

Abstract
The saturated ring and secondary amine of proline spawn equilibria between pyrrolidine ring puckers as well as peptide bond isomers. These conformational equilibria can be modulated by alterations to the chemical architecture of proline. For example, Cγ in the pyrrolidine ring can be replaced with sulfur, which can be oxidized either stereoselectively to yield diastereomeric S-oxides or completely to yield a sulfone. Here, the thiazolidine ring and peptide bond conformations of 4-thiaproline and its S-oxides were analyzed in an Ac-Xaa-OMe system by using NMR spectroscopy, X-ray crystallography, and hybrid density functional theory. The results indicate that the ring pucker of the S-oxides is governed by the gauche effect, and the prolyl peptide bond conformation is determined by the strength of the n→π* interaction between the amide oxygen and the ester carbonyl group. These findings, which are consistent with those for isologous 4-hydroxyprolines and 4-fluoroprolines, substantiate the importance of electron delocalization in amino-acid conformation.
Keywords: collagen, n→π* interaction, stereoelectronic effect, thiazolidine
Introduction
Proline is unique amongst the proteinogenic amino acids. Only proline has a saturated ring, and only proline is a secondary amine (Fischer 1906). These attributes give rise to two equilibria (Fig. 1). First, the pyrrolidine ring adopts a pucker in which Cγ is out of the plane of the other ring atoms but flips between exo and endo conformers. Secondly, the prolyl peptide bond has a nearly equal preference for its trans (Z) and cis (E) isomers, and interconverts readily between these states.
Fig. 1.

Conformations of proline-like residues. (A) Pyrrolidine ring pucker. (B) Amide-bond isomerization state. lp = lone pair.
In proline residues, the Cγ-endo ring pucker of proline residues is favored marginally over the Cγ-exo ring pucker (Improta et al. 2001; DeRider et al. 2002). Interestingly, substitutions at Cγ modulate the ring-pucker preference. For example, replacement of the pro-R hydrogen with an electron-withdrawing group changes the preferred ring pucker from endo to exo. In contrast, an analogous replacement of the pro-S hydrogen amplifies the preference for the endo ring pucker over the exo ring pucker. These ring-pucker preferences have been explained on the basis of the gauche effect (Eberhardt et al. 1996), which arises when two vicinal atoms bear electronegative substitutents. These substituents prefer to reside gauche to each other (that is, with a dihedral angle of ±60°) to maximize overlap between their σ* orbitals and σ orbitals involving more electropositive substituents.
Other substitutions enable modulation of the conformational properties of proline (Kern et al. 1997; Holmgren et al. 1998; Wittelsberger et al. 2000; Zhu et al. 2002; Melis et al. 2009). (2R)-4-Thiaproline (Thp) is an especially interesting analogue in which sulfur replaces the out-of-plane Cγ of the pyrrolidine ring. Recently, we reported that this analogue is a substrate for human prolyl 4-hydroxylase (P4H), the enzyme that catalyzes the post-translational hydroxylation of proline residues in protocollagen strands (Gorres et al. 2008). In a peptidic substrate, P4H oxidizes Thp to its S-oxide stereoselectively.
The electronic structure of a thiazolidine ring differs from that of a pyrrolidine ring, and its ring conformations could be stabilized by different donor–acceptor orbital interactions. Moreover, stereoselective oxidation of sulfur in Thp can yield one of two diastereomeric S-oxides (2 and 3; Fig. 2). This chemical transformation is analogous to the installation of an electron-withdrawing group at the Cγ of the pyrrolidine ring. Although low-resolution structures of 1–3 are available (Goodman et al. 1970a; Goodman et al. 1970b; Nachtergaele and Anteunis 1980), we sought to determine the atomic resolution structures of 1–3 using X-ray crystallography and/or hybrid density functional theory (DFT). Such a high-resolution picture is indispensable for deciphering the various donor–acceptor orbital interactions that dictate the ring-pucker preferences and accurately mapping the structure of P4H active site.
Fig. 2.

Crystal structures of compounds (A) 2, (B) 3, and (C) 4. Left, thermal ellipsoids; right, ball-and-stick representations.
In an Xaa–Pro peptidic system, a well-known correlation exists between the pyrrolidine ring pucker and the cis–trans equilibrium of the Xaa–Pro amide bond (Fig. 1) (Milner-White et al. 1992). The stabilization of the exo ring pucker over the endo ring pucker increases the population of the trans conformation, while the stabilization of the endo ring pucker over the exo ring pucker increases the population of the cis conformation. In addition to characterizing thiazolidine ring pucker, we searched for a correlation between that ring pucker and the isomerization state of the amide bond.
The Ac-Xaa-OMe model system provides an accurate and simple way to determine the conformational preferences of proline and its analogues (Fig. 1). The use of a methyl ester instead of an amide obviates the intramolecular hydrogen bonding that has been observed in Ac-Pro-NHMe (Matsuzaki and Iitaka 1971; Higashijima et al. 1977; Liang et al. 1992; Benzi et al. 2002). Accordingly, we synthesized Ac-Thp-OMe (1) and its two S-oxides (2, 3). Herein, we report the structure and the conformational preferences of 1–3 using NMR spectroscopy, X-ray crystallography, and ab initio calculations. Our findings inform a wide spectrum of activities in protein chemistry as well as medicinal chemistry, as the thiazoldine ring is a privileged scaffold in drug design (Prabhakar et al. 2006).
Materials and methods
Synthesis
Compounds 1–4 were synthesized by using procedures reported previously (Nachtergaele and Anteunis 1980; Aitken et al. 1997).
Computational analyses
Geometry optimization and frequency calculations were carried on four conformations for each model compound (1–4) at the B3LYP/6-311+G(2d,p) level of theory with Gaussian ’03 (Gaussian 03). The frequency calculations indicated that these conformations were indeed true stationary points on the potential energy surface. Each of these optimized geometries was then analyzed with NBO 5.0 at B3LYP/6-311+G(2d,p) level of theory to determine the contribution of specific donor–acceptor orbital interaction to conformational stability.
X-ray crystallography
The desired compounds were dissolved in hexanes with a minimal amount of EtOAc. Slow evaporation afforded crystals suitable for X-ray analysis after ~2 weeks. X-ray intensity data were collected on a Bruker CCD-1000 diffractometer with Mo Kα (λ = 0.71073 Å) radiation at 105(2) K with the diffractometer to crystal distance of 4.9 cm. Preliminary indexing was carried out to determine the cell constants. This indexing consisted of three series of ω scans at different initial angles with each series consisting of 20 frames at intervals of 0.3° with an exposure time of 10 s per frame. The reflections were indexed using an automated indexing routine built in the SMART program. Data were collected with the full-sphere data collection routine to a resolution of 0.80 Å. The intensity data were then corrected for absorption and Lorentz and polarization effects. Structure solution and refinement were carried out using SHELXTL V.6.10 (Bruker-AXS 2000–2003).
NMR spectroscopy
For the measurement of Ktrans/cis values, each compound (5–10 mg) was dissolved in D2O with enough CD3OD added to solubilize the compound (<20% of total volume). 1H Spectra were acquired and analyzed with the software package NUTS (Acorn NMR). The values of Ktrans/cis were determined from the relative areas of the trans and cis peaks. NOEDIFF experiments were carried out to confirm the proton assignments.
Results and Discussion
The structures of 1–3 were determined at atomic resolution with hybrid density functional theory (1–3) and X-ray crystallography (2, 3; Fig. 2). The atomic coordinates of compounds 1–4 are listed in Tables S2, S4, S10, and S16, respectively, of the Supporting Information. After determining the structure of Ac-Thp-OMe (1) and its S-oxides (2 and 3) at atomic resolution, we compared their ring parameters with those of the analogous proline derivatives: Ac-Pro-OMe (5), Ac-Hyp-OMe (6) (Panasik et al. 1994), and Ac-hyp-OMe (7) (Shoulders 2009) (Table 1; Fig. 3A and 3B), where “Hyp” refers to (2S,4R)-4-hydroxyproline and “hyp” refers to (2S,4S)-4-hydroxyproline. Compound 2, like 6, adopts the exo ring pucker, whereas compound 3, like 7, adopts the endo ring pucker. The ring torsion angles of the S-oxides and the hydroxyprolines are similar. The only notable differences include a larger ring for the S-oxides compared to that for the hydroxyprolines, and a larger φ angle for 2. Hence, we put forth the 4R diastereomer of Thp S-oxide as a surrogate for Hyp, which is the product of the most prevalent post-translational modification in animals (Gorres and Raines 2010).
Table 1.
Ring parameters for compounds 1–7
| Torsion angle | Compound |
|||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 2b | 3b | 4b | 5b | 6b,c | 6b,c | 7b | |
| Cδ–X–Cβ–Cα | −20.62 | 30.66 | −43.12 | 15.18 | −37.22 | 36.36 | 40.21 | −38.27 |
| N–Cδ–X–Cβ | −1.73 | −39.23 | 31.37 | −28.16 | 25.18 | −30.41 | −32.87 | 23.59 |
| Cα–N–Cδ–X | 26.46 | 39.51 | −10.71 | 35.58 | −3.24 | 13.59 | 13.77 | 0.31 |
| Cβ–Cα–N–Cδ | −43.13 | −16.60 | −20.75 | −25.84 | −19.69 | 8.81 | 10.70 | −23.99 |
| X–Cβ–Cα–N | 37.90 | −13.89 | 43.02 | 2.46 | 34.51 | −27.67 | −31.12 | 37.77 |
From DFT calculations.
From X-ray diffraction analysis of the crystalline compounds.
There are two independent molecules in the unit cell.
Fig 3.
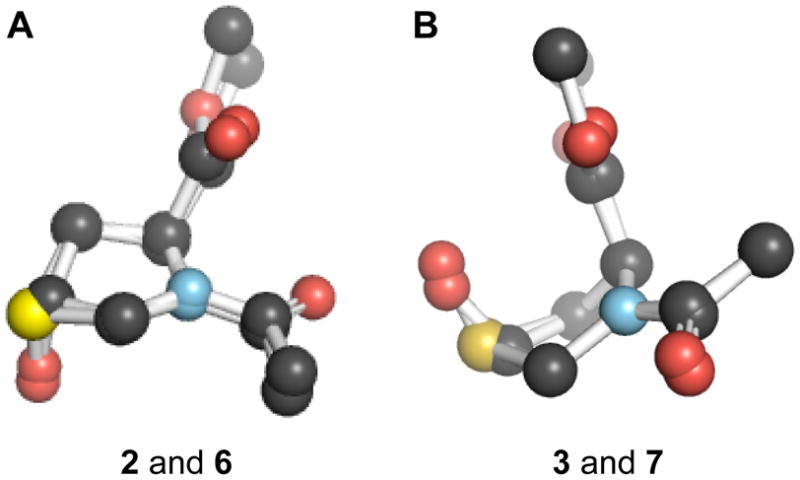
Overlay of isologous crystal structures. (A) Compounds 2 and 6, which are in the Cγ-exo conformation. (B) Compounds 3 and 7, which are in the Cγ-endo conformation.
Hybrid density functional theory (DFT) and Natural Bond Orbital (NBO) analyses (Weinhold 1998; Glendening et al. 2001; Weinhold and Landis 2005) were used to determine the donor–acceptor orbital interactions that dictate the ring-pucker preferences of compounds 1–3. These computational analyses indicate that the most stable conformation in the gas phase for 1 is trans endo, whereas for 2 it is trans exo. In the exo conformation of 2, the antibonding orbital of the S–O bond is positioned for an extensive overlap with the σ(Cβ–H) and σ(Cδ–H) bonding orbitals (Fig. 4A). This extensive overlap of the orbitals cannot be attained in the endo conformation of 2. Similar electronic delocalization takes place in the endo conformation of 3 and cannot occur in its exo conformation. These electronic delocalizations are the primary cause for the stabilization of the exo and endo conformations for 2 and 3. Analogous electronic delocalization has been reported before (Tshuchishashi et al. 1973; Ulshöfer and Podlech 2009).
Fig 4.

Quantum mechanical origin of the stereoelectronic effects in the preferred conformation of compounds 2 and 6. (A) Overlap of σ (or n) and σ* orbitals that gives rise to the gauche effect. (B) Overlap of the lone pair and π* orbital that gives rise to the n→π* interaction; depictions were generated with NBOView 1.1 (Wendt and Weinhold 2001).
A strong correlation exists between the ring pucker and the Ktrans/cis (Scheme 1). This correlation is due to an n→π* interaction in which a lone pair (n) of an amide oxygen (Oi−1) overlap with the antibonding orbital (π*) of Ci=Oi of the subsequent amide (Fig. 4B) (DeRider et al. 2002; Hinderaker and Raines 2003). Such an interaction differentially stabilizes the trans conformation over the cis conformation, as it occurs in the former but not the latter. In the exo pucker, the donor oxygen and the acceptor carbonyl group are closer than in the endo pucker. Thus, compounds with a high population of the exo ring pucker have a stronger n→π* interaction and exhibit a larger Ktrans/cis value. The preference for the exo conformation decreases in the order 2 > 1 > 3, which is parallel to the decrease in the value of Ktrans/cis (Table 2).
Scheme 1.

Correlation between ring pucker and Ktrans/cis in proline and its analogs.
Table 2.
Conformational parameters for compounds 1–4
| Compound | Conformational data | |||||
|---|---|---|---|---|---|---|
| Ktrans/cisa | Ring puckerb | d (Å)b | θ (o)b | Δ (Å)b | Θ (°)b | |
| 1 | 2.8 | endo | ND | ND | ND | ND |
| 2 | 4.0 | exo | 3.01 | 91.9 | 0.025 | 2.88 |
| 3 | 1.6 | endo | ND | ND | −0.054 | −6.28 |
| 4 | 2.8 | exo | 2.99 | 94.8 | 0.018 | 2.14 |
In D2O at 25 °C; values are ±10%.
From X-ray diffraction analysis of the crystalline compound. d is the Oi−1···C′i distance; θ is the Oi−1···C′i =O′i angle; Δ is the C′i···plane(Cαi Oi′ Oi+1) distance; and Θ is the O′i···plane(Cαi Ci′ Oi+1) angle (Choudhary et al. 2009). ND, not determined.
Compound 4 offers an opportunity to test our hypothesis regarding the basis for the correlation of ring pucker and Ktrans/cis (Scheme 1). The sulfur in compound 2 can be oxidized further to form a sulfone. The antibonding orbital of the new S–O bond is positioned to stabilize the endo conformation of 4. An increase in the population of the endo conformation should lower the value of Ktrans/cis. The Ktrans/cis of 4 is indeed lower than that of 2 and equal to that of 1 (Table 2). This finding aligns compounds 2–4 with analogous 4-fluoroprolines, in which the 4R diastereomer has a high Ktrans/cis value, the 4S diasteomer has a low value, and the unsubstituted and doubly substituted derivatives have equivalent values that are intermediate (Shoulders et al. 2009). These Ktrans/cis values for eight compounds in two distinct classes (Fig. 1: X = S, R1 = lp or O, R2 = lp or O; X = C, R1 = H or F, R2 = H or F) are consistent with Scheme 1 and its underlying quantum mechanical origin.
The crystal structure of compound 3 offers additional insight into the nature of the forces that dictate the conformational preferences of these molecules. In crystal structures, carbonyl groups often form short contacts with electron-pair donor groups in a manner reminiscent of the nucleophilic attack along the Bürgi–Dunitz trajectory. The origin of these short contacts has been attributed to dipole–dipole interactions (Paulini et al. 2005; Fischer et al. 2008). Recently, we reported that such short contacts arise instead from n→π* electronic delocalization (Choudhary and Raines 2009; Choudhary et al. 2009). In the crystal structure of 3, a short contact exists between the sulfoxide oxygen and the ester carbonyl group. This short contact cannot arise from a dipole–dipole interaction, and the two interacting dipoles can be in a repulsive orientation. One can, however, envision an n→π* electronic delocalization from a lone pair (n) of the sulfoxide oxygen into the antibonding orbital (π*) of the ester carbonyl group (Lesarri et al. 2005). This explanation is consistent with the observed pyramidalization of the carbonyl carbon (Table 2), which is an unambiguous indication of covalency (Choudhary and Raines 2009; Choudhary et al. 2009).
Conclusions
We have examined the conformational preferences of compounds 1–4 using NMR spectroscopy, X-ray crystallography, and ab initio calculations. Our data indicate that whereas 1, 3, and 4 prefer the endo conformation and have a low value of Ktrans/cis, 2 prefers the exo conformation and has a high value of Ktrans/cis. These findings on thiazolidine rings are in accord with previous findings on pyrrolidine rings (Scheme 1). The conformational preferences in both ring systems have a tractable quantum mechanical origin.
Supplementary Material
Acknowledgments
We thank C. N. Bradford, B. R. Caes, G. A. Ellis, and K. L. Gorres for contributive discussions. This work was supported by grant R01 AR044276 (NIH). KHP was supported by a Mary Shine Peterson award.
References
- Aitken RA, Mesher STE, Ross FC, Ryan BM. Effect of added benzoic acid on the phase-transfer catalysed permanganate oxidation of organosulfur compounds. Synthesis. 1997;7:787–791. [Google Scholar]
- Benzi C, Improta R, Scalmani G, Barone V. Quantum mechanical study of the conformational behavior of proline and 4R-hydroxyproline dipeptide analogues in vacuum and in aqueous solution. J Comput Chem. 2002;23:341–350. doi: 10.1002/jcc.10015. [DOI] [PubMed] [Google Scholar]
- Choudhary A, Gandla D, Krow GR, Raines RT. Nature of amide carbonyl–carbonyl interactions in proteins. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A, Raines RT. A donor–acceptor perspective on carbonyl–carbonyl interactions in proteins. In: Lebl M, editor. Breaking Away: The Proceedings of the Twenty-First American Peptide Symposium. Prompt Scientific Publishing; San Diego, CA: 2009. pp. 347–349. [Google Scholar]
- DeRider ML, Wilkens SJ, Waddell MJ, Bretscher LE, Weinhold F, Raines RT, Markley JL. Collagen stability: Insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations. J Am Chem Soc. 2002;124:2497–2505. doi: 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- Eberhardt ES, Panasik N, Jr, Raines RT. Inductive effects on the energetics of prolyl peptide bond isomerization: Implications for collagen folding and stability. J Am Chem Soc. 1996;118:12261–12266. doi: 10.1021/ja9623119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. Untersuchungen über Aminosaüren, Polypeptide und Proteine. Ber Deutsch Chem Ges. 1906;39:530–610. [Google Scholar]
- Fischer FR, Wood PA, Allen FH, Diederich F. Orthogonal dipolar interactions between amide carbonyl groups. Proc Natl Acad Sci USA. 2008;105:17290–17294. doi: 10.1073/pnas.0806129105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. NBO 5.0. Theoretical Chemistry Institute, University of Wisconsin–Madison; Madison, WI: 2001. [Google Scholar]
- Goodman M, Niu GC-C, Su K-c. Conformational aspects of polypeptide structure. XXXII. Helical poly[(S)-thiazolidine-4-carboxylic acid]. Theoretical results. J Am Chem Soc. 1970a;92:5219–5220. doi: 10.1021/ja00720a038. [DOI] [PubMed] [Google Scholar]
- Goodman M, Su K-c, Niu GC-C. Conformational aspects of polypeptide structure. XXXII. Helical poly[(S)-thiazolidine-4-carboxylic acid]. Experimental results. J Am Chem Soc. 1970b;92:5220–5222. doi: 10.1021/ja00720a039. [DOI] [PubMed] [Google Scholar]
- Gorres KL, Edupuganti R, Krow GR, Raines RT. Conformational preferences of substrates for human prolyl 4-hydroxylase. Biochemistry. 2008;47:9447–9855. doi: 10.1021/bi8009373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima T, Tasumi M, Miyazawa T. 1H Nuclear magnetic resonance studies of N-acetyl-L-proline N-methylamide. Molecular conformations, hydrogen bondings, and thermodynamic quantities in various solvents. Biopolymers. 1977;16:1259–1270. doi: 10.1002/bip.1977.360160608. [DOI] [PubMed] [Google Scholar]
- Hinderaker MP, Raines RT. An electronic effect on protein structure. Protein Sci. 2003;12:1188–1194. doi: 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren SK, Taylor KM, Bretscher LE, Raines RT. Code for collagen’s stability deciphered. Nature. 1998;392:666–666. doi: 10.1038/33573. [DOI] [PubMed] [Google Scholar]
- Improta R, Benzi C, Barone V. Understanding the role of stereoelectronic effects in determining collagen stability. 1. A quantum mechanical study of proline, hydroxyproline, and fluoroproline dipeptide analogues in aqueous solution. J Am Chem Soc. 2001;123:12568–12577. doi: 10.1021/ja010599i. [DOI] [PubMed] [Google Scholar]
- Kern D, Schutkowski M, Drakenberg T. Rotational barriers of cis/trans isomerization of proline analogues and their catalysis by cyclophilin. J Am Chem Soc. 1997;119:8403–8408. [Google Scholar]
- Lesarri A, Cocinero EJ, López JC, Alonso JL. Shape of 4S- and 4R-hydroxyproline in gas phase. J Am Chem Soc. 2005;127:2572–2579. doi: 10.1021/ja045955m. [DOI] [PubMed] [Google Scholar]
- Liang G-B, Rito CJ, Gellman SH. Variations in the turn-forming characteristics of N-acyl proline units. Biopolymers. 1992;32:293–301. doi: 10.1002/bip.360320309. [DOI] [PubMed] [Google Scholar]
- Matsuzaki T, Iitaka Y. The crystal structure of acetyl-L-proline-N-methylamide. Acta Crystallogr. 1971;B27:507–516. [Google Scholar]
- Melis C, Bussi G, Lummis SCR, Molteni C. Trans–cis switching mechanisms in proline analogues and their relevance for the gating of the 5-HT3 receptor. J Phys Chem B. 2009;113:12148–12153. doi: 10.1021/jp9046962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner-White JE, Bell LH, Maccallum PH. Pyrrolidine ring puckering in cis and trans-proline residues in proteins and polypeptides. J Mol Biol. 1992;228:725–734. doi: 10.1016/0022-2836(92)90859-i. [DOI] [PubMed] [Google Scholar]
- Nachtergaele WA, Anteunis MJO. Ring conformational aspects of cis- and trans-N-acetyl-4-carbomethoxy-1,3-thiazolidine-1-oxide. Bull Soc Chim Belg. 1980;89:749–758. [Google Scholar]
- Panasik N, Jr, Eberhardt ES, Edison AS, Powell DR, Raines RT. Inductive effects on the structure of proline residues. Int J Pept Protein Res. 1994;44:262–269. doi: 10.1111/j.1399-3011.1994.tb00169.x. [DOI] [PubMed] [Google Scholar]
- Paulini R, Müller K, Diederich F. Orthogonal multipolar interactions in structural chemistry and biology. Angew Chem Int Ed. 2005;44:1788–1805. doi: 10.1002/anie.200462213. [DOI] [PubMed] [Google Scholar]
- Prabhakar YS, Solomon VR, Gupta MK, Katti SB. QSAR studies on thiazolidines: A biologically priviliged scaffold. Top Heterocycl Chem. 2006;4:161–249. [Google Scholar]
- Shoulders MD. PhD thesis. University of Wisconsin; Madison: 2009. [Google Scholar]
- Shoulders MD, Kamer KJ, Raines RT. Origin of the stability conferred upon collagen by fluorination. Bioorg Med Chem Lett. 2009;19:3859–3962. doi: 10.1016/j.bmcl.2009.03.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tshuchishashi G-i, Mitamura S, Inoue S, Ogura K. Asymmetric synthesis using α,β-unsaturated sulfoxides. A highly selective Michael reaction. Tetrahedron Lett. 1973:323–326. [Google Scholar]
- Ulshöfer R, Podlech J. Stereoelectronic effects in vinyl sulfoxides. J Am Chem Soc. 2009;131:16618–16619. doi: 10.1021/ja904354g. [DOI] [PubMed] [Google Scholar]
- Weinhold F. Natural bond orbital methods. In: Schleyer PvR, Allinger NL, Clark T, Gasteiger J, Kollman PA, Shaefer HF, III, et al., editors. Encyclopedia of Computational Chemistry. Vol. 3. John Wiley & Sons; Chichester, UK: 1998. pp. 1792–1811. [Google Scholar]
- Weinhold F, Landis CR. Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective. Cambridge University Press; Cambridge, UK: 2005. [Google Scholar]
- Wendt M, Weinhold F. NBOView 1.1. Theoretical Chemistry Institute, University of Wisconsin–Madison; Madison, WI: 2001. [Google Scholar]
- Wittelsberger A, Keller M, Scarpellino L, Patiny L, Orbea-Acha H, Mutter M. Pseudoprolines: Targeting a cis conformation in a mimetic of the gp120 V3 loop of HIV-1. Angew Chem Int Ed. 2000;39:1111–1115. doi: 10.1002/(sici)1521-3773(20000317)39:6<1111::aid-anie1111>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Zhu W, Gincherman Y, Docherty P, Spilling CD, Becker DF. Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch Biochem Biophys. 2002;408:131–136. doi: 10.1016/s0003-9861(02)00535-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.