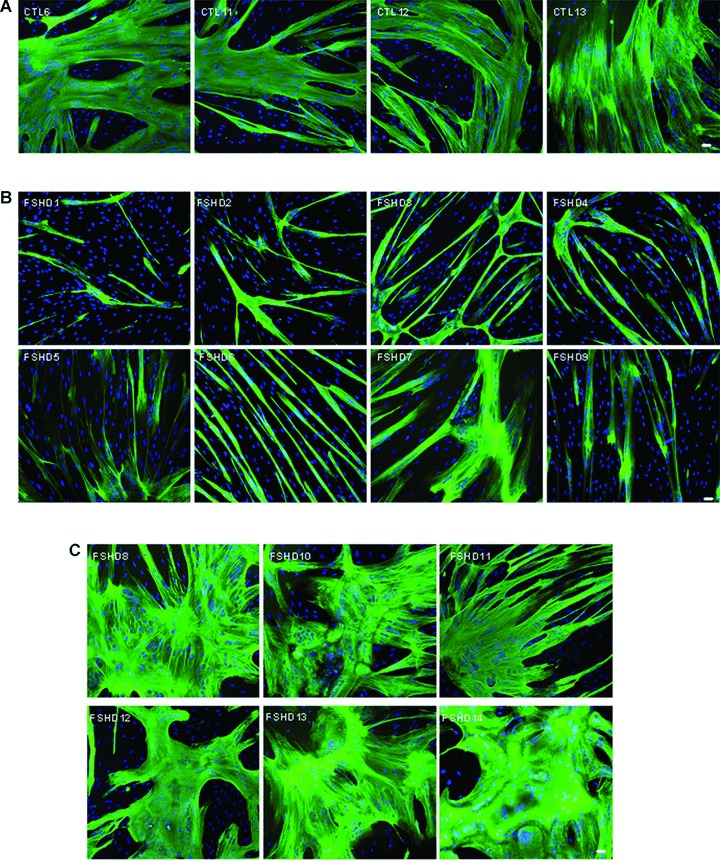
Specific myotube phenotypes. Immunofluorescence with an anti-troponinT antibody (green) and DAPI nuclear staining (blue), 10× magnification, Bar = 20 μm. (A) CTL myotubes (only 4 out of 14 controls are depicted) showing normal human myotube morphology. (B) Myotubes from the FSHD atrophic group showing thinner myotubes. (C) Myotubes of the FSHD disorganized group showing larger myotubes with abnormal nuclei repartition.
Fig 8.
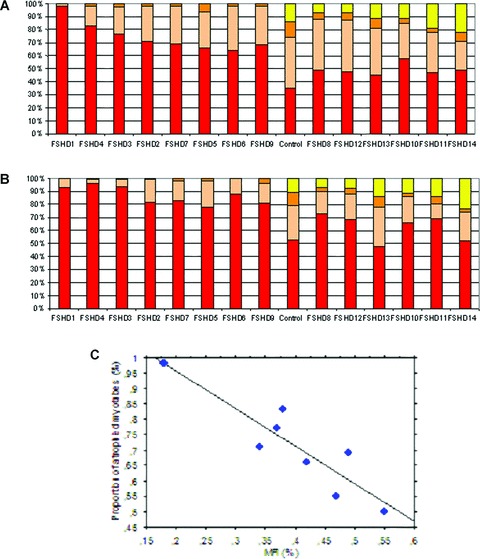
Myotube characterization. (A) Bar graph representing the repartition of the number of nuclei per myotube for each FSHD culture and a representative control. Yellow: more than 50 nuclei; Orange: from 31 to 50 nuclei; Pink: from 11 to 30 nuclei; Red: from 3 to 10 nuclei. (B) Bar graph representing the repartition of the myotube diameters for each FSHD culture and a representative control. Yellow: more than 100 μm; orange: from 61 to 100 μm; pink: from 21 to 60 μm; red: from 0 to 20 μm. (C) Statistical analysis of FSHD cultures showing correlations between the proportion of atrophic myotubes and the MFI value (D).
In the other FSHD cell cultures (FSHD 8, 12, 13, 10, 11 and 14), we did not detect marked differences in nuclei number and cell diameter in comparison to control cells. However, we observed a significant higher proportion of deformed myotubes characterized by abnormal repartition of nuclei. To quantify this phenotype, we used the deformed myotubes index (DMI), previously developed by Yip and Picketts (2003) [30]. The DMI determined the proportion of myotubes in the culture with abnormal phenotype. This proportion ranged from 13% to 40% whereas the DMI is null in controls (Fig. 9). We found a positive relationship between the MFI and the proportion of deformed myotubes. Indeed, the average MFI of FSHD cell cultures which have deformed myotubes was of 57% while those presenting no deformation were of 38%. These data might suggest that the more the FSHD myoblasts are able to fuse, the more they tend to form deformed myotubes. These defects were not correlated with the overall disease severity and the degree of histo-pathological abnormalities of the muscle of origin.
Fig 9.
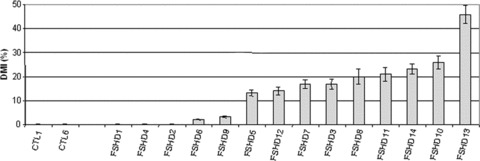
Deformed Myotube Index (DMI) of FSHD and control cultures. DMI values were plotted and ranged from 17% to 40% in FSHD cultures.
When myoblasts fuse into myotubes, the organization of the cytoskeleton changes dramatically [31]. To address this question, we analyzed the actin cytoskeleton and the microtubules organization using, respectively, phalloidin staining and α and β tubulin immuno-localization in both control and FSHD myotubes (Fig. 10). In controls, actin and microtubules are organized into a filamentous network running parallel to the long axis of the syncitium. A similar pattern is observed in FSHD myotubes, which belong to the ‘atrophied’ group of FSHD cell cultures. In contrast, in FSHD cultures, which present a high proportion of deformed myotubes, microtubules and actin networks were highly disorganized. These data confirm that FSHD myoblasts present morphological differentiation defects.
Fig 10.
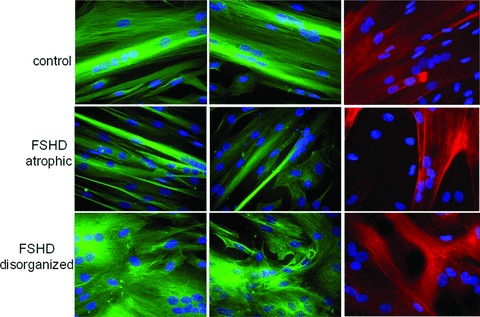
Cytoskeleton status in controls and FSHD myotubes. Myotubes from control (CTL6), FSHD. (FSHD 2, a representative culture from atrophic group), and FSHD (FSHD13, a representative culture from disorganized group) were stained for α tubulin (green; left panels); β tubulin (green; middle panels) and actin using phalloidin (red; right panels). Nuclei were revealed using DAPI (blue) staining, 40× magnification, bar = 10 μm. Experiments were performed on two controls and three FSHD cultures in each group.
Discussion
The pathogenetic mechanism of facioscapulohumeral muscular dystrophy remains largely unknown, despite 14 years of intense studies after the discovery of the genetic defect, a partial reduction of the D4Z4 repeat array on chromosome 4 [1, 2]. Recent attempts to understand this disorder have focused on identifying cellular pathways involved in FSHD and on unravelling the genetic and epigenetic mechanisms of the disease [10, 11, 32]. The results obtained are often heterogeneous and contradictory. This is in part due to the heterogeneity of the studied material, which ranges from biopsies of the affected muscles to white blood cells; in some studies the material was not sufficiently genotyped and cultured myoblasts contaminated with fibroblasts.
In this study, we have isolated large quantities of highly purified myoblasts from small muscle biopsies (50 mg) of patients with FSHD and control individuals, and described their morphology, proliferation and differentiation characteristics. Proliferating myoblasts from both clinically affected and unaffected FSHD muscles did not differ from control cells in terms of morphology, phenotype, and proliferation rate, but were more sensitive to oxidative stress. Upon induction of terminal differentiation, although all cell cultures presented similar rates of differentiation, pathological morphological features were observed in the FSHD cells, which were independent from the clinical and histo-pathological characteristics of the muscles of origin.
Differentiated myotubes from affected and unaffected FSHD muscles show clear morphological differences in comparison to control cells
While proliferating FSHD myoblasts appeared morphologically normal, FSHD myotubes appeared either thinner or deformed with random nuclei repartition, compared to the large branched myotubes with aligned nuclei of control cultures. The presence of a large proportion of deformed myotubes with randomly localized nuclei was never observed in other neuromuscular dystrophies, such as Steinert [33–35] and DMD [36]. However, our results in primary muscle cells are similar to those reported by Yip and Picketts [30] in C2C12 myoblasts, which were transiently or stably transfected with D4Z4 repeat constructs. In these cells normal differentiation is affected and myotubes are characterized by abnormal morphology and aberrant nuclei localization whilst the expression of MyoD, MEF2 and MF20 is normal. These authors proposed that the D4Z4 repeat number may be squelching some transcription factors important for muscle differentiation.
We detected morphological defects in myotubes derived from both unaffected FSHD muscles (i.e. with normal clinical and histo-pathological parameters) and affected FSHD muscles. In spite of these abnormalities, all myoblasts from patients with FSHD fully differentiated with no differences in the modulation of the myogenic pathways explored. Defects in skeletal muscle differentiation in patients affected with FSHD have been proposed by different studies, which showed deregulation of genes involved in myogenesis (such as MyoD or MEF2), cell differentiation and cell-cycle control in FSHD muscles [11, 20] as well as morphological alterations, such as irregular cytoplasmic shape and a ‘vacuolar/necrotic’ phenotype in cultured FSHD myoblasts [20]. Recently, Vilquin et al.[21] reported that myoblasts derived from unaffected FSHD muscles behave like healthy myoblasts with no changes in cell proliferation and differentiation. This report contradicts our observation that myoblasts from unaffected FSHD muscles also differentiate into morphologically abnormal myotubes. We are unable to satisfactorily explain this discrepancy, because many variables could be involved like, for example, the different anatomical location of the muscle biopsy and the cell culture parameters.
On the other hand, another recent study has shown that it is not possible to make a clear-cut distinction between myoblasts derived from affected and unaffected FSHD muscles in terms of morphological abnormalities and differentiation abilities [29]. We, therefore, propose that, in the case of FSHD, caution should be used in categorizing as ‘uninvolved’ by the disease a given muscle district even when the clinical examination is complemented by morphological investigations by MRI and histology [29]. We performed functional assays on patients with FSHD to evaluate the maximal isometric strength of quadriceps muscle compared to the controls. We observed that all patients present decreased in maximal isometric strength of the FSHD quadriceps (manuscript in preparation). Moreover, it remains to be fully clarified whether primary or secondary defects in the muscle regenerative capability involving satellite cells play a pathogenic role in the disease. Indeed, our study indicates that myoblasts derived from satellite cells of clinically affected and unaffected FSHD muscles will differentiate into morphologically abnormal myotubes whose phenotype might explain, at least in part, the muscle weakness typical of FSHD.
FSHD myoblasts from affected and unaffected muscles are hypersensitive to an induced oxidative stress
In this study, we also found that myoblast from affected and unaffected FSHD muscles are hypersensitive to oxidative stress induced by hydrogen peroxide. In accordance with a previous study [20] that showed an increased susceptibility to oxidative stress in FSHD myoblasts at early stages of muscle differentiation, this result suggests that oxidative stress might underlie the FSHD muscle patho-physiological mechanism. The idea that in FSHD there may be a defect in the oxidative stress response is also strengthened by the analysis of all available data [20, 37–39] Indeed, among genes that are deregulated in FSHD muscle, many are involved in the oxidative stress response, and, furthermore, it has been shown that these genes are not deregulated in other muscular dystrophies suggesting that aberrations in this pathway could be specific to FSHD. In a previous study, we have observed an up-regulation of ANT1 at the protein level in both affected and unaffected muscle [38]. ANT1 encodes for the adenine nucleotide translocator, which facilitates the export of ATP over the mitochondrial membrane. We are currently testing the hypothesis whether a potential correlation with the level of ANT-1 expression and the mitochondrial function exists in muscle cells.
Several papers support a role for oxidative stress as a pathological cause in FSHD [38, 39]. Free radicals may be part of a cascade, and may provide a biochemical tool by which the pathological process can be inhibited. More studies on the action of radical oxygen species (ROS) and their sources may lead to a better understanding of the basis of FSHD. Indeed, the demonstration that alterations in specific oxidant species or in their cognate antioxidant systems are a triggering event that leads to abnormalities in FSHD satellite cells would then have pathological value and represent molecular targets for therapeutic and diagnostic development.
In conclusion, this study shows that myoblasts derived from both clinically unaffected and affected muscles of patients with FSHD are more susceptible to oxidative stress than control myoblasts. Moreover, although myoblasts from patients affected with FSHD fully differentiated into multi-nucleated myotubes, they fused to form either thin and branched myotubes with aligned nuclei or large myotubes with random nuclei distribution. These two phenotypes might be the consequence of differences in oxidative stress sensitivity. Alternatively, additional signalling pathways may contribute either independently or co-operatively with oxidative stress in FSHD. It has been shown that overexpression of FRG1 in transgenic mice induces skeletal muscle atrophy [9]. By comparing genome-wide gene expression data from muscle biopsies of patients with FSHD to those of 11 other neuromuscular disorders, paired-like homeodomain transcription factor 1 (PITX1) was found specifically up-regulated in patients with FSHD [14]. Since DUX4 protein can activate PITX 1 promoter, both DUX4 and PITX1 in FSHD muscles may play critical roles in the molecular mechanisms of the disease [14].
Therefore, these abnormalities could be responsible for the muscle weakness observed in patients with FSHD and provide an important marker for FSHD myoblasts.
Aknowledgements
We are grateful to the patients from the Association FSHD Europe who continuously supported this study. We thank the Banque de Tissus pour la Recherche of the Association Française contre les Myopathies (AFM). This work was supported by the Association Française contre les Myopathies (AFM), the Centre National de la Recherche Scientifique (CNRS) and the Institut National de la Santé et de la Recherche Médicale (INSERM). M. Barro was supported by the Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche and the AFM. We thank Henri-Alexandre Michaud (Institut de Génétique Moléculaire de Montpellier CNRS-UMR5535, France) for his help and advices on FACS experiments, Dr. Camille Auziol and Dr. Krzysztof Rogowski (Centre de Recherche de Biochimie Macromoléculaire CRBM-CNRS-UMR5237) for their advices with cytoskeletal stainings, Sylvain De Rossi and the MRI crew at the CRBM, for their constant help with imaging.
References
- 1.Van Deutekom JC, Wijmenga C, Van Tienhoven EA, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2:2037–42. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 2.Lee JH, Goto K, Matsuda C, et al. Characterization of a tandemly repeated 3.3-kb KpnI unit in the facioscapulohumeral muscular dystrophy (FSHD) gene region on chromosome 4q35. Muscle Nerve. 1995;2:S6–13. [PubMed] [Google Scholar]
- 3.Lunt PW, Jardine PE, Koch M, et al. Phenotypic-genotypic correlation will assist genetic counseling in 4q35-facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995;2:S103–9. [PubMed] [Google Scholar]
- 4.Wijmenga C, Van Deutekom JC, Hewitt JE, et al. Pulsed-field gel electrophoresis of the D4F104S1 locus reveals the size and the parental origin of the facioscapulohumeral muscular dystrophy (FSHD)-associated deletions. Genomics. 1994;19:21–6. doi: 10.1006/geno.1994.1006. [DOI] [PubMed] [Google Scholar]
- 5.Lunt PW, Jardine PE, Koch MC, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD) Hum Mol Genet. 1995;4:951–8. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 6.Tawil R, Forrester J, Griggs RC, et al. Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. The FSH-DY Group. Ann Neurol. 1996;39:744–8. doi: 10.1002/ana.410390610. [DOI] [PubMed] [Google Scholar]
- 7.Van Overveld PG, Enthoven L, Ricci E, et al. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol. 2005;58:569–76. doi: 10.1002/ana.20625. [DOI] [PubMed] [Google Scholar]
- 8.Gabellini D, Green MR, Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–48. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 9.Gabellini D, D’Antona G, Moggio M, et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–7. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 10.Jiang G, Yang F, Van Overveld PG, et al. Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum Mol Genet. 2003;12:2909–21. doi: 10.1093/hmg/ddg323. [DOI] [PubMed] [Google Scholar]
- 11.Winokur ST, Chen YW, Masny PS, et al. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet. 2003;12:2895–907. doi: 10.1093/hmg/ddg327. [DOI] [PubMed] [Google Scholar]
- 12.Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 13.Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–23. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA. 2007;104:18157–62. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding H, Beckers MC, Plaisance S, et al. Characterization of a double homeodomain protein (DUX1) encoded by a cDNA homologous to 3.3 kb dispersed repeated elements. Hum Mol Genet. 1998;7:1681–94. doi: 10.1093/hmg/7.11.1681. [DOI] [PubMed] [Google Scholar]
- 16.Petrov A, Allinne J, Pirozhkova I, et al. A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: implications for the facio-scapulo-humeral dystrophy. Genome Res. 2008;18:39–45. doi: 10.1101/gr.6620908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrov A, Pirozhkova I, Carnac G, et al. Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystrophy patients versus normal human myoblasts. Proc Natl Acad Sci USA. 2006;103:6982–7. doi: 10.1073/pnas.0511235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemmers RJ, Wohlgemuth M, Van Der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–94. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Der Maarel SM, Frants RR, Padberg GW. Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta. 2007;1772:186–94. doi: 10.1016/j.bbadis.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Winokur ST, Barrett K, Martin JH, et al. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul Disord. 2003;13:322–3. doi: 10.1016/s0960-8966(02)00284-5. [DOI] [PubMed] [Google Scholar]
- 21.Vilquin JT, Marolleau JP, Sacconi S, et al. Normal growth and regenerating ability of myoblasts from unaffected muscles of facioscapulohumeral muscular dystrophy patients. Gene Ther. 2005;12:1651–62. doi: 10.1038/sj.gt.3302565. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Forner J, Fournet S, et al. Improved characterization of FSHD mutations. Ann Genet. 2001;44:105–10. doi: 10.1016/s0003-3995(01)01075-9. [DOI] [PubMed] [Google Scholar]
- 23.Thomas C, Perrey S, Lambert K, et al. Monocarboxylate transporters, blood lactate removal after supramaximal exercise, and fatigue indexes in humans. J Appl Physiol. 2005;98:804–9. doi: 10.1152/japplphysiol.01057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas C, Sirvent P, Perrey S, et al. Relationships between maximal muscle oxidative capacity and blood lactate removal after supramaximal exercise and fatigue indexes in humans. J Appl Physiol. 2004;97:2132–8. doi: 10.1152/japplphysiol.00387.2004. [DOI] [PubMed] [Google Scholar]
- 25.Kitzmann M, Bonnieu A, Duret C, et al. Inhibition of Notch signaling induces myotube hypertrophy by recruiting a subpopulation of reserve cells. J Cell Physiol. 2006;208:538–48. doi: 10.1002/jcp.20688. [DOI] [PubMed] [Google Scholar]
- 26.Baecker V, Travo P. Proceedings of the ImageJ User and Developer Conference: MRI Cell Image Analyzer–-A visual scripting interface for ImageJ and its usage at the microscopy facility Montpellier RIO Imaging; pp. 105–10. [Google Scholar]
- 27.Kaufman SJ, Foster RF. Replicating myoblasts express a muscle-specific phenotype. Proc Natl Acad Sci USA. 1988;85:9606–10. doi: 10.1073/pnas.85.24.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sinanan AC, Hunt NP, Lewis MP. Human adult craniofacial muscle-derived cells: neural-cell adhesion-molecule (NCAM; CD56)-expressing cells appear to contain multipotential stem cells. Biotechnol Appl Biochem. 2004;40:25–34. doi: 10.1042/BA20030185. [DOI] [PubMed] [Google Scholar]
- 29.Morosetti R, Mirabella M, Gliubizzi C, et al. Isolation and characterization of mesoangioblasts from facioscapulohumeral muscular dystrophy muscle biopsies. Stem Cells. 2007;25:3173–82. doi: 10.1634/stemcells.2007-0465. [DOI] [PubMed] [Google Scholar]
- 30.Yip DJ, Picketts DJ. Increasing D4Z4 repeat copy number compromises C2C12 myoblast differentiation. FEBS Lett. 2003;537:133–8. doi: 10.1016/s0014-5793(03)00110-8. [DOI] [PubMed] [Google Scholar]
- 31.Pizon V, Gerbal F, Diaz CC, et al. Microtubule-dependent transport and organization of sarcomeric myosin during skeletal muscle differentiation. Embo J. 2005;24:3781–92. doi: 10.1038/sj.emboj.7600842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–7. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 33.Furling D, Coiffier L, Mouly V, et al. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet. 2001;10:2079–87. doi: 10.1093/hmg/10.19.2079. [DOI] [PubMed] [Google Scholar]
- 34.Furling D, Lemieux D, Taneja K, et al. Decreased levels of myotonic dystrophy protein kinase (DMPK) and delayed differentiation in human myotonic dystrophy myoblasts. Neuromuscul Disord. 2001;11:728–35. doi: 10.1016/s0960-8966(01)00226-7. [DOI] [PubMed] [Google Scholar]
- 35.Timchenko NA, Iakova P, Cai ZJ, et al. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol. 2001;21:6927–38. doi: 10.1128/MCB.21.20.6927-6938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blau HM, Webster C, Chiu CP, et al. Differentiation properties of pure populations of human dystrophic muscle cells. Exp Cell Res. 1983;144:495–503. doi: 10.1016/0014-4827(83)90431-7. [DOI] [PubMed] [Google Scholar]
- 37.Macaione V, Aguennouz M, Rodolico C, et al. RAGE-NF-kappaB pathway activation in response to oxidative stress in facioscapulohumeral muscular dystrophy. Acta Neurol Scand. 2007;115:115–21. doi: 10.1111/j.1600-0404.2006.00724.x. [DOI] [PubMed] [Google Scholar]
- 38.Laoudj-Chenivesse D, Carnac G, Bisbal C, et al. Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J Mol Med. 2005;83:216–24. doi: 10.1007/s00109-004-0583-7. [DOI] [PubMed] [Google Scholar]
- 39.Celegato B, Capitanio D, Pescatori M, et al. Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics. 2006;6:5303–21. doi: 10.1002/pmic.200600056. [DOI] [PubMed] [Google Scholar]