

Abstract
In humans, two major β-hexosaminidase isoenzymes exist: Hex A and Hex B. Hex A is a heterodimer of subunits α and β (60% identity), whereas Hex B is a homodimer of β-subunits. Interest in human β-hexosaminidase stems from its association with Tay–Sachs and Sandhoff disease; these are prototypical lysosomal storage disorders resulting from the abnormal accumulation of GM2-ganglioside (GM2). Hex A degrades GM2 by removing a terminal N-acetyl-D-galactosamine (β-GalNAc) residue, and this activity requires the GM2–activator, a protein which solubilizes the ganglioside for presentation to Hex A. We present here the crystal structure of human Hex B, alone (2.4 Å) and in complex with the mechanistic inhibitors GalNAc-isofagomine (2.2 Å) or NAG-thiazoline (2.5 Å). From these, and the known X-ray structure of the GM2–activator, we have modeled Hex A in complex with the activator and ganglioside. Together, our crystallographic and modeling data demonstrate how α and β-subunits dimerize to form either Hex A or Hex B, how these isoenzymes hydrolyze diverse substrates, and how many documented point mutations cause Sandhoff disease (β-subunit mutations) and Tay–Sachs disease (α-subunit mutations).
Keywords: hexosaminidase, Sandhoff, Tay–Sachs, anchimeric assistance, X-ray crystal structure
Introduction
The existence of an N-acetyl-β-D-glucosaminidase (NAGase) was first reported in 1936.1 It was later found that mammals express lysosomal NAGases capable of efficiently catalyzing the removal of β (1 → 4) linked N-acetyl-D-glucosamine (GlcNAc) and N-acetyl-D-galactosamine (GalNAc) from the non-reducing end of oligosaccharides and glycoconjugates. Thus, the mammalian enzyme is classified as an N-acetyl-β-D-hexosaminidase (Hex).
The early interest in human Hex was derived from its association with Tay–Sachs disease, first described in 1881.2 Since that time an immense amount of biomedical research has been carried out with the goal of understanding the disease mechanism(s) through characterizing the structure and function of human Hex isozymes. This large body of research has made Hex the primary model for lysosomal glycosidases. The “classical” infantile or acute form of Tay–Sachs disease (TSD) is a devastating neurological disorder that has been found in a diverse range of ethnic groups throughout the world. The highest incidence of the disease occurs in the Ashkenazi Jewish population, with a carrier rate of approximately 1 in 35, while the carrier rate in the general population is about 1 in 300.3
The disease is caused from the heritable deficiency of one of two major human Hex isoenzymes; Hex A, an isoenzyme that removes the terminal β-linked GalNAc residue from GM2 ganglioside (GM2). Its absence results in the accumulation of GM2 primarily in the lysosomes of neuronal cells (where gangliosides are principally synthesized), and is believed to eventually cause apoptosis by an as yet undefined mechanism (reviews3,4).
Both biochemical and genetic data have confirmed that in order to hydrolyze β-GalNAc from GM2, three different gene products must first undergo proper synthesis, intracellular transport, post-translational processing, and then associate into a GM2-containing quaternary complex. Two of the genes, HEXA and HEXB, are evolutionarily related and encode the α and β-subunits of Hex A, whose primary structures are 60% identical. The third gene, GM2A, encodes a small glycolipid transport protein, the GM2 activator protein (activator) that serves as a substrate (GM2)-specific cofactor for Hex A. The activator extracts GM2 from the intralysosomal membrane as a 1:1 soluble complex and then presents the terminal GalNAc residue of the ganglioside to Hex A for removal. The structural elements of each of the three proteins that interact to form the catalytic complex, and the means by which the activator binds and presents GM2 to Hex A for hydrolysis have long been objects of scientific interest (reviews4,5). Data presented here, coupled with the previously reported crystal structure of the activator,6 have allowed us to obtain new insight into the formation of the active quaternary complex.
Mutations in any of the three genes described above can cause GM2 gangliosidosis, a group of clinically similar devastating neurological diseases. These are: Tay–Sachs (HEXA, α-subunit mutations), Sandhoff (HEXB, β-subunit mutations), and the rare AB variant form (GM2A, activator mutations). To date, there are 100 mutations in HEXA, 25 mutations in HEXB and five mutations in GM2A that have been reported to cause GM2 gangliosidosis, and much biomedical research has been done to try and explain the observed phenotype based on the biochemistry of these known mutations†. The most common and severe acute forms of GM2 gangliosidosis result from mutations that prevent any one of the three gene products from being made and/or reaching the lysosome. However, mutations that allow as little as 1–5% of wild-type Hex A activity, produce a milder subacute or much milder chronic disease phenotype. Interestingly, several missense mutations have been found in the HEXA and HEXB genes that allow the production of <10% residual Hex A activity in individuals who are asymptomatic for the disease.7 These findings set a “critical threshold” of Hex A activity; only 5–10% of normal Hex A activity is necessary to prevent the accumulation of GM2 in the lysosome.8
Although Hex A (αβ) is the only Hex isozyme necessary for life, two other isozymes exist. In normal human tissue Hex B (ββ) is present at comparable amounts to Hex A, and in Sandhoff disease tissue lacking β-protein, a small amount of Hex S (αα) can be detected.9 All three isozymes can hydrolyze the β-linked GalNAc or GlcNAc residues from artificial and some natural substrates, but only Hex A and Hex S can hydrolyze substrates containing a negatively charged, terminal non-reducing β-linked ,10,11 and only Hex A can hydrolyze GM2 (also negatively charged because of its sialic acid moiety) in the presence of the activator. Interestingly, GM2 can also be hydrolyzed by Hex S (αα), but not Hex B (ββ), when detergent is substituted for the activator. Furthermore, it has recently been demonstrated that mouse Hex A and Hex S, but not Hex B, are involved in the degradation of sulfatide SM2a.12
As no reports of active α or β-monomers exist, the above observations produce the following three conclusions. (1) The α and β-subunits each contain an active site; however, dimer formation is somehow necessary to confer functionality.13 (2) Although the two subunits have similar active sites, only the α-subunit active site can accommodate negatively charged substrates such as containing keratan sulfate and sialic acid-containing GM2.14,15 (3) Despite the necessity of the α-subunit active site for GM2 hydrolysis, the presence of the β-subunit is necessary for the binding of the GM2–activator complex to Hex A.16
The human Hex isoenzymes belong to the sequence-related family 20 glycoside hydrolases (glycosidases).17,18 Like other family 20 members, stereochemical outcome studies demonstrate that Hex B is a configuration-retaining β-glycosidase.19 Only two other members of this family have had their three-dimensional structures determined; a chitobiase from Serratia marcescens (SmCHB)20 and a β-hexosaminidase from Streptomyces plicatus (Sp HEX).21 These bacterial enzymes do not dimerize but instead exist as active monomers. Comparative molecular modeling based on the SmCHB structure was used to model the candidate active site regions in human Hex;20 however, the percentage identity of SmCHB with either subunit of human Hex is <30% in the active site region and virtually non-existent elsewhere. Hence, the resulting subunit homology models do not address any of the above questions concerning the mechanism of GM2 hydrolysis or the protein–protein interactions involved in isoenzyme dimer formation.
Nonetheless, the structural studies on SmCHB20 and Sp HEX21–23 in conjunction with kinetic studies using inhibitors,24–26 provide strong evidence that the catalytic mechanism for all family 20 glycosidases involves the participation of the neighboring C2-acetamido group of the substrate. The vast majority of retaining β-glycosidases use an enzyme carboxyl group as a nucleophile in the catalytic mechanism; whereas, family 20 glycosidases do not contain such a residue and use instead the carbonyl oxygen atom of the 2-acetamido group on the substrate as the nucleophile. Thus, instead of forming a glycosyl-enzyme intermediate, family 20 glycosidases catalyze the formation of a cyclized oxazolinium ion intermediate by rotating the 2-acetamido group of the terminal sugar molecule to be removed into a position suitable for nucleophilic attack at its anomeric center. In a manner analogous to the mechanism for “normal” retaining β-glycosidases, the cyclized intermediate undergoes base catalyzed attack from an incoming water molecule to produce a product with a retained anomeric configuration. Our present structural studies confirm that human Hex also uses this mechanism (Figure 1).
Figure 1.

Proposed catalytic mechanism for family 20 β-hexosaminidase. The Hex B residues Glu355 (Hex A; Glu323) and Asp354 (Hex A; Asp322) are shown. Glu355 acts as a general acid–base residue, whereas Asp354 acts primarily to help in orienting the C2-acetamido group into a position for nucleophilic attack and subsequently to stabilize the positive charge on the oxazolinium ion intermediate. No attempt has been made to indicate the true positions of these residues. The hydroxyl groups and C6 have been removed from the pyranose ring of the substrate for clarity.
Like secretory, plasma membrane, and other lysosomal proteins, the α and β subunits of Hex and the activator monomer are synthesized on the rough endoplasmic reticulum (ER). Each nascent prepropolypeptide is directed there by an N-terminal signal peptide (Table 1). In the ER, the signal peptides are removed by signal peptidase, and the propolypeptides are glycosylated at selected Asn-X-Ser/Thr.27 For many enzymes destined for the lysosomal compartment, glycosylation is followed by the addition of one or two phosphate markers to one or more high mannose-type oligosaccharide28,29 (Table 1). This allows them to be separated from secretory proteins through interactions with one of two mannose-6-phosphate receptors in the trans Golgi.30 The initial recognition of lysosomal enzymes in the ER is believed to be based on a common three-dimensional signal previously characterized in Cathepsin D.31–33
Table 1.
Previously characterized structures of the mature, lysosomal, forms of the α and β-subunits of human Hex and the GM2 activator protein
| Protein | Initial signal peptidea | Polypeptidesb | Asn-linked glycosylationc | Disulfide bondsd |
|---|---|---|---|---|
| α-Subunit | M1 → A22 | αp: L23 → G73e; αm: T88 → T528e | N: 114, 156, 294f | C58–C104; C277–C328; C505-C522 |
| β-Subunit | M1 → A42 | βp: A50 → G107e; βb: T122 → S311; βa: L316 → M556e | N: 84f, 142, 190, 327f | C93–C137; C309–C360; C534–C551 |
| GM2 activator | M1 → A23 | Am: S32 → I193 | N: 63 | C39–C183; C99–C106; C112–C138; C125–C136 |
In the lysosome, a complex series of proteolytic and glycosidic processing events occur converting the single pro-α and β-polypeptides into two or three smaller polypeptides, respectively, held together by disulfide bonds in the mature subunits (Table 1). These processing events have been viewed as removing non-essential loop structures (needed only during initial protein folding) when exposed to the harsh lysosomal environment.34,35 Our modeling studies presented here suggest that at least one of these events may play a biological role in forming part of a structural binding epitope that is unique to the α-subunit and to which the activator associates with to form the quaternary complex.
Results and Discussion
Structure of human Hex B
Mature Hex B (Mr 112,000) was purified from human placenta and crystallized in its native mature (lysosomal) form36 (Table 1). Its crystal structure was determined using the multiple isomorphous replacement (MIR) method with data to 2.8 Å resolution. A model of the Hex B homodimer was readily built into the experimental electron density and subsequently refined with data to 2.4 Å resolution (Table 2).
Table 2.
Crystallographic statistics
| A. Crystal information | |||||
| Space group | P6122 | ||||
| Matthews coefficient | 3.24 (2 molecules/asymmetric unit) | ||||
| Solvent content (%) | ,62 | ||||
| B. Data collection (values in parentheses refer to the high-resolution shell) | |||||
| Data set | Native | Pt | Hg | NGT complex | IFG complex |
| Unit cell dimensions (Å) | a = b = 112.47, c = 397.87 | a = b = 111.52, c = 398.23 | a = b = 111.70, c = 396.80 | a = b = 112.40, c = 397.23 | a = b = 112.39, c = 397.30 |
| Wavelength (Å) | 1.00 | 1.00 | 1.00 | 0.900 | 0.900 |
| Resolution range (Å) | 35.0–2.40 | 35.0–3.00 | 35.0–2.90 | 37.0–2.50 | 35.0–2.20 |
| High-resolution (Å) | 2.44–2.40 | 3.05–3.00 | 2.95–2.90 | 2.57–2.50 | 2.26–2.20 |
| Total observations | 2,772,717 | 800,088 | 769,328 | 784,049 | 1,153,759 |
| Unique reflections | 58,844 (2894) | 33,068 (1460) | 30,367 (1644) | 52,315 (4235) | 74,245 (6161) |
| 〈I/σ〉 | 21.9 (4.89) | 22.9 (7.73) | 20.8 (6.31) | 23.2 (5.08) | 22.3 (4.93) |
| Completenessa (%) | 99.5 (99.7) | 99.5 (99.9) | 97.0 (98.6) | 99.7 (100.0) | 99.4 (100.0) |
| Rsymb | 0.090 (0.314) | 0.071 (0.272) | 0.081 (0.314) | 0.077 (0.367) | 0.094 (0.457) |
| Risoc | 0.207 (0.209) | 0.175 (0.199) | |||
| Heavy-atom sites | 2 | 2 | |||
| Phasing power | |||||
| Centric | 0.46 | 0.48 | |||
| Acentric | 0.55 | 0.52 | |||
| Overall figure of merit | |||||
| Solve | 0.29 (Overall Z-score = 14.75) | ||||
| Resolve | 0.57 (with 2-fold non-crystallographic symmetry restraints) | ||||
| C. Refinement | |||||
| Rworkd | 0.201 | 0.202 | 0.193 | ||
| Rfreed | 0.231 | 0.229 | 0.218 | ||
| Number of atoms | |||||
| Protein | 7762 | 7762 | 7762 | ||
| Heterogen | 79 | 107 | 107 | ||
| Water | 226 | 244 | 321 | ||
| Average B (Å2) | 25.1 | 24.2 | 20.9 | ||
| r.m.s.d. from ideal geometry | |||||
| Bond lengths (Å) | 0.011 | 0.009 | 0.011 | ||
| Bond angles (deg.) | 1.59 | 1.56 | 1.57 | ||
| Ramachandran plote | |||||
| Most favored (%) | 91.7 | 91.0 | 91.6 | ||
| Additionally allowed (%) | 8.3 | 9.0 | 8.4 | ||
Calculated by treating Bijvoet pairs as equivalent.
Rsym = Σh Σi (|Ii(h) − 〈I(h)〉|)/Σh Σi Ii(h), where Ii(h) is the ith intensity measurement and 〈I(h)〉 is the weighted mean of all measurements of I(h).
Riso = Σh ||F(h)deriv| − |F(h)native||/|F(h)native|
Rwork and Rfree = Σh ||F(h)obs | − |F(h)calc ||/|F(h)obs| for reflections in the working and test sets (5% of all data), respectively.
Regions defined by PROCHECK.79
Two β-subunits, related by non-crystallographic 2-fold symmetry, comprise the asymmetric unit of the crystals used to determine the Hex B structure. The subunits share a buried surface area of 1612 Å2, and when aligned structurally, their Cα atoms have an r.m.s. difference of 0.3 Å; however, the contacts made between these subunits are the result of crystal packing and do not represent the biological homodimer interface of the Hex B enzyme. Instead, two biologically relevant Hex B homodimers were found to lie side-by-side on a common crystallographic 2-fold axis (space group P6122), such that each biological homodimer contributes one β-subunit to the asymmetric unit. The β-subunits of one of the biological homodimers share a buried surface area of 2694 Å2, whereas the subunits of the other share a buried surface area of 2737 Å2. By choosing either of these crystallographically related subunit pairs as the biologically relevant Hex B homodimer, the structural basis for much of the observed biochemistry of this enzyme became apparent.
Subunit structure
Each β-subunit of Hex B is a kidney-shaped, two-domain protein (Figure 2). The three poly-peptides present in the lysosomal form of each mature β-subunit, βp, βb, βa (Table 1 and Figure 4), could be traced as independent chains through the electron density. However, residues 50–54 of βp and 553–556 of βa, which constitute the extreme N and C termini of the mature amino acid (aa) sequence, respectively, could not be modeled due to insufficient density. The post-translational proteolytic cleavages that produce these three chains from their single precursor remove two surface accessible loops (as predicted34,37) from each β-subunit, resulting in the three disulfide-linked polypeptides that comprise the mature subunit (Table 1 and Figure 4). The locations of the disulfide bonds are consistent with results obtained by MALDI-MS38 (Table 1). Although N-glycan electron density was observed on Asn residues 84, 142, 190 and 327, only two sugar molecules (di-GlcNAc) were built into density extending from position 190; the remaining sugar molecules extending from this position and all the other N-linked glycan groups were too unstructured to model.
Figure 2.
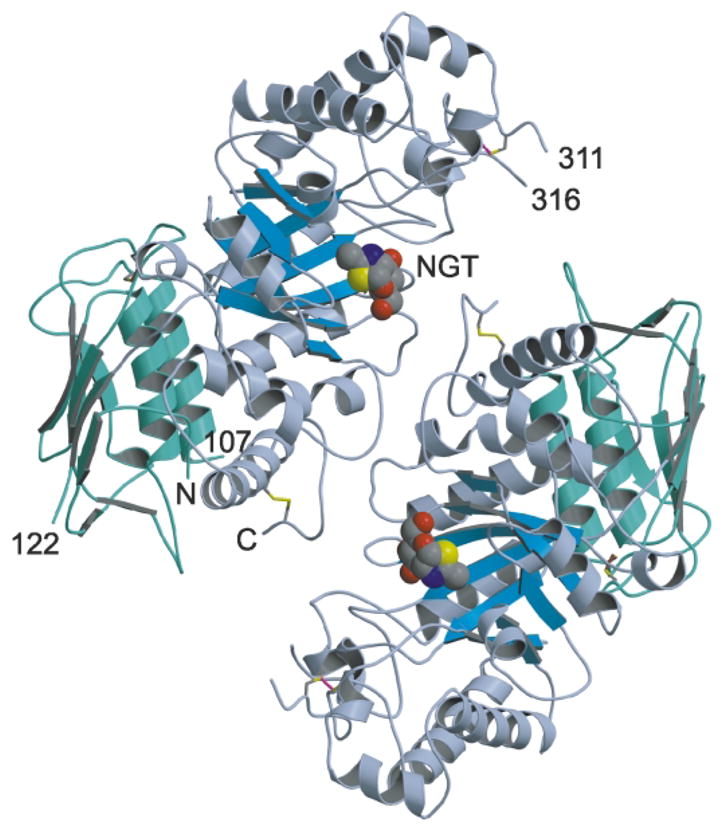
Ribbon diagram of human β-hexosaminidase B. The β-subunits of the Hex B homodimer are colored with domain I in green and domain II in blue (the eight parallel strands of the β-barrel of domain II is colored sky blue). What appear to be common structural features of family 20 glycosidases is the absence of regular α-helices at positions α5 and α7 of the (β/α)8-barrel structure of domain II and an additional C-terminal helix following helix α8. This additional helix packs between domains I and II, spatially orienting the two domains relative to each other. Helix α7 consists of only two turns and is part of an extended loop that forms a major portion of the dimer interface. The subunits are related at the dimer interface by a crystallographic 2-fold symmetry axis running perpendicular to the page. The N and C termini created as a result of post-translational processing are numbered by residue. The labels N and C denote the extreme N (residue 55) and C (residue 552) termini visible within the electron density. The disulfide bonds Cys91-Cys137, Cys309-Cys360 and Cys534-Cys551 are drawn in brown, magenta and yellow, respectively. The analogue of the reaction intermediate NAG-thiazoline, bound in the active site of each subunit is drawn as a space-filling model with carbon atoms in gray, oxygen in red, nitrogen in blue and sulfur in yellow. The active sites of each subunit are located 37 Å apart. All ribbon diagrams were drawn with Molscript82 and rendered with Raster3D83 unless otherwise indicated.
Figure 4.

Pair-wise sequence alignment and secondary structure of subunits α and β. Residues colored in light blue are removed during post-translational processing, and residues in italics compose the ER signal peptides of each subunit (Table 1). Sites (N-X-S/T) known to contain N-linked oligosaccharides are underlined, and glycan sites that receive the mannose-6-phosphate lysosomal targeting moiety are doubly underlined (Table 1). Primary sequence corresponding to the mature, lysosomal αp and βp chains are surrounded by square brackets, sequence comprising chains αm and βb are in curly brackets, and the sequence for chain βa is surrounded by normal brackets.4 Secondary structural elements are as follows: α-helices are drawn as green boxes, β-strands are drawn as blue arrows and disulfide bridges are shown by blue–gray lines connecting Cys residues. Residues boxed in yellow are involved in subunit dimerization as determined from the Hex B crystal structure and also predicted for the Hex A isozyme. The unique mature α-subunit loops 280–283 (GSEP) and 396–398 (IPV) are colored magenta and are predicted to interact directly with the bound activator protein. β-Subunit point mutations known to cause GM2-gangliosidosis are indicated directly above the β-subunit sequence in purple.
Lysosomal proteins, including human Hex, are believed to contain a conserved three-dimensional motif that is recognized by the UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase.31–33 The phosphotransferase phosphorylates N-linked oliogosaccharides of lysosomal proteins so that they can be subsequently recognized by the mannose 6-phosphate receptor and redirected from the Golgi to the lysosome. A detailed understanding of the molecular structure of the phosphotransferase recognition motif on lysosomal proteins is lacking; however, it appears that surface lysine residues may play a significant role in the recognition process.39–41 For lysosomal cathepsins at least, two surface lysine residues, spaced ~34 Å apart, appear to comprise an important component of the phosphotransferase recognition motif.40 Human Hex B contains 48 surface lysine residues, multiple pairs of which are ~34 Å apart. Thus, Hex B may indeed contain a similar lysine-based phosphotransferase recognition motif as suggested for the cathepsins, and our new structural data provide the basis with which to determine, through functional studies, which surface lysine residues on Hex B may be involved in the putative phosphotransferase recognition motif.
Domain I (residues 50–201) of each subunit consists of a six-stranded anti-parallel β-sheet that buries two, parallel α-helices against domain II. Domain II (202–556) is a (β/α)8-barrel structure that houses the active site within loops extending from the C termini of the strands that constitute the β-barrel. The β-subunits of Hex B have a fold remarkably similar to homologous domains found in the two other family 20 glycosidases that have had their structures determined; the 506 aa β-hexosaminidase from S. plicatus (Sp HEX)21 and the 818 aa chitobiase from S. marcescens (SmCHB).20 Sp HEX is a monomeric, two-domain protein with a fold nearly identical to one mature β-subunit of Hex B; 373 Cα atoms of the two proteins have an r.m.s. difference of only 1.6 Å. SmCHB consists of four domains, where domains II and III have 344 Cα atoms with an r.m.s. difference of only 1.4 Å compared to a mature β-subunit of Hex B. These structural comparisons, along with multiple sequence alignments, provide sufficient evidence to suggest that the two-domain structure observed for the mature β-subunit represents a fundamental fold present in all family 20 glycosidases found in species ranging from prokaryotes to humans.
Dimer interface
The α and β-subunits of Hex are believed to be enzymatically inactive as monomers. The crystal structure of Hex B not only reveals why dimerization is crucial for catalytic activity, but also comparative molecular modeling studies of Hex A suggest that the dimer interface forms the docking site for the GM2–activator complex (see below). The β-subunits of Hex B dimerize with their active sites facing towards one another, but are offset by ~120° about an axis perpendicular to the crystallographic 2-fold (Figures 2 and 3). This creates a large dimer (79 Å × 90 Å × 87 Å) with a continuous “U” shaped cleft between the two active sites of the enzyme, providing each active site unobstructed access to the solvent.
Figure 3.

Electrostatic potential surface map and dimer interface of human Hex B. (a) A solvent-accessible surface, drawn over one β-subunit and colored with regions of positive charge in blue and negative charge in red, reveals an overall negative charge about the active site. Note, however, that the electrostatic surface was calculated using the following charge profile only: Lys atom Nζ (charge 1.0); Arg atom NH1 (charge 0.5) and NH2 (charge 0.5); Glu atom Oε1 (charge −0.5) and Oε2 (charge −0.5); Asp atom Oδ1 (charge −0.5) and Oδ1 (charge −0.5); His atom Nδ1 (charge 0.5) and Nε2 (charge 0.5); OXT (charge −1.0). Due to the acidic environment of the lysosome, the electrostatic surface potential of Hex B in the lysosome may be slightly different from what is represented here, potentially being less negatively charged about the active site due to protonation of Glu and Asp carboxyl groups (surface created using the program GRASP84). The other subunit of the homodimer is represented by a ribbon diagram with domain I in green and the catalytic (β/α)8 domain II in yellow. The intermediate analogue NAG-thiazoline, bound in the active site of each subunit is shown as a space-filling model with carbon atoms in gray, oxygen in magenta, nitrogen in blue and sulfur in yellow. (b) Surface rendering of a single β-subunit showing the extensive surface area buried at the dimer interface as determined using the CNS program.74 Polar side-chains are colored blue, hydrophobic side-chains in yellow, backbone atoms in forest green, charged residues in magenta and residues not involved in dimerization are colored gray. The active site pocket is colored red ((b) was drawn using the program PyMOL85). (c) Active site residues (gray) stabilized by interactions from residues of the partnering subunit (yellow). The 2-fold symmetry at the dimer interface results in both active sites experiencing the same stabilizing effects from the associated monomer. The crystallographically determined position of GalNAc–isofagomine (IFG) in the active site of each subunit demonstrates that four of the six hydrogen bonds between the enzyme and inhibitor depend on stabilizing interactions from the partnering subunit. In the absence of the protein–protein interactions that are formed upon dimerization, Arg211, Glu491, Asp452 and Tyr450 are most likely too unstructured to be catalytically acitve.
Because of the crystallographic 2-fold symmetry in the Hex B dimer, each subunit experiences identical protein–protein interactions at the dimer interface. The extensive interface, formed exclusively between the catalytic (β/α)8-barrel domains, covers a patch on the monomer surface adjacent to the active site of each subunit, and several residues from one subunit structurally complete and stabilize active site residues of the other subunit (Figure 3(c)). The family 20 glycosidase inhibitor GalNAc-isofagomine22 was soaked into crystals of Hex B to form a complex and its position within the active site was used to distinguish important catalytic residues of Hex B. Figure 3(c) shows precisely how residues from the partnering subunit stabilize and arrange the active site residues that interact with the inhibitor. This arrangement is identical for both subunits and without these cooperative interactions at the dimer interface, numerous van der Waals contacts and hydrogen-bonding interactions required to stabilize more than half of each active site would be absent, rendering the lone subunits inactive.
In particular, Tyr456 and Tyr547, along with neighboring residues, create a pocket complementary to the active site residues Glu491 and Tyr492 of the partnering subunit. Glu491 selectively stabilizes sugars of galacto-configuration within the active site pocket of Hex B as evidenced by the hydrogen-bonding interaction formed between the carboxyl group of this residue and O4 of Gal-NAc-isofagomine. Hence, interactions from the partnering subunit are indirectly involved in stabilizing the galacto pyranoside configuration most readily catalyzed by human Hex A and B. The functional importance of Tyr456 is evidenced by the naturally occurring mutation Tyr456Ser, a mutation that affects dimerization, creating a transport deficiency from the ER. The patient with this mutation experienced a mild chronic form of Sandhoff disease, which was likely made possible through his second unidentified mutant allele.42– 44
Moreover, just two residues from Tyr547 lies Gly549, the backbone nitrogen atom of which donates a stabilizing hydrogen bond to the carbonyl oxygen atom of Arg211 of the partnering subunit. This Arg residue is conserved in all known family 20 glycosidases and has been shown to be crucial for substrate binding and the stabilization of reaction intermediates (Figures 3(c) and 5).20,45,46 The importance of this Arg residue to the catalytic mechanism of human Hex A is underscored by a series of naturally occurring mutations of this residue that results in a transport-competent, mature Hex A with an inactive α, but an active β-active site. Thus, patients with mutations at αArg178 (aligns with βArg211) have near normal levels of Hex A protein and activity when assayed with artificial β-GlcNAc-containing substrates, but lack the ability to hydrolyze artificial substrates containing a β-GlcNAc-6-SO4 residue or GM2. This novel bio-chemical phenotype defines what has been called the B1-variant form of Tay–Sachs disease. When present along with a null mutation, the most severe form of Tay–Sachs, the infantile/acute form, is observed. Patients who are homozygous for the most common αArg178His B1-mutation experience only a slightly milder phenotype.47–52 A more conservative mutation (made in vitro), βArg211Lys, at this site produces a Hex B variant with a Km value elevated tenfold and a kcat value reduced 500-fold over wild-type.46
Figure 5.

Hex B in complex with the transition state mimic GalNAc-isofagomine (IFG) (a–c) or the intermediate analogue NAG-thiazo-line (NGT) (d–f). (a) Unrefined 2.2 Å resolution sigma-A weighted |Fo| − |Fc|, αc electron density map containing the refined model of IFG bound in the active site of a Hex B β-subunit (αc are the calculated phases and |Fo| and |Fc| are the measured and calculated structure factor amplitudes, respectively). Carbon atoms of IFG are colored green, nitrogen blue, oxygen red. (b) The β-subunit active site showing the extensive hydrogen-bonding interactions between IFG and the enzyme. The magenta hydrogen bond between Glu355 and the ring nitrogen atom of IFG is believed to compensate for the missing hydrogen bond that occurs between an isofagomine inhibitor and a “normal” β-retaining glycosidase containing an enzyme nucleophile.22,54 Tyr456 (yellow) comes into the active site from the partnering subunit to stabilize the position of a water molecule linking Asp452 and Glu491, two residues involved in determining substrate specificity. (c) Tryptophan residues create a hydrophobic pocket in the active site into which the C2-acetamido group becomes appropriately positioned for intramolecular nucleophilic attack at the anomeric center of the terminal sugar molecule being removed from the substrate. The water molecule located above the β-face of the azasugar ring of IFG may represent the incoming water that undergoes base catalyzed (activated by Glu355) nucleophilic attack at the anomeric center of the cyclized intermediate to produce a product with retained configuration. (d) Unrefined 2.5 Å resolution sigma-A weighted |Fo| − |Fc|, αc electron density map containing the refined model of NGT bound in the active site of a Hex B β-subunit (αc are the calculated phases and |Fo| and |Fc| are the measured and calculated structure factor amplitudes, respectively). Carbon atoms of NGT are colored green, nitrogen blue, oxygen red and sulfur yellow. (e) Hydrogen-bonding interactions within the active site that stabilize the cyclized enzyme intermediate. Tyr456 (yellow) comes into the active site from the partnering subunit. (f) Once cyclization has occurred, the enzyme intermediate is protected from solvolysis via unwanted pathways within the tryptophan-lined pocket. A water molecule, activated by Glu355, can only attack from above the β-face, ensuring net retention of anomeric configuration of the product.
Both Gly549 and Tyr547 reside on a loop that terminates at the C terminus of polypeptide βa. This loop forms numerous interactions at the dimer interface and is structurally stabilized by a previously reported disulfide bond between Cys534 and Cys551.34 All residues downstream of Cys551 to the C terminus of the subunit (residue 556) are disordered. The stabilizing effect of this disulfide bond must allow for efficient and stable dimerization to take place. The loss of this disulfide bond by the natural missense mutation βCys534Tyr results in the acute form of Sandhoff disease.53 Residues buried at the dimer interface of Hex B are highlighted in the pair-wise alignment between the α and β-subunits shown in Figure 4. Assuming Hex A and B dimerize in an analogous manner, the alignment demonstrates that not all residues at the dimer interface of Hex A and B are identical, which may explain the differences in dimer stability observed between the isozymes (ββ > αβ > αα) (review4).
Active site structure and catalytic mechanism
By soaking Hex B crystals with the transition state mimic GalNAc-isofagomine22 and with the reaction intermediate analogue NAG-thiazoline,21,24 enzyme–inhibitor complexes (Figure 5) were obtained, the structures of which clearly demonstrate that human Hex B uses a substrate-assisted catalytic mechanism in accordance with results previously described for other family 20 glycosidases (Figure 1).20,21
Hex B–GalNAc-isofagomine complex
Although crystallographic studies confirm that the piperidinium ring of 1-N-azasugars of the isofagomine class do not mimic the planar conformation for atoms C1, C2, O5 and C5 of a pyranoside ring expected during the oxocarbenium ion-like transition state, they do mimic the electrostatic nature of the transition state via a protonated and positively charged nitrogen atom as their “anomeric” center.22,54 The protonated endocyclic nitrogen atom of isofagomine azasugars typically form a strong electrostatic interaction with the enzyme nucleophile of β-retaining glycosidases, thereby making them potent inhibitors of these enzymes. For Hex, however, the interaction is different; in the absence of an enzyme nucleophile, the protonated nitrogen atom of the ring forms instead a strong electrostatic, hydrogen-bonded interaction with the general acid–base residue (Figure 5).22 This interaction imparts sufficient binding energy to make isofagomines potent competitive inhibitors of family 20 glycosidases, and the interaction can be observed in the human Hex B–GalNAc-isofagomine complex as a 2.8 Å hydrogen bond donated from the ring nitrogen of the inhibitor to the carboxyl group of Glu355, a residue previously suspected of being the general-acid–base residue of Hex B.55,56 The mutation βGlu355Gln reduces the kcat value 5000-fold with only a small effect of Km, an observation consistent with its role as the acid/base catalyst in the mechanism of Hex B.57
As expected, the C2-acetamido group of the Gal-NAc-isofagomine is rotated underneath the α-face (i.e. the same face of the pyranose ring where an axial O1 would lie) of the azasugar ring and locked into position by two hydrogen-bonding interactions from residues Asp354 and Tyr450 that flank either side of a tryptophan residue lined pocket in which the C2-acetamido is seated (Figure 5). Finally, there exists a 2.6 Å intramolecular hydrogen bond between the carbonyl oxygen atom of the C2-acetamido group and the protonated ring nitrogen atom of the inhibitor; this enzyme-induced interaction represents the trajectory of nucleophilic attack that would lead to the cyclized oxazolinium ion intermediate on the hydrolytic pathway of a good substrate (Figure 1).
Hex B–NAG-thiazoline complex
NAG-thiazoline is a relatively stable analogue of the hydrolytically unstable oxazoline intermediate that is generated along the reaction coordinate of family 20 glycosidases.24 Indeed, it has been demonstrated that jack bean Hex will synthesize NAG-thiazoline from a precursor molecule in which the C2-acetamindo carbonyl oxygen atom of the terminal, non-reducing sugar was replaced by a sulfur atom.24 The thiazoline ring of NAG-thiazoline is held within the tryptophan lined pocket of the Hex B active site by hydrogen-bonding interactions to Asp354 and Tyr450 in a manner similar to the C2-acetamido group of GalNAc-isofagomine (Figure 5). Prior to cyclization, Asp354 and Tyr450 are thought to polarize the 2-acetamido amide group, thereby increasing the charge density and nucleophilicity of the carbonyl oxygen atom and promoting nucleophilc attack.22 Indeed, the mutation Asp354Asn reduces the kcat value of Hex B 2000-fold while leaving the Km value essentially unchanged.57 Upon cyclization, the carboxylate of Asp345 adopts an additional function by stabilizing the positive charge that develops on N2 of the oxazoline ring.21 Structural and kinetic analysis of the equivalent Asp residue in Sp HEX (Asp313)23 and SmCHB (Asp539)58 demonstrate that this residue not only stabilizies the transition states leading to and from the oxazoline intermediate, it also helps to orient the C2-acetamido during catalysis.
The pyranose ring conformation of NAG-thiazoline is best described as a 4C1 chair; this is in contrast to the high-energy boat conformation seen for the terminal sugar residue in the Michaelis complex of SmCHB bound to chitobiose.20 This difference in conformation indicates that the anomeric carbon atom C1 undergoes the greatest nuclear motion during the catalytic reaction and is consistent with the electrophilc migration of C1 recently described for HEW-lysozyme.59
In both inhibitor complexes of Hex B, a water molecule is held into position above the β-face (i.e. the pyranose ring face opposite to where an axial O1 would lie) of each inhibitor by the general acid–base residue, βGlu355, and may represent the incoming water molecule that is activated by βGlu355 to attack the anomeric center of the bound sugar, producing a product with β-configuration (Figure 5). However, in order for this water molecule to be in an ideal position to attack the anomeric C1 atom, it would need to be associated with, and activated by the other carboxyl oxygen atom of βGlu355. In the presence of a natural oxazolinium ion intermediate, it is believed that this water molecule would move into this ideal position so that βGlu355 could abstract a proton from it and the resulting hydroxide ion could attack the anomeric center and complete the reaction (Figure 1). The hydrophobic pocket in which the oxazoline ring sits, appears to protect the intermediate from solvolysis via unwanted pathways; water can only attack from the β-face of the intermediate, effectively re-inverting the anomeric configuration of the intermediate to produce a product with retained anomeric configuration (Figure 1).21
Finally, having inhibitors of both gluco and galacto-configuration bound in the Hex B-active site demonstrates how, through a joint effort by βArg211 and βGlu491, Hex B can accommodate both sugar configurations within its active site. Because Arg211 appears to play the dominant role for binding sugars of gluco-configuration (Figure 5), it is not surprising to find that the mutation βGlu491Gln does not significantly change the kinetic profile of Hex B when assayed for activity using substrates of gluco-configuration;57 however, the GalNAc-isofagomine·Hex B complex (Figure 5) indicates that such a mutation would have deleterious kinetic effects when catalyzing the hydrolysis of substrates of galacto-configuration.
Predictive modeling of Hex A and the HexA–GM2-ganglioside·activator complex
The primary structures of the α and β-subunits are 60% identical, making the α-subunit an excellent candidate for comparative molecular modeling using the β-subunit structure as a template (Figure 4). Most of the conservation between the α and β-subunits is found within their catalytic domains (Figures 4 and 6), and residues of the β-subunit involved in dimerization are highly conserved in the mature α-subunit. With dimerization being a common functional requirement for the human isozymes, the αβ heterodimer almost certainly dimerizes the same way as observed for the Hex B (ββ) homodimer. Given a similar dimer interface, the Hex A heterodimer model is essentially identical in overall shape to Hex B.
Figure 6.

Predicted model of human Hex A–GM2–activator quaternary complex. (a and b) Two views of the predicted quaternary complex. Residues of the α-subunit identical to those of the β-subunit are colored blue, non-identical residues are colored light brown. Most of the conserved amino acids in the α and β-subunits are located in (β/α)8-barrel of domain II. The β-subunit is colored gray, with residues of the active site distinguished in orange. The GM2–activator protein complex (GM2–AP) docks into a large groove between the two subunits so that the terminal non-reducing GalNAc sugar on GM2 can be presented to the α-subunit active site and removed. Two surface loops (magenta and green), present only on the α-subunit, interact with the docked activator protein and appear to be involved in creating a docking site unique to the α-subunit. The magenta colored loop is proteolytically removed from the β-subunit during post-translational processing and may represent a modification that regulates the metabolic function of this subunit. (c) Model of the GM2 oligosaccharide (yellow) bound to the α-subunit active site (gray). The distorted boat conformation of the terminal GalNAc to be removed (Gal, labeled in blue) and the pseudoaxial orientation of the scissile bond and leaving group are based on crystallographic observations of the Michaelis complex of chitobiose bound to SmCHB.20 By incorporating these conformational restraints into the model, only one reasonable position could be found for the sialic acid residue (labeled SIA) within the active site pocket. Once positioned, the negatively charged carboxylate of the sialic acid, which can only be accommodated by the α-subunit, was found to come within hydrogen bonding distance of Arg424, a positively charged residue that is unique to the α-subunit (the β-subunit contains a Leu at this position). αGlu394 and αAsn423 (which are both Asp residues in the β-subunit) are believed to help hold Arg424 into position. Arg424, in turn, stabilizes the negatively charged caboxylate of the sialic acid of the substrate via electrostatic and hydrogen-bonding interactions. The general acid–base residue, Glu323 (Glu355 in the β-subunit), can be seen interacting with the glycosidic oxygen atom of the scissile bond.
Interestingly, the predicted αβ dimer interface forms a large groove into which the activator structure6 can dock. The novel β-cup topology of the activator consists primarily of β-sheet structure forming a hollow hydrophobic cavity that is accessible to the solvent through a hole at only one end of the protein.6 The activator is believed to form a 1:1 complex with GM260 and two hypotheses have been made for how the activator presents GM2 to Hex A.61 In the first hypothesis, the activator binds GM2 in the intra-lysosomal membrane or vesicle and lifts it a few angstrom units out of the membrane to allow Hex A access to its terminal β GalNAc. In the second hypothesis, the activator fully removes GM2 from the membrane forming a soluble complex which then interacts with Hex A. Clearly, the closed β-cup formation of the activator indicates that the ceramide tail must be fully bound within the cup’s hydrophobic interior (hypothesis 2), presumably leaving the oligosaccharide portion of the glycosphingolipid exposed through the opening so that the terminal GalNAc sugar can be presented to the α-subunit active site and removed. Indeed, our results from an automated docking study found that the optimal surface and electrostatic complementarity between the Hex A model and the activator occurs when the entrance to the hydrophobic cavity of the activator is positioned directly over the α-subunit active site (Figure 6). Our predicted model of the active complex is consistent only with hypothesis 2, discussed above; thus, it appears that GM2 must first be fully removed from its membrane environment by the activator prior to docking onto Hex A.
The predicted GM2–activator–Hex A docking interactions are supported by previous biochemical findings demonstrating that the activator interacts with the middle section of the α-subunit and the carboxyl half of the β-subunit of Hex A.62,63 Our predicted model of the quaternary complex indicates that the docked activator interacts with discrete patches on the α-subunit surface comprised of groups of residues found between α280 and 400 and with patches of residues between β465 and 545 of the β-subunit sequence. This latter observation is consistent with the finding that elements of the β-subunit are required for effective binding of the activator to Hex A.64 Furthermore, two small loops unique to the mature α-subunit (α280–283 and α396–398), were found to interact directly with the docked activator (Figure 6). Loop 280–283 (Gly-Ser-Glu-Pro) aligns with the β-subunit residues 312–315 (Arg-Gln-Asn-Lys) that are removed during post-translational processing, suggesting that the selective removal of this loop from β-subunit helps to specify to which active site the activator presents substrate.
Two natural mutations, Cys138Arg of the activator, and Pro504Ser of the β-subunit, are believed to specifically disrupt activator binding to Hex A.65,66 Cys138 forms a disulfide bond to Cys112 within the activator structure and stabilizes the position of an α-helix spanning residues 111–120.6 Our model of the quaternary complex suggests that residues of this α-helix form part of a surface epitope that binds directly to the α-subunit of Hex A. It appears that the activator mutation Cys138Arg could severely compromise this intermolecular interaction without affecting its ability to form a complex with GM2-ganglioside. This was exactly the biochemical phenotype found after analysis of the recombinant activator mutant (Cys138Arg) produced from bacteria.65
Residue Pro504 of the β-subunit introduces a kink into helix α8 of the (β/α)8-barrel. This kink is required for proper packing of helix α8 against two loops that, according to our model of the quaternary complex, interact directly with the docked activator protein. Thus, disruption of the interactions between these two loops and helix α8 via the mutation Pro504Ser appears to adversely affect this portion of the binding epitope between the β-subunit of Hex A and the activator. This prediction fits the observed biochemical phenotype for the Hex A mutant (βPro504Ser), which displays a compromised ability to associate with the GM2–activator complex, but can still hydrolyze simple water soluble substrates.66
The active site structures of the α and β-subunits are essentially identical; however, three amino acid changes: βAsp426 → αGlu394, βAsp452 → αAsn423 and βLeu453 → αArg424 appear to provide the α-subunit with the distinct characteristic of being able to accommodate a negative charge near the terminal non-reducing β-linked hexosamine of its substrates. A model of the Michaelis complex between the GM2 oligosaccharide and the α-subunit active site suggests that the negatively charged carboxylate of the sialic acid on GM2 is stabilized by the positively charged guanidino group of Arg424, which is in turn positioned by Glu394 (Figure 6). Removal of the positively charged guanidino group (αArg424Gln) results in an α-subunit with a ninefold increase in Km value, and a slight decease in VMAX value relative to wild-type, when assayed for its ability to catalyze the removal of from an artificial substrate.67 These kinetic data indicate that the positively charged guanidino group of Arg424 can stabilize the negatively charged 6-sulfate group on the terminal that is removed from the substrate by Hex A. Furthermore, the mutation αAsn423Asp results in α-subunit activity having a Km value elevated sixfold relative to wild-type when assayed for activity toward the same 6-sulfated substrate.67 This mutation introduces a negatively charged carboxylate group into the α-subunit active site at position 423 and this negative charge appears to reduce the binding affinity of the negatively charged terminal 6-sulfated sugar molecule due to electrostatic repulsion of the substrate by the carboxylate of Asp423. Together, these kinetic results indicate that disruption of the electrostatic environment provided by Arg424 and Asn423, reduces the ability of the α-subunit active site to accommodate negatively charged substrates, and provides biochemical evidence to substantiate the model presented here. The cleft that accommodates the sialic acid residue of GM2, is not present in other family 20 glycosidases,20,21 but is instead occupied by protein structure comprising sugar binding sub-sites that for GM2 at least, appear to be present on the activator protein. Furthermore, the conformation of the modeled GM2 substrate places the negatively charged carboxylate of its sialic acid in close proximity to the hydroxymethyl group of the terminal GalNAc residue to be removed. The close spatial arrangement of these two functional groups suggests that αArg424 and αAsn423 stabilize not only the negatively charged sialic acid moiety of GM2, they also stabilize natural and artificial substrates that have the hydroxymethyl group of their non-reducing sugar molecule replaced by a negatively charged 6-sulfate group. Biochemical confirmation of this hypothesis has been obtained using a αArg424Gln Hex A variant, which exhibited an increase in Km value for the GM2–activator complex of three- to fourfold (R. Sharma & D.J.M., unpublished results). Substrates containing a 6-sulfate group are used to selectively measure α-subunit activity; however, it has been unclear how the α-subunit of Hex A could accommodate natural and artificial substrates containing negatively charged functional groups that appear to be in spatially distant positions from each other, i.e. GlcNac-6SO4-X and GalNAc-Gal(SA)-Glc-Ceramide (GM2).
Conclusions
Our structural data provide explanations for the mechanisms behind many of the known biochemical properties of human Hex, including the structural basis for how many of the naturally occurring point mutations in HEXA and HEXB cause Tay–Sachs and Sandhoff disease, respectively. The X-ray crystal structure of Hex B demonstrates clearly why dimerization is necessary for enzymatic function. Determining the structure of Hex B in complex with the mechanism based inhibitors GalNAc-isofagomine and NAG-thiazoline not only confirm that human Hex uses a substrate-assisted catalytic mechanism in accordance with other family 20 glycosidases, but also the complexes reveal the structural basis for how the enzyme binds terminal amino sugar molecules of both gluco and galacto configuration. Furthermore, the comparative molecular modeling of Hex A and subsequent activator docking studies provide insight into why only the α-subunit active site can stabilize negatively charged GlcNAc-6-sulfate containing substrates, and the negatively charged sialic acid moiety on GM2, as well as insight into the structural basis for how Hex A associates with the GM2–activator complex.
Materials and Methods
Purification and crystallization
Hex B was purified from human placenta13 and crystallized from ammonium sulfate solutions using the vapor diffusion method as described.36 The protein was used in its native glycosylation state for all crystallization experiments. Ellipsoidal crystals having a hexagonal cross-section perpendicular to the longest crystal axis (space group P6122) appeared after about three weeks from 6 μl hanging drops and were used as seeds in fresh crystallization systems that produced large, diffraction quality crystals within six months.
Structure determination
All diffraction data were collected at the Advanced Photon Source beamline 14-BM-C (BioCars) from single crystals that were briefly soaked (ten seconds) in cryosolvent (30%(v/v) glycerol, 60% (w/v) ammonium sulfate, 50 mM potassium phosphate (pH 8.0)) then flash-cooled to 100 K within a N2 (g) stream. Intensity data were processed using DENZO and SCALEPACK.68 Structure factor phases were determined experimentally using the multiple isomorphous replacement (MIR) method. Crystals were derivatized by soaking them overnight in solutions of 3.6 mM methyl mercury acetate (CH3HgCOCH3) or 2.6 mM potassium platinum tetrachloride (K2PtCl4). Difference Patterson and Fourier searches of diffraction data collected from native and heavy-atom derivitized Hex B crystals were carried out using the program SOLVE.69 Two sites were located within the asymmetric unit for each heavy-atom type. Each derivative was of sufficient quality to calculate an MIR map that clearly defined the overall shape of the Hex B subunits to a resolution of 2.8 Å. Statistical density modification using the program RESOLVE70 greatly improved the quality of the experimental map (see Table 2). The density-modified map was used to obtain a crude estimate of the non-crystallographic 2-fold symmetry (NCS) operator relating the two β-subunits that comprise the asymmetric unit. This NCS operator was refined by RESOLVE and used to produce a high quality, 2-fold averaged map into which the molecular model of Hex B was built using the program O.71
Model building and refinement of native Hex B
Prior to the refinement process, the program UNIQUE72 was used to calculate a complete data set to 2.0 Å resolution using the space group P6122 and unit cell dimensions determined for crystals of native Hex B (Table 2). A random subset of hkl indices (5%) was selected from this calculated data set using FREERFLAG72 and set aside for cross-validation using the free R factor.73 A hand-built model of native Hex B was refined using a high-temperature, simulated annealing protocol guided by a maximum-likelihood target function within the CNS program.74 Due to the high temperature simulation, molecular dynamics were restricted to torsion angles only.75 This initial round of model refinement included experimental phase information as output from SOLVE and was restrained to the 2-fold NCS operator refined by RESOLVE. Following simulated annealing, several rounds of conjugate-gradient minimization were carried out, resulting in a combined reduction of Rwork (Rfree) from 0.41 (0.40) to 0.29 (0.34). Subsequent iterative rounds of model building and TLS refinement76 were carried out using O and REFMAC5,72 respectively, until R-factor convergence was reached (Table 2). NCS restraints and experimental phases were not included during the later rounds of refinement using REFMAC5. Water molecules were picked using ARP/wARP.72
Model building and refinement of the NAG-thiazoline·Hex B and GalNAc-isofagomine–Hex B complexes
NAG-thiazoline and GalNAc-isofagomine were separately soaked into Hex B crystals to create complexes isomorphous with native Hex B crystals. Using a maximum likelihood target function within REFMAC5, rigid body refinement and several rounds of conjugate gradient minimization were sufficient to position a solvent-free molecular model of native Hex B into the asymmetric unit of each crystallographic complex. Initial sigma-A weighted |Fo| − |Fc|, αc and 2|Fo| − |Fc|, αc electron density maps (where αc are the calculated phases and |Fo| and |Fc| are the measured and calculated structure factor amplitudes, respectively), computed prior to solvent modeling, unambiguously defined the conformation of both GalNAc-isofagomine and NAG-thiazoline bound in the −1 sub-sites of the Hex B active sites. Models of the inhibitors were built into the appropriate electron density of each complex using the program O (Figure 5) and the coordinates were subjected to iterative rounds of TLS refinement and solvent modeling until R-factor convergence was reached (Table 2).
Crystallographic coordinates
Coordinates and structure factors have been deposited into the RCSB Protein Data Bank (accession codes: native Hex B, 1NOU; Hex B·NAG-thiazoline complex, 1NPO; Hex B·GalNAc–isofagomine complex, 1NOW).
Comparative molecular modeling of Hex A and docking of the GM2-ganglioside/activator protein
Using the program Swiss-PDBviewer,77 a pair-wise alignment was generated between the mature α and β-subunit amino acid sequences. This alignment was used as a guide for substituting the amino acid sequence of the α-subunit onto the Cα coordinates of a β-subunit from the Hex B structure. Loops 280–283 and 396–398, not present in the β-subunit structure, were modeled using the program MODELLER.78 Side-chain positions in the α-subunit model were optimized manually. Although not energy minimized, the α-subunit model has acceptable stereochemistry as determined by PROCHECK (Ramachandran plot: 90.5% within limits, 8.3% allowed, 1.2% generous, 0.0% disallowed).79 Because the α-subunit model replaces one of the β-subunits in the Hex B crystal structure, the remaining β-subunit of Hex B was used to create the complete Hex A αβ heterodimer with a dimer interface resembling closely that of Hex B.
The activator crystal structure contains three copies of the protein per asymmetric unit.6 A mobile loop (Val153-Leu163) forms part of the opening to the hydrophobic cavity of the activator. For two of the three copies in the asymmetric unit (monomers A and B), the loop is in an “open” conformation, whereas in the third copy (monomer C) the loop is in a “closed” conformation.6 All three models of the activator (monomers A, B and C) were individually docked onto the Hex A homology model using the program suite 3D-DOCK.80,81 Interestingly, when based on optimal electrostatics and surface complementarity, only monomer A docked onto the Hex A model such that the opening to the hydrophobic cavity aligned with the α-subunit active site (Figure 6). This suggests that the open conformation of the activator is the form that binds to Hex A. Inspection of the complex revealed that the mobile loop of the docked activator in its open conformation occluded part of the active site so that GM2-ganglioside could not be modeled into the complex without numerous steric clashes with the activator. Due to the apparent mobility of this loop and lack of crystallographic information about how the activator interacts with GM2-ganglioside, the geometry of the loop was not adjusted, and space for GM2-ganglioside in the active site was instead made through small manual rigid body movements of the whole activator structure relative to Hex A. Using the program O, the oligosachharide portion of a model of GM2-ganglioside was then fit into the α-subunit active site based on the Michaelis complex of chitobiose bound to SmCHB (Figure 6).20 The modeled conformation of the oligosaccharide and the manually adjusted position of the activator protein allowed for the lipid tail of GM2-ganglioside to enter the hydrophobic cavity of the activator protein (Figure 6).
Acknowledgments
We thank Amy Leung for her technical assistance in purifying Hex from human placenta, R. S. Williams for help with data collection, and D. J. Vocadlo and S. G. Withers for many helpful discussions. The work was supported by Canadian Institutes of Health Research grants (to D.M. and M.N.G.J.). B.L.M. is supported by scholarships from the C.I.H.R., the Alberta Heritage Foundation for Medical Research, and the University of Alberta. M.N.G.J. holds a Canada Research Chair in Protein Structure and Function at the University of Alberta.
Abbreviations used
- NAGase
N-acetyl-β-D-glucosaminidase
- GlcNAc
N-acetyl-D-glucosamine
- GalNAc
N-acetyl-D-galactosamine
- Hex
N-acetyl-β-D-hexosaminidase
- TSD
Tay–Sachs disease
- ER
endoplasmic reticulum
- aa
amino acid
Footnotes
References
- 1.Watanabe K. Biochemical studies on carbohydrates. J Biochem (Tokyo) 1936;24:297. [Google Scholar]
- 2.Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Ophthalmol Soc UK. 1881;1:155–157. [Google Scholar]
- 3.Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. The GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. 7. Vol. 2. Vol. 3 McGraw-Hill; New York: 1995. pp. 2839–2879. [Google Scholar]
- 4.Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455:105–138. doi: 10.1016/s0925-4439(99)00074-5. [DOI] [PubMed] [Google Scholar]
- 5.Mahuran DJ. The GM2 activator protein, its roles as a co-factor in GM2 hydrolysis and as a general glycolipid transport protein. Biochim Biophys Acta. 1998;1393:1–18. doi: 10.1016/s0005-2760(98)00057-5. [DOI] [PubMed] [Google Scholar]
- 6.Wright CS, Li SC, Rastinejad F. Crystal structure of human GM2-activator protein with a novel beta-cup topology. J Mol Biol. 2000;304:411–422. doi: 10.1006/jmbi.2000.4225. [DOI] [PubMed] [Google Scholar]
- 7.Cao ZM, Petroulakis E, Salo T, Triggs-Raine B. Benign HEXA mutations, C739T(R247W) and C745T(R249W), cause beta-hexosaminidase A pseudo-deficiency by reducing the alpha-subunit protein levels. J Biol Chem. 1997;272:14975–14982. doi: 10.1074/jbc.272.23.14975. [DOI] [PubMed] [Google Scholar]
- 8.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 9.O’Dowd BF, Klavins MH, Willard HF, Gravel R, Lowden JA, Mahuran DJ. Molecular heterogeneity in the infantile and juvenile forms of Sandhoff disease (O-variant GM2 Gangliosidosis) J Biol Chem. 1986;261:12680–12685. [PubMed] [Google Scholar]
- 10.Kresse H, Fuchs W, Glossl J, Holtfrerich D, Gilberg W. Liberation of N-acetylglucosamine-6-sulfate by human beta-N-acetylhexosaminidase A. J Biol Chem. 1981;256:12926–12932. [PubMed] [Google Scholar]
- 11.Hepbildikler ST, Sandhoff R, Kolzer M, Proia RL, Sandhoff K. Physiological substrates for human lysosomal beta-hexosaminidase S. J Biol Chem. 2002;277:2562–2572. doi: 10.1074/jbc.M105457200. [DOI] [PubMed] [Google Scholar]
- 12.Sandhoff R, Hepbildikler ST, Jennemann R, Geyer R, Gieselmann V, Proia RL, et al. Kidney sulfatides in mouse models of inherited glycosphingolipid disorders: determination by nano-electrospray ionization tandem mass spectrometry. J Biol Chem. 2002;277:20386–20398. doi: 10.1074/jbc.M110641200. [DOI] [PubMed] [Google Scholar]
- 13.Mahuran DJ, Lowden JA. The subunit and polypeptide structure of hexosaminidase from human placenta. Can J Biochem. 1980;58:287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 14.Hou Y, Tse R, Mahuran DJ. The direct determination of the substrate specificity of the α-active site in heterodimeric β-hexosaminidase A. Biochemistry. 1996;35:3963–3969. doi: 10.1021/bi9524575. [DOI] [PubMed] [Google Scholar]
- 15.Kytzia HJ, Sandhoff K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J Biol Chem. 1985;260:7568–7572. [PubMed] [Google Scholar]
- 16.Meier EM, Schwarzmann G, Fürst W, Sandh-off K. The human GM2 activator protein A substrate specific cofactor of β-hexosaminidase A. J Biol Chem. 1991;266:1879–1887. [PubMed] [Google Scholar]
- 17.Henrissat B, Bairoch A. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1993;293:781–788. doi: 10.1042/bj2930781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henrissat B, Bairoch A. Updating the sequence-based classification of glycosyl hydrolases. Biochem J. 1996;316:695–696. doi: 10.1042/bj3160695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai ECK, Withers SG. Stereochemistry and kinetics of the hydration of 2-acetamido-D-glucal by beta-N-acetylhexosaminidases. Biochemistry. 1994;33:14743–14749. doi: 10.1021/bi00253a012. [DOI] [PubMed] [Google Scholar]
- 20.Tews I, Perrakis A, Oppenheim A, Dauter Z, Wilson KS, Vorgias CE. Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay–Sachs disease. Nature Struct Biol. 1996;3:638–648. doi: 10.1038/nsb0796-638. [DOI] [PubMed] [Google Scholar]
- 21.Mark BL, Vocadlo DJ, Knapp S, Triggs-Raine BL, Withers SG, James MNG. Crystallographic evidence for substrate-assisted catalysis in a bacterial beta-hexosaminidase. J Biol Chem. 2001;276:10330–10337. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 22.Mark BL, Vocadlo DJ, Zhao D, Knapp S, Withers SG, James MNG. Biochemical and structural assessment of the 1-N-azasugar GalNAc-isofagomine as a potent family 20 β-N-acetylhexosaminidase inhibitor. J Biol Chem. 2001;276:42131–42137. doi: 10.1074/jbc.M107154200. [DOI] [PubMed] [Google Scholar]
- 23.Williams SJ, Mark BL, Vocadlo DJ, James MN, Withers SG. Aspartate 313 in the Streptomyces plicatus hexosaminidase plays a critical role in substrate-assisted catalysis by orienting the 2-acetamido group and stabilizing the transition state. J Biol Chem. 2002;277:40055–40065. doi: 10.1074/jbc.M206481200. [DOI] [PubMed] [Google Scholar]
- 24.Knapp S, Vocadlo D, Gao Z, Kirk B, Lou J, Withers SG. NAG-thiazoline, an N-acetyl-beta-hexosaminidase inhibitor that implicates acetamido participation. J Am Chem Soc. 1996;118:6804–6805. [Google Scholar]
- 25.Drouillard S, Armand S, Davies GJ, Vorgias CE, Henrissat B. Serratia marcescens chitobiase is a retaining glycosidase utilizing substrate acetamido group participation. Biochem J. 1997;328:945–949. doi: 10.1042/bj3280945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Legler G, Bollhagen R. (±)-6-Acetamido-1,2-anhydro-6-deoxy-myo-inositol: a tight-binding inhibitor and pseudosubstrate for N-acetyl-beta-glucosaminidases. Carbohydr Res. 1992;233:113–123. [Google Scholar]
- 27.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 28.Bach G, Bargel R, Cantz M. I-cell disease: deficiency of extracellular hydrolase phosphorylation. Biochem Biophys Res Commun. 1979;91:976–981. doi: 10.1016/0006-291x(79)91975-2. [DOI] [PubMed] [Google Scholar]
- 29.Kornfeld S. Trafficking of lysosomal enzymes in normal and disease states. J Clin Invest. 1986;77:1–6. doi: 10.1172/JCI112262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. The mannose 6-phosphate receptor and the biogenesis of the lysosomes. Cell. 1988;52:329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- 31.Baranski TJ, Cantor AB, Kornfeld S. Lysosomal enzyme phosphorylation. I. Protein recognition determinants in both lobes of procathepsin D mediate its interaction with UDP-GlcNAc:lysosoma l enzyme N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 1992;267:23342–23348. [PubMed] [Google Scholar]
- 32.Cantor AB, Baranski TJ, Kornfeld S. Lysosomal enzyme phosphorylation. II. Protein recognition determinants in either lobe of procathepsin D are sufficient for phosphorylation of both the amino and carboxyl lobe oligosaccharides. J Biol Chem. 1992;267:23349–23356. [PubMed] [Google Scholar]
- 33.Cantor AB, Kornfeld S. Phosphorylation of Asn-linked oligosaccharides located at novel sites on the lysosomal enzyme cathepsin D. J Biol Chem. 1992;267:23357–23363. [PubMed] [Google Scholar]
- 34.Sagherian C, Poroszlay S, Vavougios G, Mahuran DJ. Proteolytic processing of the prob chain of β-hexosaminidase occurs at basic residues contained within an exposed disulfide loop structure. Biochem Cell Biol. 1993;71:340–347. doi: 10.1139/o93-051. [DOI] [PubMed] [Google Scholar]
- 35.Sagherian C, Thorner P, Mahuran D. The pro-peptide of the probeta-polypeptide chain of human beta-hexosaminidase is necessary for proper protein folding and exit from the endoplasmic reticulum. Biochem Biophys Res Commun. 1994;204:135–141. doi: 10.1006/bbrc.1994.2436. [DOI] [PubMed] [Google Scholar]
- 36.Church WB, Swenson L, James MNG, Mahuran D. Crystallization of human β-hexosaminidase B. J Mol Biol. 1992;227:577–580. doi: 10.1016/0022-2836(92)90911-3. [DOI] [PubMed] [Google Scholar]
- 37.Hubbes M, Callahan J, Gravel R, Mahuran D. The amino-terminal sequences in the pro-α and -β polypeptides of human lysosomal β-hexosaminidase A and B are retained in the mature isozymes. FEBS Letters. 1989;249:316–320. doi: 10.1016/0014-5793(89)80649-0. [DOI] [PubMed] [Google Scholar]
- 38.Schuette CG, Weisgerber J, Sandhoff K. Complete analysis of the glycosylation and disulfide bond pattern of human beta-hexosaminidase B by MALDI-MS. Glycobiology. 2001;11:549–556. doi: 10.1093/glycob/11.7.549. [DOI] [PubMed] [Google Scholar]
- 39.Tikkanen R, Peltola M, Oinonen C, Rouvinen J, Peltonen L. Several cooperating binding sites mediate the interaction of a lysosomal enzyme with phosphotransferase. EMBO J. 1997;16:6684–6693. doi: 10.1093/emboj/16.22.6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuozzo JW, Tao K, Cygler M, Mort JS, Sahagian GG. Lysine-based structure responsible for selective mannose phosphorylation of cathepsin D and cathepsin L defines a common structural motif for lysosomal enzyme targeting. J Biol Chem. 1998;273:21067–21076. doi: 10.1074/jbc.273.33.21067. [DOI] [PubMed] [Google Scholar]
- 41.Warner JB, Thalhauser C, Tao K, Sahagian GG. Role of N-linked oligosaccharide flexibility in mannose phosphorylation of lysosomal enzyme cathepsin L. J Biol Chem. 2002;277:41897–41905. doi: 10.1074/jbc.M203097200. [DOI] [PubMed] [Google Scholar]
- 42.Banerjee P, Siciliano L, Oliveri D, McCabe NR, Boyers MJ, Horwitz AL, et al. Molecular basis of an adult form of beta-hexosaminidase B deficiency with motor neuron disease. Biochem Biophys Res Commun. 1991;181:108–115. doi: 10.1016/s0006-291x(05)81388-9. [DOI] [PubMed] [Google Scholar]
- 43.Banerjee P, Boyers MJ, Berry-Kravis E, Dawson G. Preferential beta-hexosaminidase (Hex) A (alpha beta) formation in the absence of beta-Hex B (beta beta) due to heterozygous point mutations present in beta-Hex beta-chain alleles of a motor neuron disease patient. J Biol Chem. 1994;269:4819–4826. [PubMed] [Google Scholar]
- 44.Redonnet-Vernhet I, Mahuran D, Salvayre R, Dubas F, Levade T. Significance of two point mutations present in each HEXB allele of patients with adult GM2 gangliosidosis (Sandhoff disease): homozygosity of Ile207 → Val substitution is not associated with a clinical or biochemical phenotype. Biochim Biophys Acta. 1996;1317:127–133. doi: 10.1016/s0925-4439(96)00044-0. [DOI] [PubMed] [Google Scholar]
- 45.Mark BL, Wasney GA, Salo TJ, Khan AR, Cao Z, Robbins PW, et al. Structural and functional characterization of Streptomyces plicatus beta-N-acetylhexosaminidase by comparative molecular modeling and site-directed mutagenesis. J Biol Chem. 1998;273:19618–19624. doi: 10.1074/jbc.273.31.19618. [DOI] [PubMed] [Google Scholar]
- 46.Hou Y, Vocadlo D, Withers S, Mahuran D. The role of beta-Arg211 in the active site of human beta-hexosaminidase B. Biochemistry. 2000;39:6219–6227. doi: 10.1021/bi992464j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akerman BR, Zielenski J, Triggs-Raine BL, Prence EM, Natowicz MR, Lim-Steele JS, et al. A mutation common in non-Jewish Tay–Sachs disease: frequency and RNA studies. Hum Mutat. 1992;1:303–309. doi: 10.1002/humu.1380010407. [DOI] [PubMed] [Google Scholar]
- 48.Brown CA, Neote K, Leung A, Gravel RA, Mahuran DJ. Introduction of the α subunit mutation associated with the B1 variant of Tay–Sachs disease into the β subunit produces a β-hexosaminidase B without catalytic activity. J Biol Chem. 1989;264:21705–21710. [PubMed] [Google Scholar]
- 49.dos Santos MR, Tanaka A, sa Miranda MC, Ribeiro MG, Maia M, Suzuki K. GM2-gangliosidosis B1 variant: analysis of beta-hexosaminidase alpha gene mutations in 11 patients from a defined region in Portugal. Am J Hum Genet. 1991;49:886–890. [PMC free article] [PubMed] [Google Scholar]
- 50.Nakano T, Muscillo M, Ohno K, Hoffman AJ, Suzuki K. A point mutation in the coding sequence of β-hexosaminidase α gene results in defective processing of the enzyme in an unusual GM2-gangliosidosis variant. J Neurochem. 1988;51:984–987. doi: 10.1111/j.1471-4159.1988.tb01836.x. [DOI] [PubMed] [Google Scholar]
- 51.Nakano T, Nanba E, Tanaka A, Ohno K, Suzuki Y, Suzuki K. A new point mutation within exon 5 of β-hexosaminidase α gene in a Japanese infant with Tay–Sachs disease. Ann Neurol. 1990;27:465–473. doi: 10.1002/ana.410270503. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka A, Ohno K, Suzuki K. GM2-Gangliosidosis B1 variant: a wide geographic and ethnic distribution of the specific β-hexosaminidase a chain mutation originally identified in a Puerto Rican patient. Biochem Biophys Res Commun. 1988;156:1015–1019. doi: 10.1016/s0006-291x(88)80945-8. [DOI] [PubMed] [Google Scholar]
- 53.Kuroki Y, Itoh K, Nadaoka Y, Tanaka T, Sakuraba H. A novel missense mutation (C522Y) is present in the beta-hexosaminidase beta-subunit gene of a Japanese patient with infantile Sandhoff disease. Biochem Biophys Res Commun. 1995;212:564–571. doi: 10.1006/bbrc.1995.2007. [DOI] [PubMed] [Google Scholar]
- 54.Notenboom V, Williams SJ, Hoos R, Withers SG, Rose DR. Detailed structural analysis of glycosidase/inhibitor interactions: complexes of Cex from Cellulomonas fimi with xylobiose-derived aza-sugars. Biochemistry. 2000;39:11553–11563. doi: 10.1021/bi0010625. [DOI] [PubMed] [Google Scholar]
- 55.Fernandes MJG, Yew S, Leclerc D, Henrissat B, Vorgias CE, Gravel RA, et al. Identification of candidate active site residues in lysosomal beta-hexosaminidase A. J Biol Chem. 1997;272:814–820. doi: 10.1074/jbc.272.2.814. [DOI] [PubMed] [Google Scholar]
- 56.Pennybacker M, Schuette CG, Liessem B, Hepbildikler ST, Kopetka JA, Ellis MR, et al. Evidence for the involvement of Glu-355 in the catalytic action of human beta-hexosaminidase B. J Biol Chem. 1997;272:8002–8006. doi: 10.1074/jbc.272.12.8002. [DOI] [PubMed] [Google Scholar]
- 57.Hou Y, Vocadlo D, Leung A, Withers S, Mahuran D. Characterization of Glu and Asp residues in the active site of human β-hexosaminidase B. Biochemistry. 2001;40:2201–2209. doi: 10.1021/bi002018s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prag G, Papanikolau Y, Tavlas G, Vorgias CE, Petratos K, Oppenheim AB. Structures of chitobiase mutants complexed with the substrate Di-N-acetyl-D-glucosamine: the catalytic role of the conserved acidic pair, aspartate 539 and glutamate 540. J Mol Biol. 2000;300:611–617. doi: 10.1006/jmbi.2000.3906. [DOI] [PubMed] [Google Scholar]
- 59.Vocadlo DJ, Davies GJ, Laine R, Withers SG. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature. 2001;412:835–838. doi: 10.1038/35090602. [DOI] [PubMed] [Google Scholar]
- 60.Conzelmann E, Burg J, Stephan G, Sandhoff K. Complexing of glycolipids and their transfer between membranes by the activator protein for degradation of lysosomal ganglioside GM2. Eur J Biochem. 1982;123:455–464. doi: 10.1111/j.1432-1033.1982.tb19789.x. [DOI] [PubMed] [Google Scholar]
- 61.Sandhoff K, Harzer K, Fürst W. Sphingolipid activator proteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Disease. 7. Vol. 2. Vol. 3 McGraw-Hill; New York: 1995. pp. 2427–2441. [Google Scholar]
- 62.Pennybacker M, Liessem B, Moczall H, Tifft CJ, Sandhoff K, Proia RL. Identification of domains in human beta-hexosaminidase that determine substrate specificity. J Biol Chem. 1996;271:17377–17382. doi: 10.1074/jbc.271.29.17377. [DOI] [PubMed] [Google Scholar]
- 63.Tse R, Wu YJ, Vavougios G, Hou Y, Hinek A, Mahuran DJ. Identification of functional domains within the α and β subunits of β-hexosaminidase A through the expression of α–β fusion proteins. Biochemistry. 1996;35:10894–10903. doi: 10.1021/bi960503a. [DOI] [PubMed] [Google Scholar]
- 64.Sandhoff K, Conzelmann E, Nehrkorn H. Specificity of human liver hexosaminidase A and B against glycosphingolipids GM2 and GA2: purification of the enzymes by affinity chromatography employing specific elution. Hoppe-Seyler’s Z Physiol Chem. 1977;358:779–787. doi: 10.1515/bchm2.1977.358.2.779. [DOI] [PubMed] [Google Scholar]
- 65.Xie B, Rigat B, Smiljanic-Georgijev N, Deng H, Mahuran DJ. Biochemical characterization of the Cys138Arg missense mutation associated with the AB-variant form of GM2 Gangliosidosis: evidence that Cys138 is required for the recognition of the GM2 activator: GM2-Ganglioside complex by β-hexosaminidase A. Biochemistry. 1998;37:814–821. doi: 10.1021/bi971211s. [DOI] [PubMed] [Google Scholar]
- 66.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. A Pro504Ser substitution in the β-subunit of β-hexosaminidase A inhibits α-subunit hydrolysis of GM2 ganglioside, resulting in chronic Sandhoff disease. J Biol Chem. 1998;273:21386–21392. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
- 67.Sharma R, Deng H, Leung A, Mahuran D. Identification of the 6-sulfate binding site unique to alpha-subunit-containing isozymes of human beta-hexosaminidase. Biochemistry. 2001;40:5440–5446. doi: 10.1021/bi0029200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 69.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallog sect D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallog sect D Biol Crystallog. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones TA, Zou JY, Cowan SW, Kjeldgaard Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallog sect A. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 72.Collaborative Computational Project Number 4. The CCP4 Suite: programs for protein crystallography. Acta Crystallog sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 73.Brunger AT. Free R-value—a novel statistical quantity for assessing the accuracy of crystal-structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 74.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallog sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 75.Rice LM, Brunger AT. Torsion angle dynamics: reduced variable conformational sampling enhances crystallographic structure refinement. Proteins: Struct Funct Genet. 1994;19:277–290. doi: 10.1002/prot.340190403. [DOI] [PubMed] [Google Scholar]
- 76.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallog sect D Biol Crystallog. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 77.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 78.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structure coordinates. J Appl Crystallog. 1993;26:283–291. [Google Scholar]
- 80.Gabb HA, Jackson RM, Sternberg MJ. Modelling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol. 1997;272:106–120. doi: 10.1006/jmbi.1997.1203. [DOI] [PubMed] [Google Scholar]
- 81.Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, Vakser IA. Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc Natl Acad Sci USA. 1992;89:2195–2199. doi: 10.1073/pnas.89.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kraulis PJ. Molscript: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallog. 1991;24:946–950. [Google Scholar]
- 83.Merritt EA, Bacon DJ. Raster3D: photo-realistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 84.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins: Struct Funct Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 85.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 86.Little LE, Lau MML, Quon DVK, Fowler AV, Neufeld EF. Proteolytic processing of the a chain of the lysosomal enzyme β-hexosaminidase, in normal human fibroblasts. J Biol Chem. 1988;263:4288–4292. [PubMed] [Google Scholar]
- 87.Neote K, Brown CA, Mahuran DJ, Gravel RA. Translation initiation in the HEXB gene encoding the β-subunit of human β-hexosaminidase. J Biol Chem. 1990;265:20799–20806. [PubMed] [Google Scholar]
- 88.Quon DVK, Proia RL, Fowler AV, Bleibaum J, Neufeld EF. Proteolytic processing of the β-subunit of the lysosomal enzyme, β-hexosaminidase, in normal human fibroblasts. J Biol Chem. 1989;264:3380–3384. [PubMed] [Google Scholar]
- 89.Stirling J, Leung A, Gravel RA, Mahuran DJ. Localization of the pro-sequence within the total deduced primary structure of human β-hexosaminidase B. FEBS Letters. 1988;231:47–50. doi: 10.1016/0014-5793(88)80699-9. [DOI] [PubMed] [Google Scholar]
- 90.Mahuran DJ, Neote K, Klavins MH, Leung A, Gravel RA. Proteolytic processing of human pro-β hexosaminidase: identification of the internal site of hydrolysis that produces the nonidentical βa and βb polypeptides in the mature β-subunit. J Biol Chem. 1988;263:4612–4618. [PubMed] [Google Scholar]
- 91.Mahuran DJ. Characterization of human placental β-hexosaminidase I2: proteolytic processing intermediates of hexosaminidase A. J Biol Chem. 1990;265:6794–6799. [PubMed] [Google Scholar]
- 92.Sonderfeld-Fresko S, Proia RL. Analysis of the glycosylation and phosphorylation of the lysosomal enzyme, β-hexosaminidase B, by site-directed mutagenesis. J Biol Chem. 1989;264:7692–7697. [PubMed] [Google Scholar]
- 93.O’Dowd BF, Cumming D, Gravel RA, Mahuran D. Isolation and characterization of the major glycopeptides from human β-hexosaminidase: their localization within the deduced primary structure of the mature a and b polypeptide chains. Biochemistry. 1988;27:5216–5226. doi: 10.1021/bi00414a041. [DOI] [PubMed] [Google Scholar]
- 94.Schütte CG, Lemm T, Glombitza GJ, Sandhoff K. Complete localization of disulfide bonds in GM2 activator protein. Protein Sci. 1998;7:1039–1045. doi: 10.1002/pro.5560070421. [DOI] [PMC free article] [PubMed] [Google Scholar]