

Abstract
Pseudomonas aeruginosa PAK (serotype O6) produces a single polar, glycosylated flagellum composed of a-type flagellin. To determine whether or not flagellin glycosylation in this serotype requires O-antigen genes, flagellin was isolated from the wild type, three O-antigen-deficient mutants wbpL, wbpO, and wbpP, and a wbpO mutant complemented with a plasmid containing a wild-type copy of wbpO. Flagellin from the wbpO mutant was smaller (42 kDa) than that of the wild type (45 kDa), or other mutants strains, and exhibited an altered isoelectric point (pI 4.8) when compared with PAK flagellin (pI 4.6). These differences were because of the truncation of the glycan moiety in the wbpO-flagellin. Thus, flagellin glycosylation in P. aeruginosa PAK apparently requires a functional WbpO but not WbpP. Because WbpP was previously proposed to catalyze a metabolic step in the biosynthesis of B-band O-antigen that precedes the action of WbpO, these results prompted us to reevaluate the two-step pathway catalyzed by WbpO and WbpP. Results from WbpO-WbpP-coupled enzymatic assays showed that either WbpO or WbpP is capable of initiating the two-step pathway; however, the kinetic parameters favored the WbpO reaction to occur first, converting UDP-N-acetyl-D-glucosamine to UDP-N-acetyl-D-glucuronic acid prior to the conversion to UDP-N-acetyl-D-galacturonic acid by WbpP. This is the first report to show that a C4 epimerase could utilize UDP-N-acetylhexuronic acid as a substrate.
Nucleotide-activated monosaccharides serve as precursors to a number of cell-surface glycoconjugates, including lipopolysaccharide (LPS),8 exopolysaccharide, and glycosylated proteins of pili and flagella. In some cases, these precursors may be shared between different systems. In Neisseria meningitidis, for example, more than three unlinked loci are required for synthesis and attachment of the pilin glycan (1, 2). One of these loci includes galE, a gene that is required for synthesis of UDP-galactose, which in turn is required both for the production of lipo-oligosaccharide and for glycosylation of the pilin subunits (3). In Pseudomonas aeruginosa strains, dTDP-L-rhamnose is channeled into synthesis of numerous L-rhamnose-containing cell-surface structures, including rhamnolipids (4), core oligosaccharide, and O-antigen polysaccharide (5). This sugar-nucleotide is synthesized by proteins encoded in the rmlBDAC gene cluster, which is unlinked to the gene clusters that are required for synthesis of these glycoconjugates.
Another example of the interrelationship between biosynthesis pathways for cell-surface glycoconjugates is pilin glycosylation in P. aeruginosa strain 1244 (serotype O7). The covalently attached pilin glycan is identical to the repeat unit from the B-band O-antigen, one of two types of O-antigen possessed by P. aeruginosa strains (6). In fact, a mutation in the gene that encodes the initiating glycosyltransferase WbpL, which transfers the first monosaccharide of the O-antigen unit from UDP-N-acetyl-D-quinovosamine (UDP-D-QuiNAc) to the lipid carrier undecaprenol phosphate, abrogates pilin glycosylation as well as O-antigen LPS.
P. aeruginosa produces a single polar flagellum that has been shown to be an important virulence factor in pulmonary disease and is important for colonization of the respiratory tract in patients with cystic fibrosis and individuals with nosocomial pneumonia (7, 8). The flagellar filament is composed of flagellin subunits encoded by fliC, and in this species flagellin is classified as either a-type or b-type based on their amino acid sequence, apparent molecular weight, and antigenicity (9–12). Like the pilin of P. aeruginosa 1244, the a-type flagellins of strain PAK (serotype O6) are glycosylated, although less is known about the nature of the glycosylation (13–15). The flagellin of strain PAK contains two variable length O-linked oligosaccharide chains, each with up to 11 monosaccharides (14). Although the exact composition of the flagellin glycans is unknown, evidence suggests that each glycan begins with an L-rhamnose residue and contains pentoses, hexoses, deoxyhexoses, hexuronic acids, and/or deoxyhexoses with amino and formyl substitutions. A cluster of 14 genes, located upstream of the flagellin gene fliC in the PAK genome, has been designated as the flagellin glycosylation island (16). Two of the genes in this cluster have been shown through individual null mutations to be required for flagellin glycosylation, but the function of the remaining 12 genes remains unknown, and because of the paucity of information about the composition of the glycans, it is not possible to propose a biosynthesis pathway at this time.
The objective of this study was to determine whether flagellin glycosylation in P. aeruginosa PAK (serotype O6) requires genes involved in B-band O-antigen biosynthesis as does pilin glycosylation in strain 1244. The genes responsible for biosynthesis of the serotype O6 O-antigen have been characterized and designated as the wbp gene locus (17), and the structure of the O-antigen of strain PAK is a tetrasaccharide repeat of [→2)-α-L-rhamnose-(1→4)-α-D-2-acetamido-3-O-acetyl-6-aminogalacturonic acid-(1–4)-α-D-6-amino-2-deoxy-2-formamido-D-galacturonic acid-(1→3)-α-D-2-acetamido-2,6-dideoxy-D-glucose-(1→] (18). We isolated and compared flagellin from wild-type PAK and three mutants, wbpL, wbpO, and wbpP. Null mutations in these genes, contained within the O6 O-antigen cluster, have been generated, and analysis of the LPS phenotypes from these mutants showed the absolute requirement of these genes for O-antigen production (17). WbpL is the initiating transferase of O-antigen biosynthesis and transfers the first sugar of the O-repeat unit, N-acetyl-Dquinovosamine (D-QuiNAc), from UDP-D-QuiNAc to the undecaprenol phosphate (C55-P) lipid carrier (Fig. 1). WbpP and WbpO are required for synthesis of the UDP-N-acetyl-D-galacturonic acid (UDP-D-GalNAcA), a precursor to the galactosaminuronic acid-derived residues of the O-antigen. WbpP has been characterized as a UDP-N-acetyl-D-glucosamine (UDP-D-GlcNAc) 4-epimerase (19), and WbpO has been characterized as a 6-dehydrogenase that accepts UDP-N-acetyl-D-galactosamine (UDP-D-GalNAc), the product of WbpP/UDPD-GlcNAc reactions, and produces UDP-D-GalNAcA (20).
FIGURE 1. Putative biosynthetic pathway for the serotype O6 B-band O-antigen repeat unit.
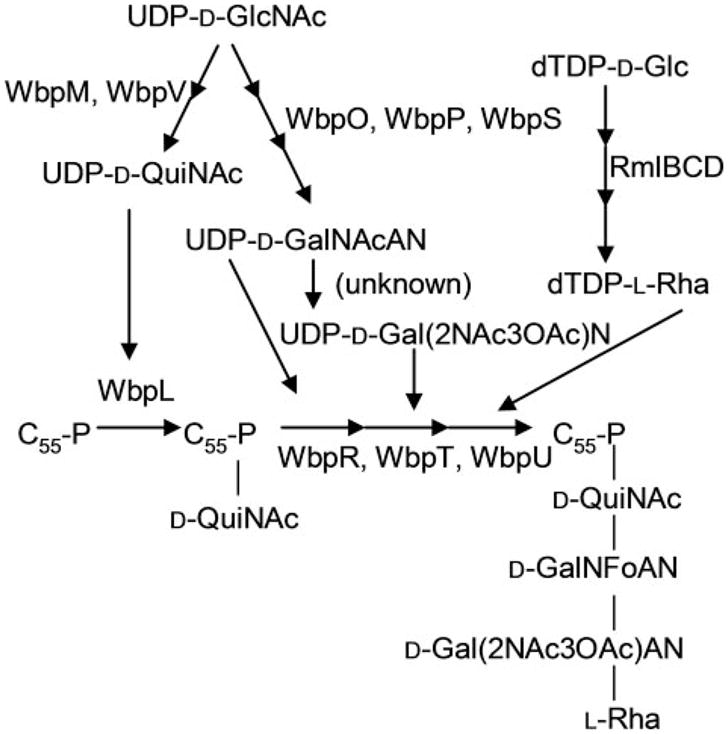
All enzymes are encoded within the B-band O-antigen gene cluster, except RmlB, RmlC, and RmlD, which are encoded in the rmlBDAC locus. UDP-D-GalNAcA, UDP-N-acetyl-D-galacturonic acid; UDP-DGal( 2NAc3OAc)N, UDP-2-acetamido-3-O-acetyl-2-deoxy-D-galactopyranosiduronamide; dTDP–D-Glc, dTDP-D-glucose; dTDP-L-Rha, dTDP-L-rhamnose; D-Gal(2NAc3OAc)N, 2-acetamido-3-O-acetyl-2-deoxy-D-galactopyranosiduronamide; D-GalNFoAN, 2-formamido-2-deoxy-D-galactopyranosiduronamide; D-QuiNAc, N-acetyl-D-quinovosamine; L-Rha, L-rhamnose.
In this study, we show that WbpO is required for flagellin glycosylation, but WbpP is not. This is surprising because WbpP was proposed to catalyze an earlier step in the metabolic pathway than WbpO, and it prompted us to conduct more in depth evaluation of the metabolic steps catalyzed by these two proteins in the O-antigen biosynthesis in P. aeruginosa strain PAK. Here we present the biochemical evidence to show that whereas either WbpP or WbpO could initiate the two-step pathway, the kinetic parameters of the two enzymes indicated a strong preference for WbpO to convert UDP-D-GlcNAc into UDP-N-acetyl-D-glucuronic acid (UDP-D-GlcNAcA) prior to catalysis of this compound by WbpP to generate UDP-D-GalNAcA.
EXPERIMENTAL PROCEDURES
Materials
UDP-D-GlcNAc, UDP-D-GalNAc, NAD+, NADP+, nitro blue tetrazolium (NBT), 5-bromo-4-chloro-3-indolylphosphate, transferrin, cytochrome c, bovine serum albumin, trypsinogen, alcohol dehydrogenase, and the antibiotics used in this study were obtained from Sigma. Isopropyl β-D-thiogalactopyranoside (IPTG), isoelectric focusing (IEF) sample loading buffer, Novex® IEF gels, and the SimplyBlue™ Safestain were purchased from Invitrogen.
Bacterial Strains
The bacterial strains used in this study have been listed in supplemental Table S1.
Analytical Techniques
BLAST (21) and ClustalW (22) were used for the analysis of nucleotide and protein sequences. SDSPAGE was performed according to the method of Hancock and Carey (23) based on the discontinuous buffer system of Laemmli (24), except that a mini-gel system was used (Bio-Rad). The gels were stained with Coomassie Brilliant Blue R-250 (Sigma). Protein concentrations were determined as described by Bradford (25).
Capillary electrophoresis (CE) analysis was performed using a P/ACE MDQ glycoprotein system with UV detection (Beckman Coulter, Inc., Fullerton, CA) as described previously (26).
IEF was performed using an XCell Surelock™ Mini-Cell (Invitrogen). Samples containing flagella were mixed with an equal volume of IEF sample loading buffer and loaded onto Novex® IEF gels with a range of pH 3–7, and electrophoresis was performed according to the manufacturer’s instructions. After electrophoresis, the IEF gel was fixed in 12% trichloroacetic acid for 30 min and then washed with distilled water over a period of 45 min with the water changed every 5 min. The IEF gel was stained with SimplyBlue™ Safestain (Invitrogen) and washed as described by the manufacturer’s instructions. For the Western immunoblotting experiment, proteins were transferred from the IEF gel after electrophoresis to Biotrace™ nitrocellulose using a Bio-Rad transfer cell for 1 h at 150 mA using Tris-glycine transfer buffer as described by the manufacturer (Invitrogen).
For mass spectrometry (MS), lyophilized flagellin protein was first resuspended in water, and then a 1:1 mixture of protein solution to matrix solution (sinapinic acid in 50% acetonitrile, 0.1% trifluoroacetic acid) was made, and 1 μl was spotted onto the MALDI plate. Some of the flagellin samples were desalted directly on the MALDI plate by pipetting 2 μl of water onto the spot and removing after 1 min using a gel-loading tip. Analysis of intact flagellin proteins was performed using a matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) MS instrument (model Reflex III, Bruker, Germany) equipped with a 337 nm nitrogen laser (Biological Mass Spectrometry Facility, University of Guelph). The instrument was externally calibrated using various protein standards, including cytochrome c, bovine serum albumin, trypsinogen, and alcohol dehydrogenase. MS analysis of intact flagellin was performed in the linear detection mode, in the positive mode, with laser energy set at 35–40%.
Isolation of Flagellin
Flagella were isolated from the different P. aeruginosa strains grown overnight in Davis minimal broth (Difco) supplemented with 0.01 mM glucose, 0.8 mM MgSO4, 0.015 mM thiamine, and 0.04 mg/ml casamino acids. Flagella were isolated and purified using the ammonium sulfate precipitation method as described by Brimer and Montie (13). The flagellin preparation was assessed for purity by SDS-PAGE analysis. Preparation of flagella from the P. aeruginosa LPS mutants was more difficult than from the wild-type strain because the LPS mutants were more prone to cell lysis during the initial steps of the isolation procedure, resulting in large amounts of contaminating protein. In the case where the sample was found to contain many contaminating proteins, the sample was brought up to a volume of 200 ml in sodium phosphate buffer, and the flagella were again recovered by repeated ammonium sulfate precipitation. Flagella prepared in this manner were about 90% pure, but trace amounts of pilin were found to be co-purified in the process.
Adsorption of Nonspecific Antibodies from Anti-FliC Antiserum
Rabbit antiserum to P. aeruginosa strain PAK flagellin, FliC, was kindly provided by Dr. Reuben Ramphal (27). To increase the specificity, the antiserum was adsorbed against proteins isolated from P. aeruginosa strain PAK-N1, a mutant that does not produce flagella. Whole cell protein lysates were prepared from strain PAK-N1 as follows: 5 ml of a bacterial culture grown overnight in Miller’s LB broth, 37 °C, was centrifuged at 13,000 × g for 2 min, and the pellet was resuspended in protein sample buffer and boiled in a water bath for 10 min. This protein preparation was resolved on a 10% glycine SDS polyacrylamide gel and transferred to Biotrace™ nitrocellulose (VWR International, Mississauga, Ontario, Canada). The nitrocellulose was cut into 0.5 × 8-cm pieces. The original antiserum (15μl) was diluted in 7.5 ml of phosphate-buffered saline (PBS; 125mMNaCl, 1.5 mM KH2PO4, 8 mM Na2HPO4·12H2O, 7 mM KCl, pH 7.0) and sodium azide (final concentration of 0.02%). Five nitrocellulose strips were incubated in 7.5 ml of antiserum for 4 h at room temperature on a CLAY ADAMS Brand Nutator (BD Biosciences). The strips were then removed, and five new strips were added, and the antiserum was again incubated as described. This process was repeated five times with fresh strips each time, and the antiserum was stored at −20 °C. The specificity of the antiserum for FliC was tested by Western immunoblotting using flagellin prepared from the wild-type PAK and PAK-N1 (serves as a negative control).
Flagellin Glycosylation Detection
Flagellins were resolved on a 10% glycine SDS-polyacrylamide gel and transblotted onto nitrocellulose. The glycosylation detection protocol of O’Shannessey et al. (28) was used with the following modifications. All incubations took place at room temperature. After transfer of the proteins to the nitrocellulose, the membrane was washed with 10 ml of PBS (9 mM sodium phosphate, 27 mM sodium chloride, pH 7.2) for 10 min. The membrane was incubated in 15 mM sodium periodate in 50 mM sodium acetate buffer, pH 5.5, for 30 min in the dark. The membrane was washed four times for 10 min with PBS followed by a 1-h incubation in 5 mM biotin hydrazide, 50 mM sodium acetate buffer. The nitrocellulose was then washed three times for 10 min in Tris-buffered saline (TBS: 50 mM Tris-HCl, 27 mM sodium chloride, pH 7.2) and then blocked for 30 min in TBS containing 3% bovine serum albumin. The membrane was washed three times for 10 min in TBS and incubated for 2 h in a 1:1000 dilution of alkaline phosphatase streptavidin (Vector Laboratories Inc., Burlingame, CA) in TBS. This was followed by three 10-min washes in TBS. The membrane was developed using a substrate consisting of 30 mg of NBT and 15 mg of 5-bromo-4-chloro-3-indolylphosphate in 100 ml of 0.1 M sodium bicarbonate buffer, pH 9.8. The nonglycosylated WbpD protein used as a negative control was purified as described previously (29).
Construction of pET-28a wbpO
The wbpO gene was cloned into the pET-28a(+) expression vector with an N-terminal polyhistidine tag. wbpO was amplified from genomic DNA (P. aeruginosa IATS O6) by PCR using the following primers: forward primer 5′-GTGCGAAGCATATGAAGGATCTGAAG-3′, which incorporates an NdeI restriction digestion site (underlined); reverse primer 5′-CAACTAAGCTTACAGGCGTAGATC-3′, which incorporates a HindIII restriction digestion site. The PCR consisted of 100 ng of genomic DNA, 0.5μM of each primer, 2.5mM of each dNTP, 1mM MgSO4, and 1 × buffer. A 5-min denaturation was carried out before the addition of 2.5 units of Pwo polymerase (Roche Applied Science), followed by 30 cycles of 30 s at 94 °C, 45 s at 60 °C, and 1 min at 72 °C. A final elongation step of 7 min at 72 °C was performed. Both the PCR product and the pET-28a vector were digested with NdeI and HindIII, purified using UltraClean™ 15 (MO BIO Laboratories, Carlsbad, CA), and ligated overnight at 15 °C using T4 DNA ligase (New England Biolabs, Pickering, Ontario, Canada). The construct obtained was analyzed by restriction digestion and sequencing, and subsequently transformed into the expression strain E. coli BL21(DE3) pLysS, using kanamycin (50 μg/ml) for selection.
Expression and Purification of Enzymes
For the expression of WbpO, 250 ml of Terrific Broth (30) containing kanamycin was inoculated with 6 ml of an overnight culture and grown at 37 °C, 200 rpm. When OD600 reached 0.6, IPTG was added to a final concentration of 1 mM, and expression was allowed to proceed for 6 h at 37 °C. Cells were harvested by centrifugation (Sorvall Evolution RC, Mandel Scientific, Guelph, Ontario, Canada) at 5,000 × g for 15 min at 4 °C, and pellets were stored at −20 °C. WbpP and WbpAO5 were expressed as described previously (19, 26). WbpAO5 from P. aeruginosa PAO1 (serotype O5) has been shown to be a UDP-D-GlcNAc 6-dehydrogenase (26).
For purification ofWbpOor WbpP, the pellet from 250 ml of induced culture was resuspended in 25 ml of binding buffer (20 mM sodium phosphate, pH 7.4, 500 mM sodium chloride). The cells were disrupted by ultrasonication on ice and cell debris, and membrane fractions were removed by ultracentrifugation (Beckman L8-M, Beckman Coulter Canada, Inc., Mississauga, Ontario, Canada) at 175,000 × g for 1 h at 4 °C. Purification using the HiTrap chelating HP column (GE Healthcare) was performed as recommended by the manufacturer, with nickel as the chelating ion and binding buffer containing 300 mM imidazole as the eluent. Following purification, the concentration of sodium chloride in purified WbpO or WbpP was reduced to 50mM using a PD-10 desalting column (GE Healthcare) according to the protocol recommended by the manufacturer. The purity of WbpO and WbpP was analyzed by SDSPAGE, and purified protein was stored at −20 °C after the addition of glycerol to a final concentration of 25%. WbpAO5 was purified as described previously (26).
Enzymatic Reactions
Coupled reactions contained 2 mM substrate (UDP-D-GlcNAc or UDP-D-GalNAc), 5 mM NAD+, 50 mM Tris-HCl, pH 8.5, 50 mM ammonium sulfate, 1.8 μg of WbpP, 18 μg of WbpO, and/or 1.8 μg of WbpAO5. Single enzyme reactions were incubated for 1 h at 37 °C, whereas coupled reactions were carried out in two steps. After addition of the first enzyme, the reaction was incubated at 37 °C for 1 h, followed by the addition of the second enzyme and further incubation for 1 h.
For the pH and temperature optima studies of WbpO, reactions contained 1 mM substrate (UDP-D-GlcNAc or UDP-DGalNAc), 2.5 mM NAD+, 100 mM buffer, 100 mM ammonium sulfate, and 5μg of WbpO and were carried out for 1 h at 37 °C. For determination of the pH optimum, MES (pKa 6.1) was used as the buffer for pH 5, 5.5, 6, and 6.5; and BisTris propane (pKa1 6.8 and pKa2 9.0) was the buffer for pH 7, 7.5, 8, 8.5, 9, 9.5, and 10. Reactions for the determination of the temperature optimum were carried out at 0, 15, 20, 30, 37, 42, 55 or 65 °C in Tris-HCl, pH 7.5. For both pH and temperature studies, substrate conversion was determined by CE. The cofactor requirements of WbpO were determined as described previously for WbpAO5 (26), using 2 μg of WbpO and 100 mM of the salts as listed in supplemental Table S2.
Synthesis and Purification of UDP-D-GlcNAcA and UDP-D-GalNAcA
Ten μmol of UDP-D-GlcNAc was converted into UDP-D-GlcNAcA in a reaction containing 25 μmol of NAD+, 100mM Tris-HCl, pH 8, 100mM ammonium sulfate, and 500 μg of WbpAO5. After removal of protein by ultrafiltration (Centriplus YM3 cartridge from Millipore), UDP-D-Glc-NAcA was purified by anion-exchange on an Econo-Pac High Q column (Bio-Rad) using a linear gradient of 0–500 mM ammonium bicarbonate, pH 8.0. The fractions containing UDP-D-GlcNAcA were pooled and lyophilized. Ten μmol of UDP-D-GalNAc was converted into UDP-D-GalNAcA in the same manner as for UDP-D-GlcNAcA, except that 1.5 mg of WbpO was used instead of WbpAO5.
Analysis of the WbpO Reaction Products by NMR Spectroscopy
The purified reaction products of the WbpO reaction were suspended in 200 μl of 99% D2O (Cambridge Isotopes Laboratories Inc., Andover, MA) and placed into 3-mm NMR tubes (Wilmad, Buena, NJ). Standard homo- and heteronuclear correlated two-dimensional 1H NMR, 13C heteronuclear single quantum correlation spectroscopy, heteronuclear multiple bond correlation spectroscopy, COSY, TOCSY, and NOESY pulse sequences from Varian were used for general assignments. Selective one-dimensional TOCSY and NOESY experiments with a Z-filter were used for complete residue assignment and characterization of individual spin systems (31, 32). NMR experiments were performed with a Varian 600 MHz (1H) spectrometer equipped with a Varian 5-mm Z-gradient triple resonance cryogenically cooled probe (cold probe) for optimal sensitivity. NMR experiments were typically performed at 25 °C with suppression of the HOD resonance at 4.78 ppm. The methyl resonance of acetone was used as an internal reference (δH 2.225 ppm and δC 31.07 ppm).
Determination of Kinetic Parameters of WbpO
All reactions contained 100 mM Tris-HCl, pH 7.5, and 100 mM ammonium sulfate in a final volume of 170 μl. For determination of kinetic parameters for UDP-D-GlcNAc as the substrate, reactions contained 3mMNAD+ and concentrations of UDP-D-GlcNAc that varied from 0.025 to 1.5 mM. For determination of kinetic parameters for NAD+ as a co-enzyme, reactions contained 1.5 mM UDP-D-GlcNAc and concentrations of NAD+ that varied from 0.05 to 3mM. For determination of kinetic parameters for NADP+ as an alternative coenzyme, reactions contained 5 mM UDP-D-GlcNAc and concentrations of NADP+ that ranged from 0.5 to 10mM. For determination of kinetic parameters for UDP-D-GalNAc, reactions contained 3mMNAD+ and concentrations of UDP-D-GalNAc that ranged from 0.1 to 1.5 mM. Reactions were incubated at 37 °C, and the progress of the reactions was followed by measuring A340 in a Varian Cary 100 spectrophotometer (Varian Instruments, Walnut Creek, CA). Subsequently, the concentration of NAD(P)H produced was determined from A340 measurements and ε340 = 6,220 M−1 cm−1. The data were evaluated using the Enzyme Kinetics Module of Sigma Plot from SPSS Science (Richmond, CA). The fit of the data to the Michaelis-Menten and Hill models by nonlinear regression were compared using the square of residuals (R2), the Akaike Information Criterion, and the standard deviation of the residuals (Sy.x). A model could be judged to be a better fit if it produced a higher R2 value, a lower Akaike Information Criterion, and/or a lower Sy.x value. Upon rejection of the Hill model, the data were subsequently fit to the Michaelis-Menten model by nonlinear regression (Sigma Plot), and the results are the average of three experiments.
Cell Extract Assays
For expression of GalE of Klebsiella pneumonia (33) or Gne of Yersinia enterocolitica (34), 50 ml of LB broth supplemented with 34μg/ml chloramphenicol and 50 μg/ml kanamycin for GalE or 15μg/ml tetracycline for Gne was inoculated with 1.25 ml of an overnight culture. Cells were grown at 37 °C for 8 h. For expression of WbgU of Plesiomonas shigelloides, 50 ml of LB broth supplemented with 100 μg/ml ampicillin was inoculated with 1.25 ml of an overnight culture. When OD600 reached 0.6, expression was induced by the addition of 0.15mMIPTG, and WbgU was expressed for 8 h at 30 °C. For expression of WbpP, 50 ml of LB broth supplemented with 100μg/ml ampicillin and 34μg/ml chloramphenicol was inoculated with 1.25 ml of an overnight culture. Expression was induced by the addition of 0.5 mM IPTG once OD600 reached 0.6, and WbpP was expressed for 6 h at 30 °C. In all cases, culture were divided into 10-ml aliquots and cells harvested by centrifugation.
To prepare cell extracts, cells from a 10-ml aliquot were resuspended in 1 ml of Na2HPO4 buffer, pH 8.0, and disrupted by ultrasonication on ice (Sonic Dismembrator model 500, Fisher). Cell debris was removed by centrifugation at 15,500 × g for 10 min at 4 °C, followed by ultracentrifugation of the supernatant at 117,000 × g for 1 h at 4 °C. For each assay, 2μl of supernatant from the ultracentrifugation step was used. As a negative control in each case, cell extracts from the appropriate host strain (without the expression vector) were prepared in the same manner as described above.
Modeling of UDP-D-GlcNAcA and UDP-D-GalNAcA in the Active Site of WbpP
Three-dimensional structure models of UDP-D-GlcNAcA and UDP-D-GalNAcA in energy-minimized conformation were generated using SYBYL 6.9 (Tripos Inc., St. Louis). These models were individually positioned in the active site of WbpP based on the crystal structures of WbpP·NAD+ ·UDP-D-GalNAc (Protein Data Bank code 1SB8) and human UDP-D-GlcNAc 4-epimerase·NADH·UDP-D-Glc-NAc (Protein Data Bank code 1HZJ subunit B) ternary complexes. Conformations of UDP-D-GlcNAcA and UDP-D-Gal-NAcA in the active site of WbpP were optimized by performing conjugate gradient energy minimization using the Crystallography and NMR System software suite (35).
RESULTS
Analysis of Flagellin
Flagellins from wild-type P. aeruginosa PAK and the mutant strains were purified and analyzed by SDSPAGE and Western immunoblotting. Flagellin from wild-type PAK, as well as the wbpL and wbpP mutants, showed an apparent molecular mass of 45 kDa, whereas flagellin from the wbpO mutant was 42 kDa (Fig. 2A). Flagellin isolated from the PAK wbpO mutant complemented with the plasmid pFV616-26a, which contains the wild-type wbpO gene, had an apparent molecular mass similar to that of the flagellin from the wildtype parent. Western immunoblotting of the PAK flagellins with rabbit polyclonal antiserum raised against purified PAK flagellin showed strong reactions with the 45-kDa flagellin proteins isolated from the parent, wbpL and wbpP mutant strains, as well as the smaller sized flagellin isolated from the wbpO mutant (Fig. 2B).
FIGURE 2. SDS-PAGE and Western immunoblot analysis of P. aeruginosa flagellins.
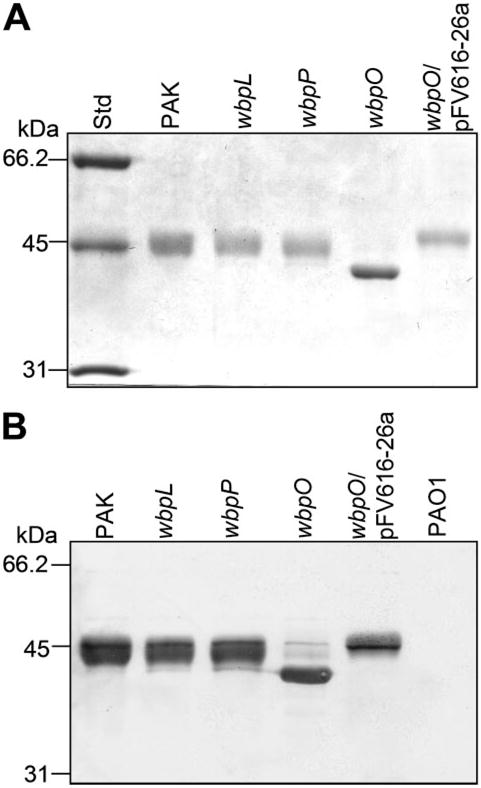
Flagellins were isolated from wild-type P. aeruginosa PAK, three PAK mutants, wbpL, wbpP, and wbpO, and wbpO mutant transformed with the plasmid pFV616-26a containing a copy of the wild-type wbpO to complement the mutation. Flagellin isolated from PAO1 was included as negative control. A, SDS-PAGE; B, Western immunoblotting using anti-FliC polyclonal antiserum.
When subjected to IEF, the flagellin isolated from the wbpO mutant exhibited an altered isoelectric point (pI) when compared with flagellins isolated from either the parent, wbpL, or wbpP mutants. Based on amino acid sequence, the pI of the FliC (flagellin) protein from PAK is predicted to be 4.9, but this does not include any charge effects that may be contributed by the glycan moiety. In IEF gel electrophoresis, flagellin from the wild type, wbpL, and wbpP mutants resolved as multiple isoforms ranging from a pI of 4.6 to 4.8, with a predominant isoform at pI 4.6 (Fig. 3A). Flagellin from the wbpO mutant also appeared to have multiple isoforms, but with the predominant isoform focusing at pI 4.8, suggesting that it is less acidic than the flagellin from the parent strain. Flagellin from the complemented mutant, wbpO/pFV616-26a, exhibited an IEF banding profile similar to that of the wild-type flagellin. Western immunoblotting of flagellin bands resolved by IEF using anti-FliC rabbit polyclonal antiserum showed a strong reaction to the predominant protein band in each lane and weaker reactions to secondary bands (Fig. 3B).
FIGURE 3. Isoelectric focusing of PAK flagellins.
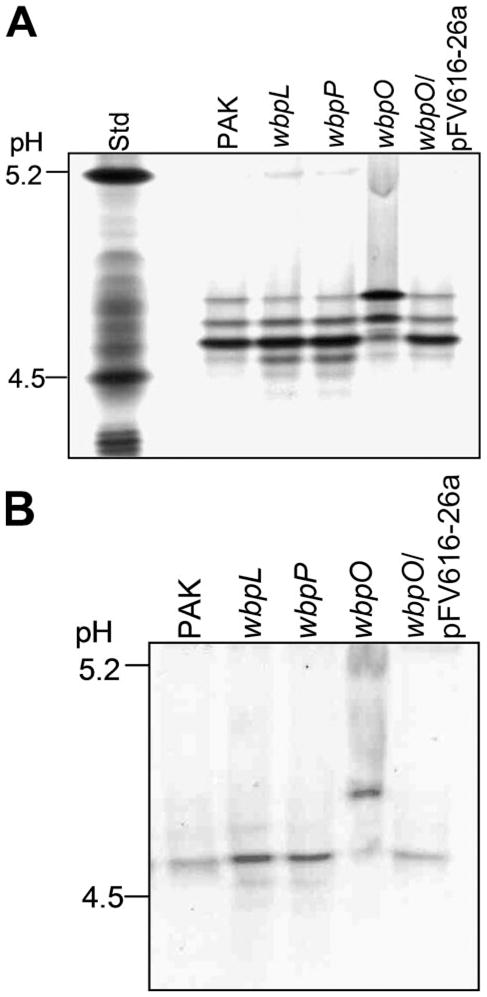
Flagellins were resolved based on their pI in an ampholyte mixture of pH 3–7. A, simply blue-stained IEF gel; B, Western immunoblot using anti-FliC antiserum.
Analysis of Flagellin Glycosylation
A biotin-hydrazide glycosylation assay of the flagellin proteins was performed, and the results showed a strongly positive reaction with the flagellins isolated from all strains except for the wbpO mutant (Fig. 4). Nonglycosylated WbpD and the glycoprotein transferrin were included in the assay as negative and positive controls, respectively.
FIGURE 4. Biotin-hydrazide glycosylation assay of PAK flagellins.
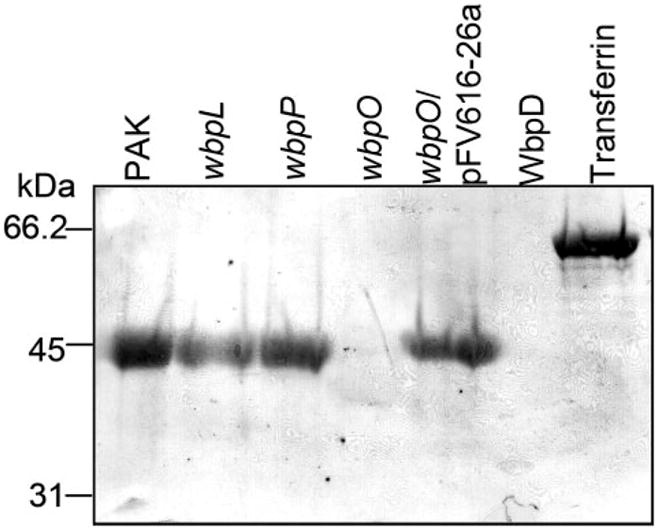
Flagellins isolated from P. aeruginosa PAK and mutant strains were examined for the presence of carbohydrate residues. Negative and positive controls included in the glycosylation assay were purified WbpD and the glycoprotein transferrin, respectively.
Mass Spectrometry Analysis of Flagellins
MALDI-TOF MS analysis of flagellin purified from strain PAK, the wbpO mutant, and the wbpO mutant carrying pFV616-26a showed peaks ([M + H]+ ion) at a mass-to-charge ratio (m/z) of 42,577, 40,197, and 41,719 (±10), respectively (supplemental Fig. S1). Because the molecular mass of nonglycosylated flagellin based on primary amino acid sequence is predicted to be 39,905 Da, these data indicate that the wild-type PAK flagellin contains a covalently bound moiety corresponding to a total mass of 2,671 Da. In contrast, flagellin from the wbpO mutant has an additional mass of 292 Da above that predicted for nonglycosylated FliC, a mass consistent with the addition of two deoxyhexose residues (146 Da each). Flagellin from the wbpO mutant carrying wbpO on a plasmid showed a broad molecular mass distribution at 41,719, indicating that wbpO on a plasmid could complement the defect in flagellin glycosylation, although not to the same glycosylation levels as the parent.
Expression and Purification of WbpO, WbpP, and WbpAO5
WbpO was expressed with an N-terminal His tag using the pET-28(+) expression vector, with WbpO making up 30–35% of total protein (as estimated from an SDS-polyacrylamide gel). WbpAO5 (serotype O5) and WbpP were expressed to high levels using the pET expression system, with yield and purity of WbpAO5 and WbpP being similar to that described previously (19, 26). For WbpO, 1.5–2.1 mg of highly purified protein (>98% pure, as estimated from the SDS-polyacrylamide gel) could be obtained from 250 ml of culture (supplemental Fig. S2). The apparent molecular mass of WbpO (46.0 kDa) as determined by SDS-PAGE correlates well with the predicted molecular mass of 46.6 kDa. The pI of the His6-WbpO fusion was predicted to be 6.0.
CE Analysis of WbpO and WbpP Reactions
CE analysis of reactions containing UDP-D-GlcNAc, NAD+, and WbpO revealed two reaction products that eluted at 15 and 19 min, respectively (Fig. 5, trace B). The product that eluted at 15 min was identified as NADH by comparison with an NADH standard, and the product at 19 min (labeled as product I in Fig. 5) was subsequently identified as UDP-D-GlcNAcA by NMR (see below). Similarly, in WbpO-catalyzed reactions containing UDP-D-GalNAc and NAD+, two product peaks eluting at 15 and 18 min were observed (Fig. 5, trace D). Again, the product eluting at 15 min was identified as NADH, and the second product (eluting at 18 min, labeled as product II in Fig. 5) was subsequently identified as UDP-D-GalNAcA by NMR (see below).
FIGURE 5. CE analysis of WbpO-catalyzed reactions.
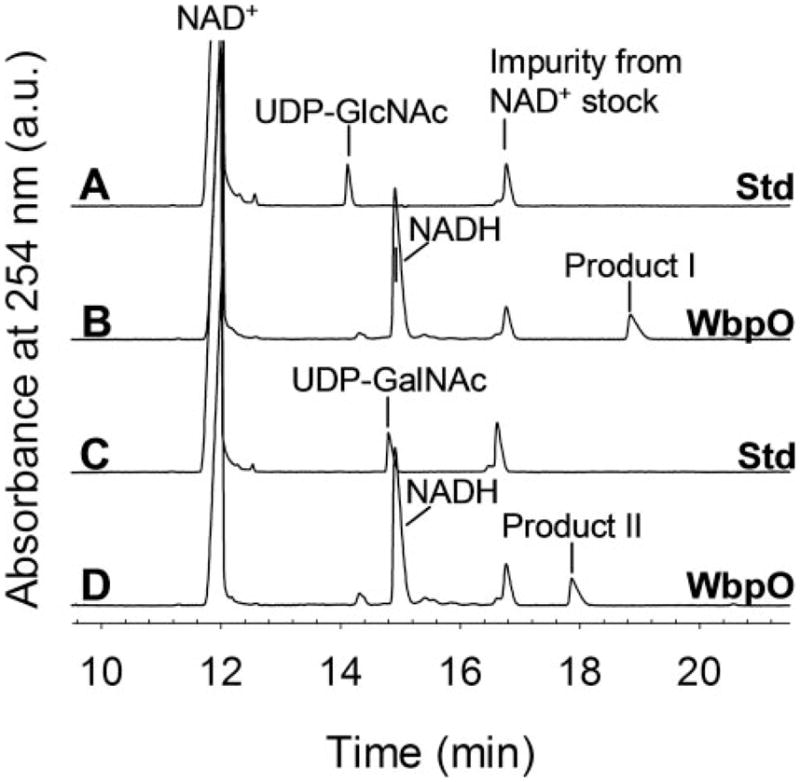
Trace A, NAD+ and UDP-D-GlcNAc standards; trace B, UDP-D-GlcNAc converted with WbpO; trace C, NAD+ and UDP-D-GalNAc standards; trace D, UDP-D-GalNAc converted with WbpO.
Because UDP-D-GlcNAcA is not commercially available, WbpAO5 (from serotype O5) was used to synthesize this sugarnucleotide for WbpP reactions (26). WbpAO5 was used to completely convert UDP-D-GlcNAc (and NAD+) into UDP-D-Glc-NAcA (and NADH; Fig. 6, trace B) prior to the addition of WbpP. CE analysis of these WbpAO5/WbpP sequential reactions showed that WbpP was capable of converting UDP-DGlcNAcA (eluted at 19 min) into a product that migrates at 18 min (labeled as product III in Fig. 6, trace C). This product was identified as UDP-D-GalNAcA by comparison to WbpO/UDP-D-GalNAc reactions (Fig. 6, trace E).
FIGURE 6. CE analysis of WbpP-catalyzed reactions.
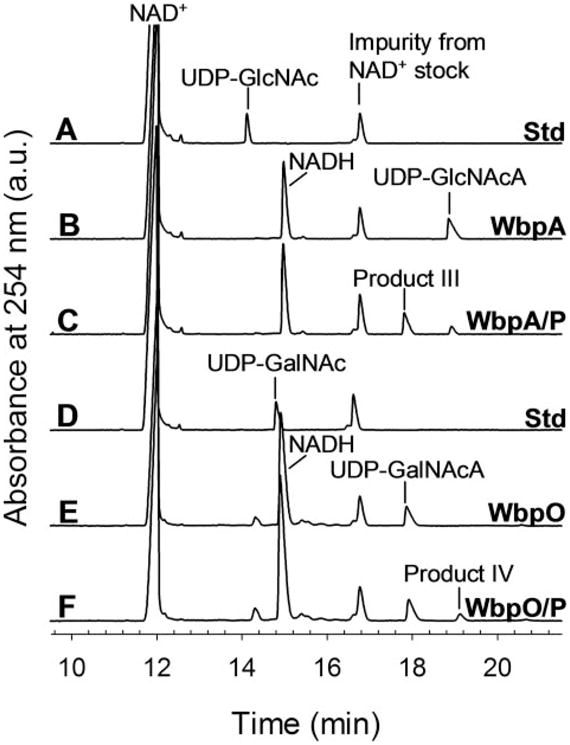
Trace A, NAD+ and UDP-D-GlcNAc standards; trace B, UDP-D-GlcNAc converted to UDP-D-Glc-NAcA by WbpAO5; trace C, UDP-D-GlcNAc completely converted to UDP-DGlcNAcA by WbpAO5, followed by the addition of WbpP; trace D, NAD+ and UDP-D-GalNAc standards; trace E, UDP-D-GalNAc converted to UDP-D-Gal-NAcA by WbpO; trace F, UDP-D-GalNAc completely converted to UDP-D-Gal-NAcA by WbpO, followed by the addition of WbpP.
UDP-D-GalNAcA is also not commercially available, and because WbpAO5 is not capable of using UDP-D-GalNAc, WbpO was used to synthesize UDP-D-GalNAcA for WbpP reactions. WbpO was used to completely convert UDP-D-Gal-NAc (and NAD+) into UDP-D-GalNAcA (and NADH; Fig. 6, trace E) prior to the addition of WbpP. CE analysis of WbpO/WbpP sequential reactions revealed that WbpP is capable of converting UDP-D-GalNAcA (eluted at 18 min) into a product that eluted at 19 min (labeled as product IV in Fig. 6, trace F). This product was identified as UDP-D-GlcNAcA by comparison with WbpAO5/UDP-D-GlcNAc reactions (Fig. 6, trace B).
Identification of the WbpO Reaction Products I and II by NMR Spectroscopy
By comparing proton chemical shifts, carbon chemical shifts, coupling constants (supplemental Table S3 and supplemental Fig. S3), and correlation patterns obtained using COSY, TOCSY, and NOESY experiments (not shown) to those reported in the literature (26), compound I was concluded to be UDP-α-D-GlcNAcA. The proton spectrum for compound II revealed that signals for sugar ring protons overlapped with those originating from the UDP moiety (supplemental Fig. S4A). A selective one-dimensional TOCSY of compound II H1 permitted the assignment of H2, H3, H4, and H5 and also the measurement of coupling constants (supplemental Fig. S4B). Large J2,3 (11.1 Hz) and small J3,4 (3.1) and J4,5 (1.1 Hz) couplings measured for compound II indicated a galacto ring configuration (supplemental Table S3). A strong H3/H5 NOE observed within the two-dimensional NOESY spectrum and no H2/H4 NOE confirmed the galacto ring configuration (not shown). Using a 13C heteronuclear single quantum correlation spectroscopy experiment, the chemical shift of C2 was determined to be δC 50.2 ppm, which indicated the presence of an N-acetyl group (supplemental Fig. S4C), and a heteronuclear multiple bond correlation spectroscopy experiment showed the chemical shift of C6 to be δC 175.4 ppm, which indicated a carboxyl group. Based on these results, compound II was concluded to be UDP-α-D-GalNAcA.
Determination of Physical Parameters and Cofactor Requirements of WbpO
WbpO is active over a pH range of 7–10 and has a pH optimum of 8.5 (data not shown). WbpO is active over a temperature range of near 0–65 °C, with optimal activity occurring between 37 and 42 °C (data not shown). WbpO requires either potassium- or ammonium-containing salts for activity (supplemental Table S2). The initial rate values are highest when the accompanying anion is kosmotropic and lowest when the accompanying anion is chaotropic, with a strong correlation between initial rate and placement within the Hofmeister series (36). Note that , and Na+-containing salts were not tested because no activity was detected when these experiments were previously conducted on WbpAO5 (26), and F− salts were not available at the time of testing.
In the assay where the activity of WbpO was examined in a range of ammonium sulfate concentrations, the initial rate of WbpO increased with increasing salt concentration throughout the range tested (5–300mM; Fig. 7). Overall, substrate conversion also increased with an increase in ammonium sulfate up to a concentration of 50 mM, above which overall conversion decreased with increasing salt concentration.
FIGURE 7. Effect of ammonium sulfate on the initial rate and overall substrate conversion of WbpO reactions.
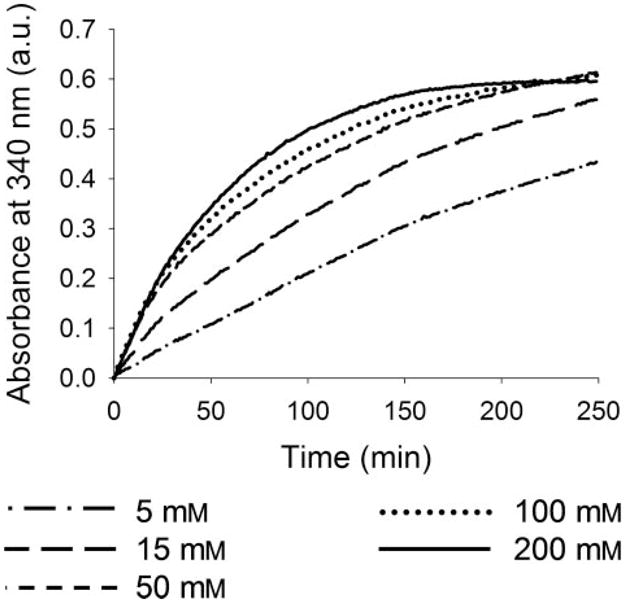
All reactions contained 1mM UDP-DGlcNAc, 2.5 mM NAD+, and 4 μg of WbpO in a final volume of 170 μl, with varying concentrations of (NH4)2SO4 as labeled.
Determination of Kinetic Parameters of WbpO
The kinetics of WbpO catalytic activity was examined by comparing the fit of kinetic data to the Michaelis-Menten and Hill models using the enzyme kinetics module of Sigma Plot from SPSS Science (Richmond, CA). The levels of fit in the Michaelis-Menten and Hill models were compared and evaluated based on the square of residuals (R2), the Akaike Information Criterion, and the standard deviation of residuals (Sy.x; supplemental Table S4). No significant difference between the two models could be observed with respect to the three criteria; therefore, the Michaelis-Menten model was subsequently chosen for further kinetic analysis because it has fewer parameters than the Hill model (Table 1). His-WbpO has a lower Km value for UDP-DGlcNAc (47μM) than for UDP-D-GalNAc (270μM). A comparison of the Km values for NAD+ (230μM) and NADP+ (3.7mM) with UDP-D-GlcNAc as the substrate indicated a preference for NAD+ as the coenzyme. His-WbpO has a higher kcat/Km ratio for UDP-D-GlcNAc (2,200 mM−1 × min−1) than for UDP-DGalNAc (2.6mM−1 × min−1), indicating a preference for UDPD-GlcNAc as the substrate.
TABLE 1. Kinetic parameters of His6-WbpO determined spectrophotometrically.
Parameters were determined by nonlinear regression, and each experiment was performed in triplicate.
| Sugar substrate | Parameters | Km | Vmax | Enzymea | kcat | kcat/Km |
|---|---|---|---|---|---|---|
| μM | pmol × min−1 | pmol | min−1 | mM−1 × min−1 | ||
| UDP-D-GlcNAc | UDP-D-GlcNAc | 47 ± 4 | 3700 ± 70 | 35 | 110 | 2200 |
| UDP-D-GlcNAc | NAD+ | 230 ± 21 | 3800 ± 97 | 35 | 110 | 470 |
| UDP-D-GlcNAc | NADP+ | 3700 ± 560 | 100 ± 6 | 140 | 0.71 | 0.19 |
| UDP-D-GalNAc | UDP-D-GalNAc | 270 ± 40 | 97 ± 4 | 140 | 0.69 | 2.6 |
The total reaction volume was 170 μl.
Equilibrium Analysis from Cell Extract Assay
When the three-dimensional structure of WbpP was determined by our group, a conceptual model was proposed for the saccharide-binding pockets of three groups of UDP-hexose 4-epimerases (37). Group 1 epimerases preferentially catalyze the conversion between UDP-Glc and UDP-Gal; group 2 epimerases do not show a preference for either UDP-Glc/UDP-Gal or UDP-Glc-NAc/UDP-GalNAc, and group 3 enzymes preferentially convert between UDP-GlcNAc and UDP-GalNAc. Because UDPD-GlcNAcA and UDP-D-GalNAcA were not available at the time of that study, cell extracts from one representative of each group were analyzed for the ability of these epimerases to utilize UDP-D-GlcNAcA and UDP-D-GalNAcA as substrates. Cell extracts from GalE, Gne, WbgU, and WbpP overexpression strains were combined with NAD+ and UDP-D-GlcNAc or UDP-D-GalNAc, purchased from Sigma, with UDP-D-Glc-NAcA purified from UDP-D-GlcNAc/WbpAO5 reactions or with UDP-D-GalNAcA purified from UDP-D-GalNAc/WbpO reactions. Each reaction was incubated until equilibrium was reached, and the relative abundance of substrate/product at equilibrium was determined by CE analysis in comparison to cell extracts from control strains (without the expression vector). Although both WbpP and WbgU were able to efficiently use either UDP-D-GlcNAcA or UDP-D-GalNAcA as substrates, the ratio of the substrate to product at equilibrium (~25–30% UDP-D-GalNAcA/~70–80% UDP-D-GlcNAcA) differed from those obtained previously for the non-uronic acid substrates, UDP-D-GlcNAc/GalNAc (~30% UDP-D-GlcNAc/~70% UDPD-GalNAc; Table 2). Gne was able to catalyze the epimerization of UDP-D-GlcNAcA to the UDP-D-GalNAcA product at low levels (14%), but no activity was detected when UDP-DGalNAcA was used as a substrate. In addition, GalE did not show any activity when either of the two UDP-uronic acids were used as a substrate indicating a narrower range of substrate specificity for this protein.
TABLE 2. Equilibrium data from cell extract assays for three groups of UDP-hexose 4-epimerases.
Each equilibrium experiment was performed once, in 100 mM Tris-HCl, pH 8.0, with 1mM substrates and 2.0μl of cell extract in a 35-μl reaction. All reactions were incubated at 37 °C for 1 h except WbgU reactions, which were incubated for 2 h.
| Substrate | Substrate product | Substrate and product composition at equilibrium |
|||
|---|---|---|---|---|---|
| Group 1, GalE | Group 2, Gne | Group 3 |
|||
| WbgU | WbpP | ||||
| %a | |||||
| UDP-D-Glc | UDP-D-Glc | 77 | 83 | 100 | 100 |
| UDP-D-Gal | 23 | 17 | 0 | 0 | |
| UDP-D-Gal | UDP-D-Gal | 25 | 79 | 100 | 91 |
| UDP-D-Glc | 75 | 21 | 0 | 9 | |
| UDP-D-GlcNAc | UDP-D-GlcNAc | 98 | 74 | 74 | 72 |
| UDP-D-GalNAc | 2 | 26 | 26 | 28 | |
| UDP-D-GalNAc | UDP-D-GalNAc | 97 | 36 | 29 | 28 |
| UDP-D-GlcNAc | 3 | 64 | 72 | 73 | |
| UDP-D-GlcNAcA | UDP-D-GlcNAcA | 100 | 86 | 24 | 28 |
| UDP-D-GalNAcA | 0 | 14 | 76 | 72 | |
| UDP-D-GalNAcA | UDP-D-GalNAcA | 100 | 100 | 75 | 73 |
| UDP-D-GlcNAcA | 0 | 0 | 25 | 27 | |
Percent composition of substrate and product for each reaction is reported for each enzyme-containing cell extract as compared with a cell extract from a control strain.
Structural Modeling of UDP-D-GlcNAcA and UDP-D-Gal-NAcA Binding in WbpP
Modeling of UDP-D-GlcNAcA in the active site of WbpP suggested that it could be accommodated in the saccharide-binding pocket of WbpP without causing steric hindrance and could potentially form six hydrogen bonds with the active site residues of WbpP (Fig. 8A). However, in the case of UDP-D-GalNAcA, there was the potential for only five hydrogen bonds between the saccharide moiety and the active site residues (Fig. 8B). In addition, there is the potential for steric hindrance because the carboxyl oxygen of UDP-D-GalNAcA and Tyr-166 of the active site are within 2.32 Å as modeled. Any rotation about the C-5′–C-6′ bond of the saccharide moiety would result in reduced hydrogen bonding potential.
FIGURE 8. UDP-D-GlcNAcA and UDP-D-GalNAcA in the saccharide-binding pocket of WbpP.
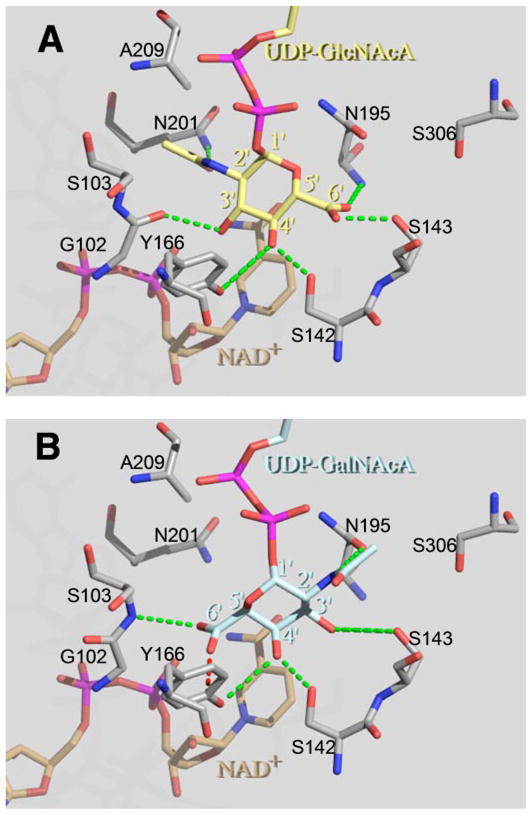
UDP-D-GlcNAcA (yellow; A) and UDP-D-GalNAcA (blue; B) were individually modeled into the active site of WbpP. The saccharide moieties of substrates were positioned in the catalytically productive orientation with respect to the cofactor NAD+ (brown) and the catalytic base Tyr-166. Although the saccharide moieties of UDP-D-GlcNAcA and UDP-D-GalNAcA are in different orientations, both substrates interact with a virtually identical set of active site residues (gray; G102/S103, S142, S143, Y166, and N195) to form five hydrogen bonds (green; 3.5 Å or less). In addition, the 2′-N-acetyl group of UDP-D-GlcNAcA forms an extra hydrogen bond with Gln-201. Note that a peptide bond between Gly-102 and Ser-103 is expected to flip in the presence of UDP-D-GlcNAcA to allow the carbonyl oxygen of Gly-102 to interact with 3′-OH of GlcNAcA (41). This figure was prepared using The PyMol Molecular Graphics System (DeLano Scientific, San Carlos, CA).
DISCUSSION
P. aeruginosa PAK produces a single polar flagellum containing a-type flagellin, which has been shown to be post-translationally modified at two sites with O-linked heterogeneous glycans (14). We investigated the potential role that O-antigen biosynthetic genes might play in the synthesis of these complex glycans by comparing flagellin proteins isolated from wild-type PAK and three mutant strains defective in O-antigen biosynthesis. Flagellins from wbpL and wbpP mutants have a similar size as the wild-type protein (45 kDa), and both were glycosylated. In contrast, flagellin from the wbpO mutant has an apparent molecular mass of 42 kDa, indicating that there is a defect in flagellin glycosylation. Complementation of the wbpO mutant with the plasmid pFV616-26a containing wbpO resulted in flagellin with increased levels of glycosylation. Based on IEF analysis, WbpO-flagellin has a less acidic pI than that of either PAK or the other mutant strains. The mass of the glycosylated wild-type flagellin in our study determined by MALDITOF MS was found to be 42,577 (±10) m/z, and this mass is consistent with that reported by Schirm et al. (14), whose results suggested that PAK flagellin was modified at two sites with a broad distribution of glycoforms, including one corresponding to 42,573 Da. Our data showed that the wbpO mutant flagellin has a mass of 40,197 (±10) Da, which would correspond to the mass of flagellin with two deoxyhexose substituents each with a mass of 146 Da.
Schirm et al. (14) have proposed a structural model for the complex, heterogeneous oligosaccharide glycan chains that are covalently linked to PAK flagellin. This model indicates that there is a central variable oligosaccharide region of two to seven sugars attached to the flagellin by a Rha residue. Although the monosaccharide constituents of the flagellin glycans were determined, the precise chemical structures of glycans have not been elucidated. Our results revealed that the nucleotide-activated sugar product of the WbpO reaction, UDP-D-GlcNAcA, is required for the synthesis of the variable oligosaccharide chain region of the flagellin glycans. The observation that flagellin from the wbpO mutant had a size consistent with the covalent attachment of two deoxyhexose sugars suggests that the sugar provided by the WbpO reaction is located in this central variable chain region. In the flagellin glycan model by Schirm et al. (14), the variable region is proposed to contain monosaccharides of various sizes, including hexuronic acids. Whether D-GlcNAcA would be incorporated into the flagellin glycans or requires modification by other enzymes encoded by genes within the flagellin glycosylation island before being incorporated into the glycans is not known at present. Furthermore, because the exact composition of the flagellin glycans is not known, it is not possible at present to propose a pathway for its biosynthesis.
The observation that mutations in wbpL and wbpP did not affect flagellin glycosylation indicates that a complete O-antigen unit is not required before flagellin glycosylation occurs. This is the same conclusion that had been reached by Schirm et al. (14) who could not detect the presence of complete O-antigen units in the flagellin glycan. This is in contrast to pilin glycosylation in P. aeruginosa strain 1244, where an O-antigen unit is O-linked to pilin (6). Therefore, it appears that P. aeruginosa utilizes distinct mechanisms to glycosylate flagellin and pilin, respectively. It is of interest to note that changes in the bacterial surface coat observed in clinical isolates obtained from cystic fibrosis patients with chronic lung infection showed a loss of B-band O polysaccharide (38, 39), the adaptation to a biofilm mode of growth, and a stable nonmotile phenotype where the bacterium is no longer producing pili or flagella (40).
In previous reports, WbpP was shown to possess UDP-DGlcNAc 4-epimerase activity (19), and WbpO was shown to be a UDP-D-GalNAc 6-dehydrogenase that also possessed low levels of UDP-D-GlcNAc 6-dehydrogenase activity (20). Based on this biochemical evidence, WbpP was proposed to act first in the conversion of UDP-D-GlcNAc into the nucleotide-activated uronic acid product. Our surprising result that flagellin glycosylation requires WbpO, but not WbpP, prompted us to re-examine the order of activity of WbpO and WbpP in producing UDP-D-GalNAcA. Furthermore, WbpO was previously expressed with a C-terminal histidine tag and had to be refolded for activity (WbpORf). Based on our recent success with expressing WbpAO5 (a WbpO homologue from serotype O5) as an N-terminal histidine fusion that is active without the need for refolding, WbpO was expressed with an N-terminal histidine tag (His-WbpO). Purified His-WbpO was active without the need for refolding and facilitated purification of sufficient amounts of purified WbpO for kinetic assays. His-WbpO possesses 6-dehydrogenase activity using either UDP-D-GlcNAc or UDP-D-GalNAc as a substrate.
To test whether WbpP could use UDP-D-GlcNAcA or UDPD-GalNAcA as a substrate, WbpP was added after the completion of UDP-D-GlcNAc/WbpAO5 or UDP-D-GalNAc/WbpO substrate-enzyme reactions. The identity of the UDP-D-Glc-NAc/WbpAO5 reaction product was previously identified by NMR as UDP-D-GlcNAcA (26), and the product of UDP-DGlcNAc/WbpO and UDP-D-GalNAc/WbpO reactions were identified in this study as UDP-D-GlcNAcA and UDP-D-Gal-NAcA, respectively, also by NMR. In this study the results showed that WbpP could interconvert UDP-D-GlcNAcA and UDP-D-GalNAcA through a C4 epimerization reaction, in addition to its previously reported activity as a UDP-D-GlcNAc/UDP-D-GalNAc 4-epimerase (19). Together with the results of His-WbpO assays, these results showed that in the overall conversion of UDP-D-GlcNAc into UDP-D-GalNAcA, WbpO, and WbpP are capable of acting in either order (Fig. 9).
FIGURE 9. Two alternative pathways for the conversion of UDP-D-GlcNAc into UDP-D-GalNAcA by WbpO and WbpP.
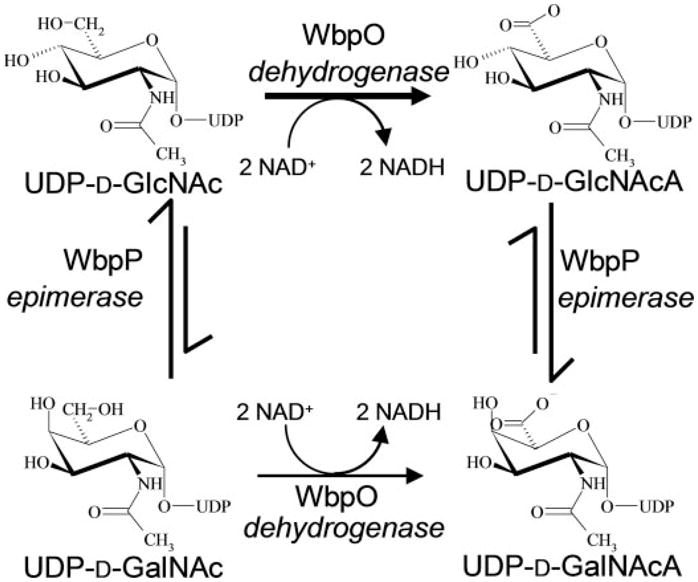
Whereas WbpO and WbpP can act in either order, kinetic analysis of WbpO and equilibrium analysis of WbpP suggest a preference for conversion of UDP-D-GlcNAc to UDP-D-GlcNAcA by WbpO before the conversion by WbpP into UDP-D-GalNAcA.
Because His-WbpO did not require refolding and could readily be purified in higher yield as an active enzyme, this has allowed us to carry out a thorough kinetic and physicochemical analysis of His-WbpO. This analysis revealed that there are a number of similarities between the activity of His-WbpO (serotype O6) and that of WbpAO5 (serotype O5). Both enzymes can catalyze a UDP-D-GlcNAc 6-dehydrogenation reaction, both have similar pH (8–9) and temperature optima (37–42 °C), and both require ammonium- or potassium-containing salt for activity. Because no significant activity was observed in reactions containing (CH3)4N+, , or Na+ (26), this suggested that the effects observed with or K+ do not result from stabilization of WbpAO5 or WbpO because of the kosmotropic nature of these cations, but rather that and K+ are activators of WbpAO5 and WbpO.
It was also observed that the anions of the or K+-containing salts appear to play a significant role in stabilizing the enzyme activity of WbpAO5 and WbpO. The effects of these anions correlate well with their placement within the Hofmeister series (36), whereby the initial reaction rate is increased by anions of increasing kosmotropicity and decreased by anions of increasing chaotropicity, as observed for WbpAO5 (26) and WbpO (supplemental Table S2).
Examinations of the effect of ammonium sulfate on the activity of WbpO revealed both a concentration-dependent activation and a salting out effect at high concentrations (Fig. 7). The activation effect was observed throughout the concentration range, because an increase in the concentration of ammonium sulfate correlated with an increase in initial rate. At high concentrations of ammonium sulfate, however, the overall conversion of substrate to product decreased with an increase in ammonium sulfate concentration, despite an increased initial rate. This effect is consistent with the salting out phenomenon characteristically observed with kosmotropic salts at high concentrations. The salting out phenomenon may also explain why reactions containing F− have lower initial rates than the equivalent reactions with as the anion, despite their ranking in the Hofmeister series.
A comparison of the kinetics of WbpO and of WbpAO5 with UDP-D-GlcNAc as a substrate revealed that WbpO has a lower K0.5 (47 μM) and higher Vmax (3.7 nmol/min−1) than WbpAO5 (K0.5= 94 μM, Vmax = 2.4 nmol/min−1), indicating that WbpO is more efficient thanWbpAO5. In addition, WbpO is capable of utilizing UDP-D-GalNAc as a substrate whereasWbpAO5 could not. The most intriguing result from the kinetic analysis of WbpO relates to its substrate specificity. WbpO has a higher kcat/K0.5 ratio for UDP-D-GlcNAc (2,200 mM−1 × min−1) than for UDP-D-GalNAc (2.6 mM−1 × min−1) and thus prefers UDP-D-GlcNAc over UDP-D-GalNAc. These results were opposite to what has been reported previously for WbpORf (20). This discrepancy is likely due to the requirement to refold the C-terminally His-tagged WbpO enzyme during purification in the previous study. Taken together these results showed that His-WbpO is more efficient than either WbpAO5 or WbpORf.
With a Hill coefficient for UDP-D-GlcNAc of 1.8, WbpAO5 displays positive cooperativity with respect to binding of UDPD-GlcNAc. In comparison, kinetic data for WbpO did not fit significantly better to the Hill model than to the Michaelis-Menten model and thus WbpO did not display positive cooperativity. This difference may be due to the fact that although WbpAO5 catalyzes the first step in the pathway for the biosynthesis of UDP-D-Man(2NAc3NAc)A, WbpO may catalyze either the first step or the second step of the pathway for UDPD-GalNAcA biosynthesis.
Results from the previous analysis of WbpP reactions showed that only 30% of UDP-D-GlcNAc was converted into UDP-D-GalNAc, whereas the yield of the reverse reaction was 70% (19). In this study, at equilibrium of the enzyme-substrate reaction using uronic acid substrates, WbpP catalyzed 72% conversion of UDP-D-GlcNAcA into UDP-D-GalNAcA, whereas the yield of the reverse reaction was 27% (Table 2). Moreover, comparison of the kcat values for UDP-D-GlcNAc (120 min−1) and UDP-D-GalNAc (271 min−1) shows that the conversion of UDP-D-GalNAc to UDP-D-GlcNAc occurred at a faster rate than the reverse reaction. Finally, a comparison of the Km value of WbpP for UDP-D-GlcNAc (224 μM) with the Km value of WbpO for the same substrate (47 μM) reveals that WbpO is capable of catalysis at lower concentrations of UDPD-GlcNAc than WbpP. Altogether, these novel observations suggested that although WbpO and WbpP can act in either order, there is a strong preference for WbpO to act first upon UDP-D-GlcNAc to produce UDP-D-GlcNAcA, before this uronic acid compound is epimerized by WbpP to UDP-D-Gal-NAcA (Fig. 9).
There is a dramatic difference at equilibrium between the pair UDP-D-GlcNAc/UDP-D-GalNAc, which favors catalysis of UDP-D-GalNAc (and production of UDP-D-GlcNAc), and their uronic acid derivatives. With the latter pair of substrates, the catalysis of UDP-D-GlcNAc to the product UDP-D-GalNAc was favored. Because our group has solved the structure and catalytic mechanism of WbpP (37), and given that there have been no reports of C4 epimerase family proteins using nucleotide-N-acetylhexuronic acid as a substrate, structural models were created to examine how WbpP might accommodate UDP-D-Glc-NAcA and UDP-D-GalNAcA into the active site. The results of this modeling showed that WbpP can accommodate these uronic acid substrates in a similar manner to their non-uronic acid derivatives (Fig. 8). The results also showed that UDP-DGlcNAc could form an extra hydrogen bond in the saccharide-binding pocket of WbpP compared with UDP-D-GalNAc. In addition, there is potential for steric hindrance between the 6′ carbonyl oxygen of UDP-D-GalNAc and C4 of the aromatic ring of Tyr-166. Because the saccharide moiety of the substrate is inherently mobile in the active site of WbpP (37), an additional hydrogen bond could help to stabilize and better align the saccharide moiety of UDP-D-GlcNAc for catalysis. This may be sufficient to promote a higher conversion rate of UDP-D-Glc-NAcA into UDP-D-GalNAcA at equilibrium than the reverse reaction.
When the three-dimensional structure of WbpP was determined, a conceptual model was proposed for the saccharide-binding pockets of three groups of UDP-hexose 4-epimerases (37). Group 1 epimerases preferentially catalyze the conversion between UDP-D-Glc and UDP-D-Gal; group 2 epimerases do not show a preference for either UDP-D-Glc/UDP-D-Gal or UDP-D-GlcNAc/UDP-D-GalNAc, and group 3 epimerases preferentially convert between UDP-D-GlcNAc and UDP-DGalNAc. In this study, cell extract assays were performed to determine which of the three groups of proteins are capable of catalyzing C4 epimerization using UDP-D-GlcNAcA and UDPD-GalNAcA as substrates. GalE, a group 1 epimerase from K. pneumoniae (33), was not capable of using either UDP-D-Glc-NAcA or UDP-D-GalNAcA, but this is not surprising because GalE could not utilize N-acetylated UDP-sugars as substrates (19, 37). Gne, a group 2 epimerase from Y. enterocolitica (34), was able to epimerize UDP-D-GlcNAcA at a rather low conversion rate, i.e. only 14% conversion to UDP-D-GalNAcA was observed at equilibrium. Also, Gne could not catalyze the reverse epimerization reaction. This is somewhat surprising because Gne is capable of catalyzing UDP-D-GlcNAc and UDPD-GalNAc as well as UDP-D-Glc and UDP-D-Gal. Both group 3 epimerases tested, WbpP (P. aeruginosa) and WbgU (Plesiomonas shigelloides), were able to utilize either UDP-D-GlcNAcA or UDP-D-GalNAcA as substrates, although, as discussed above for WbpP, the equilibrium shifts toward greater production of the galacto-epimer (and less production of the gluco-epimer) when the substrates are in the uronic acid form. Thus, the presence of the carboxylic acid group appears to have a significant effect upon the activity of the C4 epimerases resulting in reduced activity for group 2 epimerases and altered equilibria for group 3 epimerases.
In conclusion, we have shown that flagellin glycosylation in P. aeruginosa PAK depends upon the O-antigen biosynthetic gene wbpO but not wbpP or wbpL. We have also provided kinetic evidence to show that, whereas WbpO and WbpP can act in either order in the biosynthesis of UDP-D-GalNAcA, there is a strong preference for WbpO to convert UDP-D-Glc-NAc into UDP-D-GlcNAcA first before the conversion into UDP-D-GalNAcA by WbpP (Fig. 9). In addition, there are a number of similarities in the activities of WbpAO5 (from serotype O5) and WbpO (from serotype O6) as 6-dehydrogenases, but there is a difference in their substrate specificity in that WbpAO5 could not use UDP-D-GalNAc as a substrate. Given these differences and their potential for regulating key metabolic steps, the need for further analysis is clear, and studies are currently underway to determine the three-dimensional structures of WbpAO5 and WbpO.
Supplementary Material
Acknowledgments
We thank Cory Wenzel for assistance with the cloning and expression of His-WbpO. We are also grateful to Chris Whitfield, Mikael Skurnik, and Peng Wang for providing the GalE, Gne, and WbgU expression constructs, respectively, and to Reuben Ramphal for the kind gift of anti-FliC rabbit antiserum.
Footnotes
The abbreviations used are: LPS, lipopolysaccharide; CE, capillary electrophoresis; COSY, correlation spectroscopy; D-QuiNAc, N-acetyl-D-quinovosamine; IPTG, isopropyl β-D-thiogalactopyranoside; IEF, isoelectric focusing; MALDI-TOF, matrix-assisted laser desorption ionization-time of flight; MES, 2-morpholinoethanesulfonic acid; MS, mass spectrometry; NBT, nitro blue tetrazolium; NOESY, nuclear Overhauser effect spectroscopy; PBS, phosphate-buffered saline; TBS, Tris-buffered saline; TOCSY, total correlation spectroscopy; UDP-D-GalNAcA, UDP-N-acetyl-D-galacturonic acid; UDP-D-GalNAc-AN, UDP-2-acetamido-6-amino-2-deoxy-D-galactose; UDP-D-GlcNAc, UDP-N-acetyl-D-glucosamine; UDP-D-GlcNAcA, UDP-N-acetyl-D-glucuronic acid; UDP-D-QuiNAc, UDP-N-acetyl-D-quinovosamine; BisTris propane, 1,3-bis[tris(hydroxymethyl)methylamino]propane.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4, Figs. S1–S4, and additional references.
Supplemental Material can be found at: http://www.jbc.org/content/suppl/2007/12/26/M708894200.DC1.html
This work was supported in part by operating grants from the Canadian Cystic Fibrosis Foundation and Canadian Institute of Health Research Grants MOP-14687 (to J. S. L.) and MT-13107 (to A. M. B.).
References
- 1.Power PM, Roddam LF, Dieckelmann M, Srikhanta YN, Tan YC, Berrington AW, Jennings MP. Microbiology. 2000;146:967–979. doi: 10.1099/00221287-146-4-967. [DOI] [PubMed] [Google Scholar]
- 2.Power PM, Roddam LF, Rutter K, Fitzpatrick SZ, Srikhanta YN, Jennings MP. Mol Microbiol. 2003;49:833–847. doi: 10.1046/j.1365-2958.2003.03602.x. [DOI] [PubMed] [Google Scholar]
- 3.Stimson E, Virji M, Makepeace K, Dell A, Morris HR, Payne G, Saunders JR, Jennings MP, Barker S, Panico M, Blench I, Moxon ER. Mol Microbiol. 1995;17:1201–1214. doi: 10.1111/j.1365-2958.1995.mmi_17061201.x. [DOI] [PubMed] [Google Scholar]
- 4.Rahim R, Ochsner UA, Olvera C, Graninger M, Messner P, Lam JS, Soberon-Chavez G. Mol Microbiol. 2001;40:708–718. doi: 10.1046/j.1365-2958.2001.02420.x. [DOI] [PubMed] [Google Scholar]
- 5.Rahim R, Burrows LL, Monteiro MA, Perry MB, Lam JS. Microbiology. 2000;146:2803–2814. doi: 10.1099/00221287-146-11-2803. [DOI] [PubMed] [Google Scholar]
- 6.DiGiandomenico A, Matewish MJ, Bisaillon A, Stehle JR, Lam JS, Castric P. Mol Microbiol. 2002;46:519–530. doi: 10.1046/j.1365-2958.2002.03171.x. [DOI] [PubMed] [Google Scholar]
- 7.Tang HB, DiMango E, Bryan R, Gambello M, Iglewski BH, Goldberg JB, Prince A. Infect Immun. 1996;64:37–43. doi: 10.1128/iai.64.1.37-43.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, Tang H, Prince A. Infect Immun. 1998;66:43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spangenberg C, Heuer T, Burger C, Tummler B. FEBS Lett. 1996;396:213–217. doi: 10.1016/0014-5793(96)01099-x. [DOI] [PubMed] [Google Scholar]
- 10.Allison JS, Dawson M, Drake D, Montie TC. Infect Immun. 1985;49:770–774. doi: 10.1128/iai.49.3.770-774.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansorg R. Zentbl Bakteriol Orig A. 1978;242:228–238. [PubMed] [Google Scholar]
- 12.Lanyi B. Acta Microbiol Acad Sci Hung. 1970;17:35–48. [PubMed] [Google Scholar]
- 13.Brimer CD, Montie TC. J Bacteriol. 1998;180:3209–3217. doi: 10.1128/jb.180.12.3209-3217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schirm M, Arora SK, Verma A, Vinogradov E, Thibault P, Ramphal R, Logan SM. J Bacteriol. 2004;186:2523–2531. doi: 10.1128/JB.186.9.2523-2531.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Totten PA, Lory S. J Bacteriol. 1990;172:7188–7199. doi: 10.1128/jb.172.12.7188-7199.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arora SK, Bangera M, Lory S, Ramphal R. Proc Natl Acad Sci U S A. 2001;98:9342–9347. doi: 10.1073/pnas.161249198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bélanger M, Burrows LL, Lam JS. Microbiology. 1999;145:3505–3521. doi: 10.1099/00221287-145-12-3505. [DOI] [PubMed] [Google Scholar]
- 18.Knirel A, Kochetkov NK. Biokhimiia. 1994;59:1784–1851. [PubMed] [Google Scholar]
- 19.Creuzenet C, Belanger M, Wakarchuk WW, Lam JS. J Biol Chem. 2000;275:19060–19067. doi: 10.1074/jbc.M001171200. [DOI] [PubMed] [Google Scholar]
- 20.Zhao X, Creuzenet C, Belanger M, Egbosimba E, Li J, Lam JS. J Biol Chem. 2000;275:33252–33259. doi: 10.1074/jbc.M004191200. [DOI] [PubMed] [Google Scholar]
- 21.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hancock RE, Carey AM. J Bacteriol. 1979;140:902–910. doi: 10.1128/jb.140.3.902-910.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 25.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Miller WL, Wenzel CQ, Daniels C, Larocque S, Brisson JR, Lam JS. J Biol Chem. 2004;279:37551–37558. doi: 10.1074/jbc.M404749200. [DOI] [PubMed] [Google Scholar]
- 27.Frisk A, Jyot J, Arora SK, Ramphal R. J Bacteriol. 2002;184:1514–1521. doi: 10.1128/JB.184.6.1514-1521.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Shannessy DJ, Voorstad PJ, Quarles RH. Anal Biochem. 1987;163:204–209. doi: 10.1016/0003-2697(87)90114-x. [DOI] [PubMed] [Google Scholar]
- 29.Wenzel CQ, Daniels C, Keates RA, Brewer D, Lam JS. Mol Microbiol. 2005;57:1288–1303. doi: 10.1111/j.1365-2958.2004.04767.x. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 31.Brisson JR, Sue SC, Wu WG, McManus G, Nghia PT, Uhrin D. In: NMR Spectroscopy of Glycoconjugates. Jimenez-Barbero J, Peters T, editors. Wiley-VCH; Weinhem, Germany: 2002. pp. 59–93. [Google Scholar]
- 32.Uhrin D, Brisson JR. In: NMR in Microbiology: Theory and Applications. Barbotin JN, Portais JC, editors. Horizon Scientific Press; Wymondham, UK: 2000. pp. 165–190. [Google Scholar]
- 33.Frirdich E, Whitfield C. J Bacteriol. 2005;187:4104–4115. doi: 10.1128/JB.187.12.4104-4115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bengoechea JA, Pinta E, Salminen T, Oertelt C, Holst O, Radziejewska-Lebrecht J, Piotrowska-Seget Z, Venho R, Skurnik M. J Bacteriol. 2002;184:4277–4287. doi: 10.1128/JB.184.15.4277-4287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr Sect D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Collins KD, Washabaugh MW. Q Rev Biophys. 1985;18:323–422. doi: 10.1017/s0033583500005369. [DOI] [PubMed] [Google Scholar]
- 37.Ishiyama N, Creuzenet C, Lam JS, Berghuis AM. J Biol Chem. 2004;279:22635–22642. doi: 10.1074/jbc.M401642200. [DOI] [PubMed] [Google Scholar]
- 38.Lam MY, McGroarty EJ, Kropinski AM, MacDonald LA, Pedersen SS, Hoiby N, Lam JS. J Clin Microbiol. 1989;27:962–967. doi: 10.1128/jcm.27.5.962-967.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hancock RE, Mutharia LM, Chan L, Darveau RP, Speert DP, Pier GB. Infect Immun. 1983;42:170–177. doi: 10.1128/iai.42.1.170-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahenthiralingam E, Campbell ME, Speert DP. Infect Immun. 1994;62:596–605. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demendi M, Ishiyama N, Lam JS, Berghuis AM, Creuzenet C. Biochem J. 2005;389:173–180. doi: 10.1042/BJ20050263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.