

Abstract
Hyperbaric oxygen (HBO2) therapy is approved by the FDA for limited clinical indications but is reported to produce pain relief in several chronic pain conditions. However, there have been no studies to explain this apparent analgesic effect of HBO2. Research conducted in our laboratory demonstrates that four daily 60-min HBO2 treatments at 3.5 ATA induced an unparalleled antinociceptive response that consists of 1) an early phase that lasted at least six hours after the HBO2 treatment before dissipating; and 2) a late phase that emerged about 18 hours after the early phase and lasted for up to three weeks. The early phase was sensitive to antagonism by acutely intracerebroventricular (i.c.v.)-administered opioid antagonist naltrexone and the nitric oxide synthase (NOS)-inhibitor L-NAME. The late phase was inhibited by treatment with i.c.v. naltrexone or L-NAME during the four HBO2 treatments but was not antagonized by either naltrexone or L-NAME following acute pretreatment two weeks after HBO2 treatment. These experimental results implicate a novel mechanism that is activated by HBO2, resulting in an antinociceptive response of unusually long duration that is of potential interest in the clinical management of pain.
Perspective
Hyperbaric oxygen treatment of mice can induce a two-phase antinociceptive response of unusually long duration. Nitric oxide and opioid receptors appear to initiate or mediate both phases of the antinociceptive response. Further elucidation of the underlying mechanism may potentially identify molecular targets that cause long-lasting activation of endogenous analgesic systems.
Keywords: Hyperbaric oxygen, antinociception, nitric oxide, opioid receptors, mice
Introduction
Hyperbaric oxygen (HBO2) therapy is the clinical application of 100% oxygen at higher than atmospheric pressures for limited periods of time (60–90 min) to achieve therapeutic outcomes. HBO2 treatment is approved by the U.S. Food and Drug Administration (FDA) for only a limited number of conditions, which include primarily decompression sickness (the "bends"), carbon monoxide poisoning, cerebral arterial gas embolus, osteoradionecrosis and clostridial myonecrosis (gas gangrene)9.
However, there are reports that HBO2 treatment can also be effective in a number of non-approved conditions. Among these conditions that are reportedly responsive to HBO2 therapy are a variety of chronic pains, for which intermittent exposure to HBO2 reportedly causes a long-lasting analgesic effect. Patients suffering from complex regional pain syndrome (reflex sympathetic dystrophy syndrome) experienced less pain following HBO2 therapy14,21,25. Significant pain reduction was also reported in patients with generalized allodynia/hyperalgesia as a consequence of fibromyalgia syndrome (FMS)32. Patients suffering from migraine headache or cluster headache also reported pain relief following HBO2 therapy6,17,31. Pain associated with radiotherapy of cancer has also been reported to be alleviated by HBO2 therapy4,13,18.
Animal studies related to HBO2 treatment of pain have been limited, and prior studies have focused largely on the peripheral antiinflammatory effect rather than a central antinociceptive effect of HBO2 22,28. More recently, inflammatory pain induced by carrageenan in rats was evaluated after HBO2 exposure at 2.4 ATA for 90 min29. HBO2 treatment was found to reduce inflammation (as determined by reduced paw swelling) as well as mechanical hypersensitivity (as determined by increased threshold for paw withdrawal)30. The ability of HBO2 treatment to reduce inflammation and pain was comparable to that of acetylsalicylic acid treatment29. It is notable that there was a temporal dissociation of the antiinflammatory and antinociceptive effects. The antiinflammatory effect was almost immediate after carrageenan was administered to HBO2-treated rats, while the antinociceptive effect did not manifest itself until nearly two hours after the HBO2 treatment.
We recently reported that a 60-min exposure to 100% oxygen (O2) at 3.5 absolute atmospheres (ATA) produced an antinociceptive effect of at least 90 min duration that involves opioid and nitric oxide mechanisms33. Mice exposed to HBO2 for 60 min were returned to room air, after which antinociception was assessed. The antinociceptive effect was significantly attenuated by intracerebroventricular (i.c.v.) pretreatment with two inhibitors of nitric oxide synthase (NOS) enzyme, the non-selective inhibitor NG-nitro-L-arginine methyl ester (L-NAME) and the neuronally-selective inhibitor S-methyl-L-thiocitrulline (SMTC). The endothelial-selective NOS-inhibitor N5-(1-iminoethyl)-L-ornithine (L-NIO) administered i.p. just prior to the start of HBO2 treatment had no effect on the antinociceptive response. The antinociceptive effect at 90 min was also markedly antagonized by i.p. pretreatment with the opioid receptor blocker naltrexone. Confirming an involvement of endogenous opioid peptides in the antinociception, the effect was found to be sensitive to antagonism by i.c.v. pretreatment with a rabbit antiserum against rat dynorphin but not by antisera against either β-endorphin or methionine-enkephalin. The prolonged antinociceptive effect at 90 min after HBO2-induced treatment was also significantly attenuated by naltrexone — but not L-NAME — administered 75 min following HBO2 treatment but 15 min prior to nociceptive testing. Based on these experimental findings, we concluded that the HBO2-induced antinociceptive effect involves both NO and opioid mechanisms in the brain and is consistent with our hypothesis that HBO2 can stimulate an NO-dependent neuronal release of dynorphin, which, in turn, activates κ opioid receptors that mediate antinociception.
The present study grew out of an attempt to determine whether repeated sessions of HBO2 treatment might prolong the duration of the HBO2-induced antinociception and to pharmacologically characterize the antinociceptive response.
Materials and Methods
Animals
Male NIH Swiss mice, weighing 18–22 g, were purchased from Harlan Laboratories (Indianapolis, IN) and used in this study, which was approved by an institutional animal care and use committee with post-approval review and carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23, revised 1996). All measures to minimize pain or discomfort were taken by the investigators.
Mice were housed in the AAALAC-accredited Wegner Hall Vivarium with access to food and water ad libitum. The facility was maintained on a 12-h light:dark cycle (lights on 0700–1900 h) under standard conditions (22 ± 1°C room temperature, 33% humidity). Mice were kept in the holding room for at least four days after arrival in the facility for acclimation prior to experimentation.
Exposure to Hyperbaric Oxygen (HBO2)
Cages of five mice each were placed in a B-11 research hyperbaric chamber (Reimers Systems, Inc., Lorton, VA) as previously described20. The chamber was ventilated with 100% oxygen (O2), U.S.P. (A-L Compressed Gases, Inc., Spokane, WA) at a flow rate of 20 L/min to minimize carbon dioxide accumulation. The pressure within the cylindrical clear acrylic chamber (27.9 cm diameter × 55.9 cm L) was increased at a rate of 1.0 ATA/min to the desired pressure (3.5 ATA) and maintained for 60 min. The mice were allowed to breathe spontaneously during HBO2 treatment. After completion of the HBO2 exposure, mice were then decompressed at a rate of 1.0 ATA/min. Control groups of mice were exposed to compressed air (A-L Compressed Gases) circulated through the chamber at 1.0 ATA and maintained for 60 min. Decompression occurred as described above.
Mice were subjected to HBO2 treatment at 3.5 ATA for 60 min between 1000 and 1100 hr for four consecutive days. At different time intervals following the fourth HBO2 session, different groups of mice were assessed for antinociceptive responsiveness, as described in the following section, due to loss of sensitivity to acetic acid for several days following injection.
Antinociceptive Testing
Antinociceptive responsiveness was assessed using the abdominal constriction test as previously described8. At varying time intervals following HBO2 treatment, different groups of mice were treated i.p. with 0.1 ml per 10 g body weight of 0.6% glacial acetic acid and placed into an open clear Plexiglas® chamber (35 cm L × 20 cm W × 15 cm H). Exactly five min later, the number of abdominal constrictions—lengthwise stretches of the torso with concave arching of the back—in each animal was counted for six-min period for each treatment group. Multiple raters were used for some but not all experiments; at least one of the raters was blinded to the drug treatment. The control reference group was exposed to room air. The degree of antinociception (inhibition of abdominal constrictions) produced in various treatment groups of mice was calculated as:
A mean number of abdominal constrictions was determined for control mice and was used as the basis for determining the % antinociceptive response for each experimental mouse at a specific time point or in a particular pretreatment group. In the time course experiment, different groups of at least 6 mice were tested at each time interval following HBO2 treatment because the i.p. injection desensitizes for up to five days the sensory receptors that initiate the abdominal constrictions.
Drugs
The following drugs were used in this research: naltrexone hydrochloride (Tocris Bioscience, Ellisville, MO); and L-NG-nitro arginine methyl ester (L-NAME) (Research Biochemicals International, Natick, MA). Naltrexone and L-NAME were freshly prepared daily in sterile 0.9% physiological saline solution and administered acutely or chronically into the lateral cerebral ventricle. Control animals received vehicle (physiological saline solution) via the same route.
Acute Intracerebroventricular Microinjection Procedure
Acute i.c.v. microinjections of 1.0 µg naltrexone or L-NAME were made using the microinjection technique of Haley and McCormick10. Briefly, mice were anesthetized with isoflurane, U.S.P. (Abbott Laboratories, N. Chicago, IL). A short incision was made along the midline of the scalp using a scalpel, and the skin was pulled back to expose the calvarium. The i.c.v. microinjection was made using a 10-µl microsyringe (Hamilton, Reno, NV) with a 26-gauge cemented needle. The microsyringe was held vertically by hand at a point on the calvarium 2.0 mm lateral and 1.0 mm caudal from bregma to a depth of −2.0 mm from the skull surface. Penetration was controlled by a large-bore needle through which the microsyringe needle was inserted; the hypodermic needle which served as a collar to limit penetration of the microsyringe needle to 2.0 mm. A volume of 4.0 µl of drug solution was delivered directly into the lateral cerebral ventricle over 30 sec.
Chronic Intracerebroventricular Infusion Procedure
For chronic i.c.v. delivery of 1.0 µg/day naltrexone and L-NAME during the four-day HBO2 treatment, mice were anaesthetized with isoflurane and brain infusion cannulae (Alza, Cupertino, CA) were implanted into the lateral cerebral ventricle. Each brain infusion cannula was connected to a dorsally subcutaneously implanted Alzet® osmotic minipump model 2001 (200-µl reservoir, 1.0±0.04 µl/hr pumping rate, delivers for up to one week) with polyvinylchloride tubing. The concentration of naltrexone and L-NAME stored in each minipump was 0.0416 µg/µl. The osmotic minipumps were implanted prior to the first HBO2 treatment and were removed immediately following the fourth HBO2 treatment.
Statistical Analysis of Data
Percent changes in antinociception were arc-sine-transformed to normalize the distribution of percentages prior to statistical analysis. A one-way ANOVA and post-hoc Bonferroni’s multiple comparison test was used to compare HBO2-induced antinociception various pretreatment groups.
Results
Time Course of HBO2-Induced Antinociception
Fig. 1 shows the time course of the biphasic antinociceptive response following four daily HBO2 treatments at 3.5 ATA of 60 min each. HBO2 treatment produced a robust antinociceptive response (approximately 90–95% suppression of abdominal constrictions) that lasted for up to six hours following the last HBO2 session.
Fig. 1.

Time course of antinociceptive response to four daily 60-min HBO2 treatment : A, an early-phase response that lasts at least 6 hr after HBO2 treatment and dissipating by 12 hr; and B, a late-phase antinociceptive response that emerges 24 hr (1D) after HBO2 treatment and lasts for up to 21 days (21D) following HBO2 treatment. The data are expressed as the mean ± S.E.M. of at least 6 mice per group.
When antinociceptive testing was conducted after additional time intervals, it was discovered that the antinociceptive effect of HBO2 had completely dissipated by 12 hr but began to re-emerge 24 hr after the last HBO2 treatment. This delayed antinociceptive effect — now called the late-phase response to differentiate it from the 6-hr-long early-phase effect — proved to be equal in antinociceptive intensity to the earlier response and persisted for 14 days after the last HBO2 session. At 21 days after the last session, the antinociceptive response was still at 40%.
Influence of Acute Naltrexone and L-NAME Pretreatment on the HBO2-Induced Early-Phase Antinociceptive Response
Fig. 2 shows the influence of naltrexone and L-NAME pretreatments three hr after the last HBO2 treatment and 30 min prior to the glacial acetic acid challenge. I.c.v. pretreatment with 1.0 µg naltrexone caused a 50% reduction in the magnitude of the HBO2-induced antinociceptive effect. Similar i.c.v. pretreatment with 1.0 µg L-NAME reduced the HBO2-induced response by 40%.
Fig. 2.
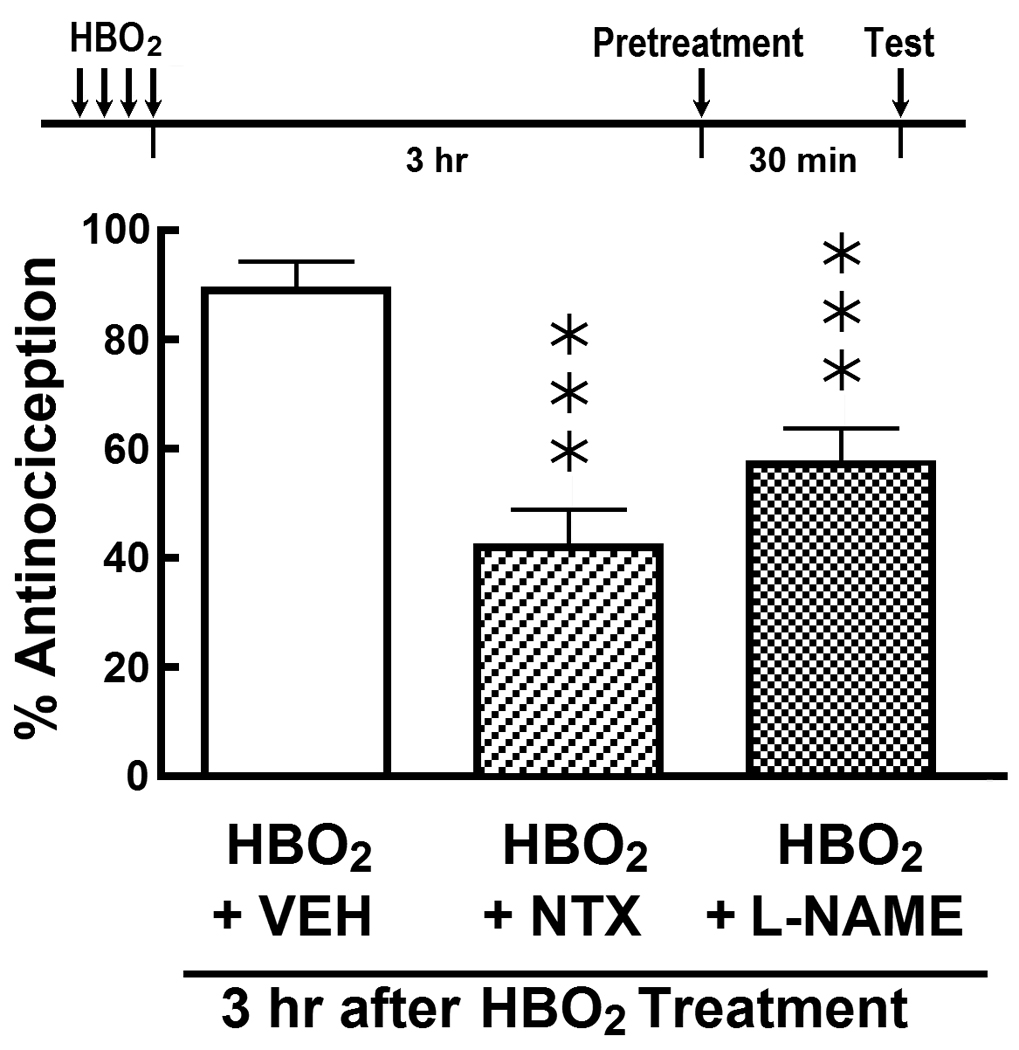
Influence of acute i.c.v. pretreatment with vehicle (VEH), 1.0 µg naltrexone (NTX) and 1.0 µg L-NAME on the HBO2-induced early-phase antinociceptive response 3 hr after the fourth HBO2 treatment. The data are expressed as the mean ± S.E.M. of 8–12 mice per group. Significance of difference: ***, p < 0.001, compared to HBO2 control group (one-way ANOVA and post-hoc Bonferroni’s multiple comparison test).
Influence of Acute Naltrexone and L-NAME Pretreatment on the HBO2-Induced Late-Phase Antinociceptive Response
We then assessed the influence of nalrexone and L-NAME on the late-phase antinociceptive response following the four-day HBO2 treatment. Fig. 3 shows the influence of i.c.v.-administered naltrexone (1.0 µg) and L-NAME (1.0 µg) 14 days after the last HBO2 treatment and 30 min prior to the glacial acetic acid challenge. Neither pretreatment had any effect on the magnitude of the HBO2-induced late-phase antinociceptive effect.
Fig. 3.
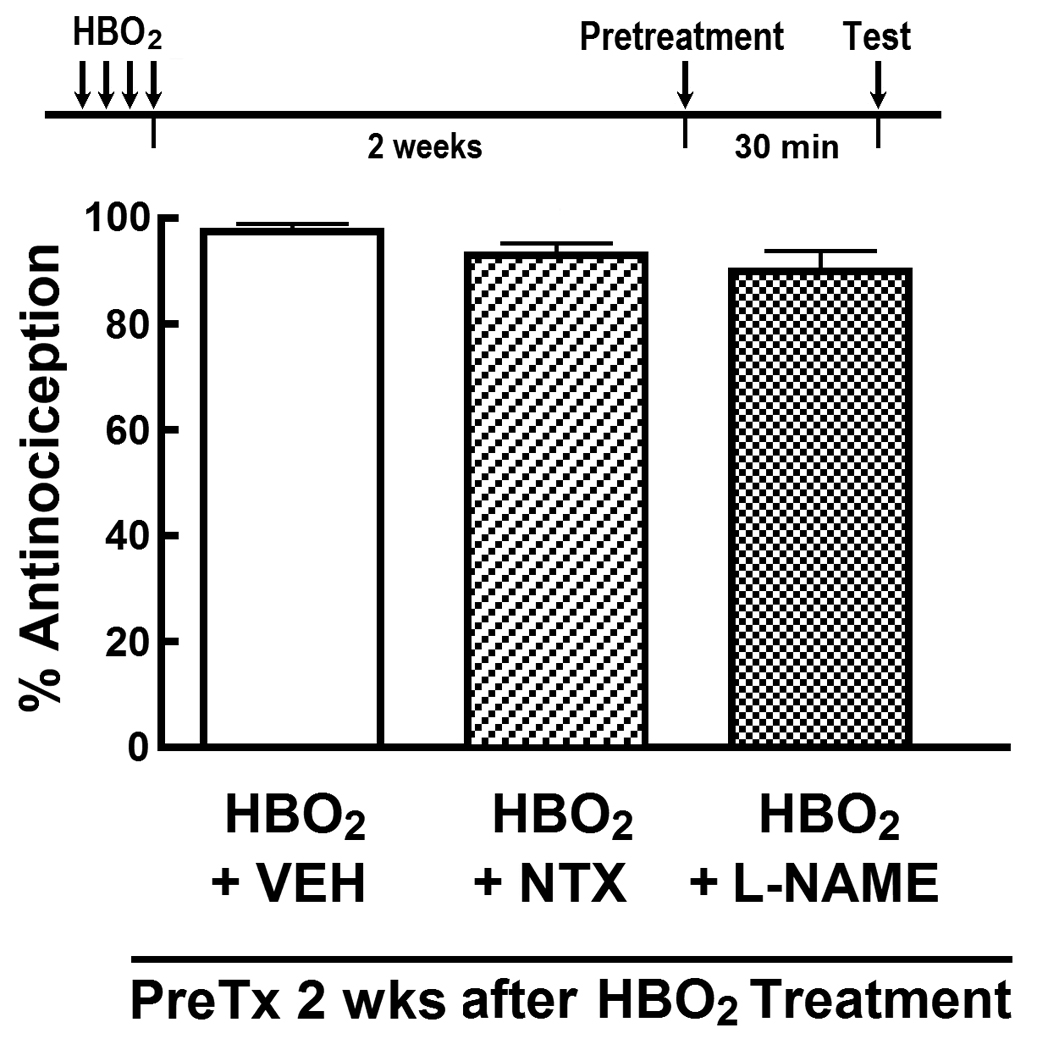
Influence of acute i.c.v. pretreatment with vehicle (VEH), 1.0 µg naltrexone (NTX) and 1.0 µg L-NAME on the HBO2-induced late-phase antinociceptive response 14 days after the fourth HBO2 treatment. The data are expressed as the mean ± S.E.M. of 8–12 mice per group. Significance of difference: ***, p < 0.001, compared to HBO2 control group (one-way ANOVA and post-hoc Bonferroni’s multiple comparison test).
Influence of Continuous Naltrexone and L-NAME Pretreatment on the HBO2-Induced Late-Phase Antinociceptive Response
Finally, we wanted to determine whether opioid receptor blockade or inhibition of NO production during HBO2 exposure had any effect on development of the late-phase antinociception. Naltrexone (1.0 µg/day) and L-NAME (1.0 µg/day) were continuously delivered into the lateral cerebral ventricle using osmotic minipumps during the four-day period in which mice were exposed to HBO2 for 60 min each day. Immediately following the fourth day of HBO2 treatment, the osmotic minipumps were removed and mice were returned to the vivarium. Two weeks later, the mice were tested for nociceptive responsiveness to the glacial acetic acid. Fig. 4 shows that both naltrexone and L-NAME treatments during HBO2 exposure significantly reduced the intensity of the HBO2-induced late-phase antinociceptive response.
Fig. 4.
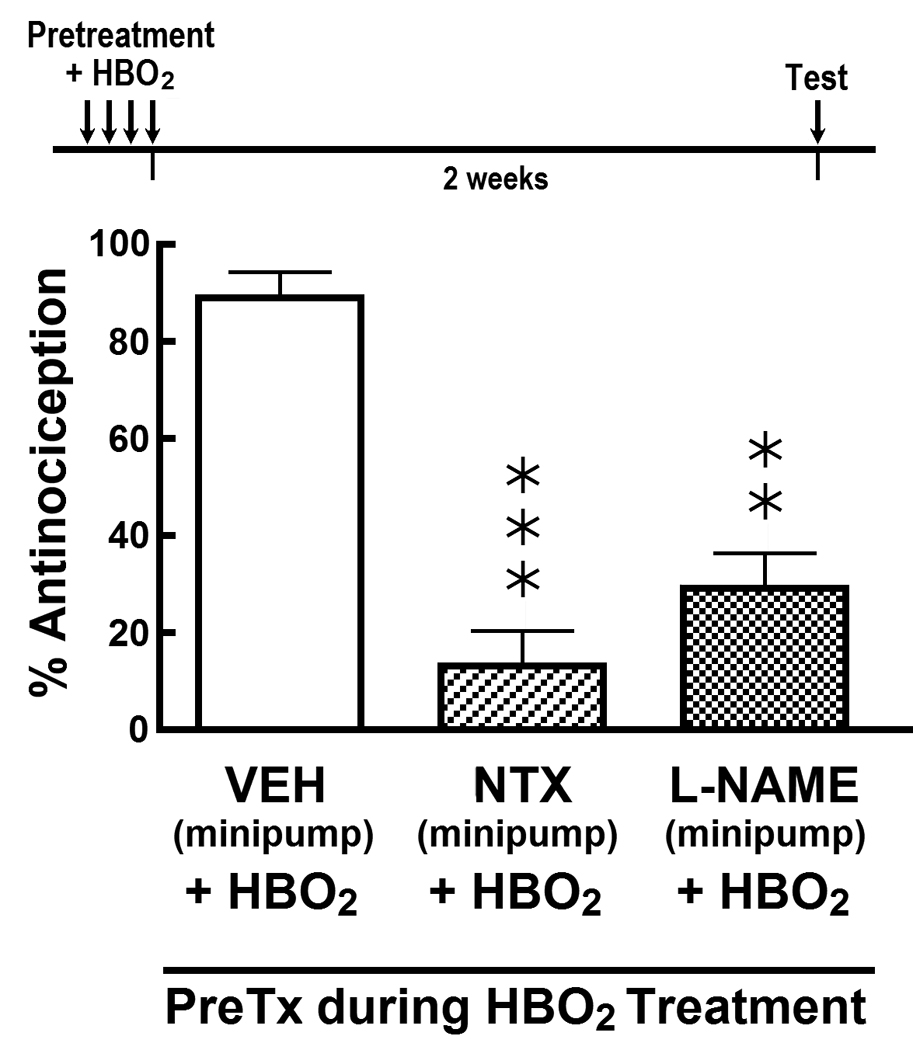
Influence of continuously delivered i.c.v. vehicle (VEH), 1.0 µg/day naltrexone (NTX) and 1.0 µg/day L-NAME on the HBO2-induced late-phase antinociceptive response 14 days after the fourth HBO2 treatment. Osmotic minipumps containing VEH, NTX or L-NAME were activated prior to the first HBO2 treatment and removed immediately after the fourth HBO2 treatment. The data are expressed as the mean ± S.E.M. of 8–12 mice per group. Significance of difference: **, p < 0.01 and ***, p < 0.001, compared to HBO2 control group (one-way ANOVA and post-hoc Bonferroni’s multiple comparison test).
Discussion
HBO2 and Nitric Oxide Function
During HBO2 therapy in humans breathing room air (2% O2) at 1.0 ATA, the alveolar pO2 (pAO2) is approximately 102 mm Hg; breathing 100% oxygen increases the pAO2 to 673 mm Hg. During HBO2 therapy, the pAO2 increases rapidly as the pressure in the hyperbaric chamber increases. At 2.0 ATA, the pAO2 rises to 1433 mm Hg; at 2.5 ATA, it is about 1813 mm Hg, a 17-fold increase as compared to breathing air at 1.0 ATA12. These partial pressures are attained within minutes, depending upon the rate of compression. Thus, it is expected that the tissue O2 concentration throughout the body will correspondingly increase in a relatively rapid manner.
This rapid increase has been shown in vivo in the rat brain24. As monitored by NO- and O2-specific electrodes implanted in the cerebral cortex of ketamine/xylazineanesthesized rats, HBO2 exposures at 2.0–2.8 ATA rapidly increased the pO2 and NO within ~2 minutes. The NO concentration increased from 36 nM at 1.0 ATA to 641 nM at 2.0 ATA and 692 nM at 2.8 ATA, the latter about a 19-fold increase. So there appears to be a clear relationship between pO2 and tissue NO concentration in the brain.
An in vivo microdialysis study in rats has also shown that HBO2 treatment at 3.0 ATA for 120 min elevated NO metabolites in the hippocampus and striatum7. In a more recent study19, a 60-min HBO2 treatment caused site-specific changes in levels of the stable NO metabolites (NOx) — nitrite (NO2 −) and nitrate (NO3 −) — in rat brain regions and spinal cord. Exposure to 100% O2 alone generally reduced regional brain and spinal cord levels of nitrite and nitrate, while exposure to compressed air at 3.5 ATA had little effect on tissue levels of NO metabolites. However, the combination of 100% O2 and pressure (i.e., HBO2) generally increased tissue levels of both nitrite and nitrate, which serve as an index of increased NO production in selected brain regions, most notably in the striatum, brainstem, cerebellum and spinal cord. Thus, these data suggest that at hyperbaric pressures, molecular oxygen can be converted to NO.
There is always the potential that the effects observed in this study may be associated to O2 toxicity. Rats exposed to 5–6 ATA HBO2 have been shown to exhibit EEG seizure patterns1,2,5. The onset of these seizures were delayed by the nNOS inhibitor, 7-nitroindazole, suggesting the involvement of NO and/or oxygen radicals. However, exposure for 75 min at 4 ATA did not demonstrate any seizure patterns5. What these data suggest that, under our experimental condition of 3.5 ATA, our results do not appear to reflect a toxic effect of HBO2.
Recent research has also shown that mice can tolerate as much as 6.0 ATA HBO2 and exhibit seizures only if there is preconditioning by 60-min twice daily HBO2 treatments at 2.5 ATA for 3 consecutive days16. Preconditioning led to an increase in levels of protein and mRNA of eNOS and nNOS in the hippocampus and hypothalamus. Our 4 daily 60-min HBO2 treatments in the present study did not appear to sensitize the mice to oxygen-induced toxicity.
Antinociceptive Responsiveness of Rats to HBO2 Treatment
All of the discussion above supports our recent report that antinociception occurs very rapidly, that is, within 5 minutes of HBO2 exposure at 3.5 ATA20,34. If the HBO2 exposure is continued for 60 min, the antinociception is extended beyond the duration of the HBO2 exposure for at least 90 min and at 150 min about 40% of the antinociception still remained33. The exact mechanism by which this HBO2-induced antinociception is prolonged is not clear at this time.
Interestingly, in the present study, administration of the 60-min HBO2 treatment for four consecutive days extends the duration of antinociception during the early-phase response four-fold to 6 hrs. It is interesting to note here that the duration of this antinociceptive effect (early-phase antinociception) is a multiple of the 90-min effect observed after a single HBO2 exposure. This may suggest that HBO2 treatment induces some type of up-regulation mechanism, which has been reported recently by others3,15,23.
Even more interesting is that after four daily 60-min exposures of HBO2, a second antinociception period (i.e., the late-phase response) is initiated 24 hours after the last HBO2 exposure. The level of antinociception increased over four days and peaked on the fifth day after the last HBO2 session. This peak level of antinociception equaled that of the early-phase response in magnitude and was maintained for another 9 days, at which time it gradually decreased to about 45% antinociception by three weeks after HBO2 treatment. What this suggests is that an antinociceptive pathway is clearly being up-regulated in the absence of HBO2 exposure. That would suggest that activation and/or inhibition of particular genes in the central nervous system, meaning that changes in gene regulation had possibly occurred under HBO2 treatment.
Recently it was reported that rats treated daily for five days with HBO2 for 60 min at 3.5 ATA showed distinct changes in gene function in the hippocampal CA1 region as assessed by DNA microarray analysis11. These HBO2-treated animals were subjected to forebrain ischemia. Ischemic neuronal damage in the hippocampal CA1 was determined and showed that the prior HBO2 treatment decreased the amount of neuronal damage. Seven genes with their respective proteins presumed to be related to the neuroprotective effect of HBO2 were found to be up-regulated. The peak gene expression occurred generally at 12 hrs and the Western blot analysis of their respective proteins peaked at 24 hrs. If gene regulation is involved in the late-phase antinociceptive response in the present study, then it would appear that the half-life of the proteins involved in the antinociception observed is much longer than in the previous study33. This requires a closer examination of the genes and proteins that might possibly be involved in the antinociceptive response.
Characterization of the HBO2-Induced Early-Phase Antinociceptive Response
The present results show that, after four daily 60-min HBO2 treatments, the early-phase antinociceptive effect is sensitive to antagonism by naltrexone administered i.c.v. three hrs after the HBO2 treatment. This suggests that the early-phase antinociceptive response was mediated by opioid receptors and is in agreement with our previous finding that naltrexone antagonized the antinociceptive effect demonstrated after a single 60-min exposure to HBO2 33. It is highly unlikely that O2 directly interacts with opioid receptors, which are not known to contain a heme group in their molecular structure. In addition, there is no evidence that hyperoxemia leads to direct activation of opioid receptors. The early-phase antinociception was also antagonized by L-NAME, a NOS-inhibitor. This also agrees with our data on antagonism by L-NAME of the 90-min antinociceptive response to a single 60 min exposure to HBO2 33. The naltrexone and L-NAME antagonisms of the early-phase antinociceptive response might imply that there might be a relationship between NO and endogenous opiate release as previously proposed.
Characterization of the HBO2-Induced Late-Phase Antinociceptive Response
Acute i.c.v. treatment with naltrexone two weeks after the four-day HBO2 treatment had no effect on the late-phase antinociceptive response as contrasted with its influence on the early-phase antinociception. Similarly, acute i.c.v. administration of L-NAME failed to have any influence on the late-phase antinociceptive effect. This appears to indicate that the early- and late-phase antinociceptive effects of HBO2 are not immediately mediated by the same mechanisms.
By using osmotic minipump technology, L-NAME and naltrexone were continuously administered i.c.v. during the four-day HBO2 treatment to determine their influence on HBO2-induced antinociception. The late-phase antinociception induced by HBO2 was inhibited ~80% by naltrexone and ~70% by L-NAME. Thus, it appears that there is an action of HBO2 at the time of exposure that requires NO and/or opioid receptor activation in order for the late-phase antinociceptive response to develop. But the final step in the antinociceptive pathway is downstream and is not immediately mediated by NO or opioid mechanisms.
The late-phase antinociceptive effect developed only after four daily 60-min HBO2 treatments and was not evident after a single 60-min HBO2 exposure. Multiple-dosing or exposures of patients or experimental animals to HBO2 appear to be very complicated. In the first instance, the usual HBO2 treatment is 60–90 min once a day, so the HBO2 exposure is relatively short over a 24-hr time period. This would necessarily imply that some endogenous mechanism is activated or invoked to sustain the pharmacological effect of HBO2 beyond a 24-hr period. Up-regulation might be at least part of the answer.
A varying multiple-dosing schedule for HBO2 therapy has been reported in the treatment of patients with retinitis pigmentosa (RP)26. In this study, the patients were exposed to HBO2 at 2.2 ATA for 90 min. The exposure or dosing schedule was the following: daily exposure for five days for four weeks followed by five exposures once a month for 11 months and, finally, five exposures once every three months for two years. The results showed a significant increase in electroretinogram measurements as compared to controls. A 10-yr study conducted by the same investigators27 employed the same dosing schedule but increasing the duration of the final regimen (five exposures every three months) from three years to 10 years. Results showed that different visual acuity measurements were significantly higher than for controls. In these studies, the turnover of the functional proteins involved appeared to be relatively long, i.e., approximately three months in duration. By comparison, in the present investigation, the turnover of the key proteins involved in the late-phase antinociceptive pathway appears to be somewhat shorter, about 2–3 weeks.
It remains to be determined whether increasing the number of HBO2 exposures or that periodic re-exposure to HBO2 will further extend the duration of the late-phase antinociceptive response analogous to the HBO2 treatment of RP cited above. This will be important to understand pharmacologically, since HBO2 may potentially be effective in treatment of different types of chronic pain.
Acknowledgements
This research was supported by NIH Grant GM-77153 (R.M.Q.) and funds from the WSU College of Pharmacy, the Allen I. White Distinguished Professorship and the Chico Hyperbaric Center. None of the authors have a conflict of interest in this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bitterman N. Bitterman H: L-arginine-NO pathway and CNS oxygen toxicity. J Appl Physiol. 1998;84:1633–1638. doi: 10.1152/jappl.1998.84.5.1633. [DOI] [PubMed] [Google Scholar]
- 2.Chavko M, Auker CR, McCarron RM. Relationship between protein nitration and oxidation and development of hyperoxic seizures. Nitric Oxide. 2003;9:18–23. doi: 10.1016/s1089-8603(03)00045-4. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Nadi NS, Chavko M, Auker CR, McCarron RM. Microarray analysis of gene expression in rat cortical neurons exposed to hyperbaric air and oxygen. Neurochem Res. 2009;34:1047–1056. doi: 10.1007/s11064-008-9873-8. [DOI] [PubMed] [Google Scholar]
- 4.Dall'Era MA, Hampson NB, Hsi RA, Madsen B, Corman JM. Hyperbaric oxygen therapy for radiation induced proctopathy in men treated for prostate cancer. J Urol. 2006;176:87–90. doi: 10.1016/S0022-5347(06)00491-5. [DOI] [PubMed] [Google Scholar]
- 5.Demchenko IT, Boso AE, Whorton AR, Piantadosi CA. Nitric oxide production is enhanced in rat brain before oxygen-induced convulsions. Brain Res. 2001;917:253–261. doi: 10.1016/s0006-8993(01)03057-8. [DOI] [PubMed] [Google Scholar]
- 6.Di Sabato F, Fusco BM, Pelaia P, Giacovazzo M. Hyperbaric oxygen therapy in cluster headache. Pain. 1993;52:243–245. doi: 10.1016/0304-3959(93)90137-E. [DOI] [PubMed] [Google Scholar]
- 7.Elayan IM, Axley MJ, Prasad PV, Ahlers ST, Auker CR. Effect of hyperbaric oxygen treatment on nitric oxide and oxygen free radicals in rat brain. J Neurophysiol. 2000;83:2022–2029. doi: 10.1152/jn.2000.83.4.2022. [DOI] [PubMed] [Google Scholar]
- 8.Emmanouil DE, Dickens AS, Heckert RW, Ohgami Y, Chung E, Han S, Quock RM. Nitrous oxide-antinociception is mediated by opioid receptors and nitric oxide in the periaqueductal gray region of the brain. Eur Neuropsychopharmacol. 2008;18:194–199. doi: 10.1016/j.euroneuro.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldmeier JJ, editor. The UHMS Hyperbaric Oxygen Therapy Committee Report. Durham, NC: Undersea and Hyperbaric Medical Society; 2003. Hyperbaric Oxygen 2003 – Indications and Results. [Google Scholar]
- 10.Haley TJ, McCormick WG. Pharmacological effects produced by intracerebral injections of drugs in the conscious mouse. Br J Pharmacol. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata T, Cui YJ, Funakoshi T, Mizukami Y, Ishikawa Y-I, Shibasaki F. Matsumoto M and Sakabe T: The temporal profile of genomic responses and protein synthesis in ischemic tolerance of the rat brain induced by repeated hyperbaric oxygen. Brain Res. 2007;1130:214–222. doi: 10.1016/j.brainres.2006.10.077. [DOI] [PubMed] [Google Scholar]
- 12.Jain KK, Jain KK. Textbook of Hyperbaric Medicine. Seattle: Hogrefe and Huber Publishers; 1999. Physical, physiological, and biochemical aspects of hyperbaric oxygenation; pp. 11–27. [Google Scholar]
- 13.Jones K, Evans AW, Bristow RG, Levin W. Treatment of radiation proctitis with hyperbaric oxygen. Radiother Oncol. 2006;78:91–94. doi: 10.1016/j.radonc.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Kiralp MZ, Yildiz S, Vural D, Keskin I, Ay H, Dursun H. Effectiveness of hyperbaric oxygen therapy in the treatment of complex regional pain syndrome. J Int Med Res. 2004;32:258–262. doi: 10.1177/147323000403200304. [DOI] [PubMed] [Google Scholar]
- 15.Lee CC, Chen SC, Tsai SC, Wang BW, Liu YC, Lee HM, Shyu KG. Hyperbaric oxygen induces VEGF expression through ERK, JNK and c-Jun/AP-1 activation in human umbilical vein endothelial cells. J Biomed Sci. 2006;13:143–156. doi: 10.1007/s11373-005-9037-7. [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Li J, Sun X, Liu K, Zhang JH, Xu W, Tao H. Repetitive hyperbaric oxygen exposures enhance sensitivity to convulsion by upregulation of eNOS and nNOS. Brain Res. 2008;1201:128–134. doi: 10.1016/j.brainres.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 17.Myers DE, Myers RA. A preliminary report on hyperbaric oxygen in the relief of migraine headache. Headache. 1995;35:197–199. doi: 10.1111/j.1526-4610.1995.hed3504197.x. [DOI] [PubMed] [Google Scholar]
- 18.Nakabayashi M, Beard C, Kelly SM, Carr-Locke DL, Oh WK. Treatment of a radiation-induced rectal ulcer with hyperbaric oxygen therapy in a man with prostate cancer. Urol Oncol. 2006;24:503–508. doi: 10.1016/j.urolonc.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Ohgami Y, Chung E, Shirachi DY, Quock RM. Influence of hyperbaric oxygen on regional brain levels and spinal cord levels of nitric oxide metabolites in rat. Brain Res Bull. 2008;75:668–673. doi: 10.1016/j.brainresbull.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Ohgami Y, Zylstra CC, Quock LP, Chung E, Shirachi DY, Quock RM. Nitric oxide in hyperbaric oxygen-induced acute antinociception in mice. NeuroReport. 2009;20:1325–1329. doi: 10.1097/WNR.0b013e3283305a49. [DOI] [PubMed] [Google Scholar]
- 21.Peach G. Hyperbaric oxygen and the reflex sympathetic dystrophy syndrome: a case report. Undersea Hyperb Med. 1995;22:407–408. [PubMed] [Google Scholar]
- 22.Sümen G, Çimşit M, Eroğlu L. Hyperbaric oxygen treatment reduces carrageenan-induced acute inflammation in rats. Eur J Pharmacol. 2001;431:265–268. doi: 10.1016/s0014-2999(01)01446-7. [DOI] [PubMed] [Google Scholar]
- 23.Takeyama N, Sakai H, Ohtake H, Mashitori H, Tamai K, Saotome K. Effects of hyperbaric oxygen on gene expressions of procollagen, matrix metalloproteinase and tissue inhibitor of metalloproteinase in injured medial collateral ligament and anterior cruciate ligament. Knee Surg Sports Traumatol Arthrosc. 2007;15:443–452. doi: 10.1007/s00167-006-0241-4. [DOI] [PubMed] [Google Scholar]
- 24.Thom SR, Bhopale V, Fisher D, Manevich Y, Huang PL, Buerk DG. Stimulation of nitric oxide synthase in cerebral cortex due to elevated partial pressures of oxygen: An oxidative stress response. J Neurobiol. 2002;51:85–100. doi: 10.1002/neu.10044. [DOI] [PubMed] [Google Scholar]
- 25.Tuter NV, Danilov AB, Poliakova LV. The treatment of a complex regional pain syndrome. Zh Nevrol Psikhiatr Im S S Korsakova. 1997;97:33–35. [PubMed] [Google Scholar]
- 26.Vingolo EM, Pelaia P, Forte R, Rocco M, Giusti C, Rispoli E. Does hyperbaric oxygen (HBO) delivery rescue retinal photoreceptors in retinitis pigmentosa? . Doc Ophthalmol. 1999;97:33–39. doi: 10.1023/a:1002015317479. [DOI] [PubMed] [Google Scholar]
- 27.Vingolo EM, Rocco M, Grenga P, Salvatore S, Pelaia P. Slowing the degenerative process, long lasting effect of hyperbaric oxygen therapy in retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2008;246:93–98. doi: 10.1007/s00417-007-0652-z. [DOI] [PubMed] [Google Scholar]
- 28.Warren J, Sacksteder MR, Thuning CA. Therapeutic effect of prolonged hyperbaric oxygen in adjuvant arthritis of the rat. Arthritis Rheum. 1979;22:334–339. doi: 10.1002/art.1780220404. [DOI] [PubMed] [Google Scholar]
- 29.Wilson HD, Toepfer VE, Senapati AK, Wilson JR, Fuchs PN. Hyperbaric oxygen treatment is comparable to acetylsalicylic acid treatment in an animal model of arthritis. J Pain. 2007;8:924–230. doi: 10.1016/j.jpain.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Wilson HD, Wilson JR, Fuchs PN. Hyperbaric oxygen treatment decreases inflammation and mechanical hypersensitivity in an animal model of inflammatory pain. Brain Res. 2006;1098:126–128. doi: 10.1016/j.brainres.2006.04.088. [DOI] [PubMed] [Google Scholar]
- 31.Wilson JR, Foresman BH, Gamber RG, Wright T. Hyperbaric oxygen in the treatment of migraine with aura. Headache. 1998;38:112–115. doi: 10.1046/j.1526-4610.1998.3802112.x. [DOI] [PubMed] [Google Scholar]
- 32.Yildiz S, Kiralp MZ, Akin A, Keskin I, Ay H, Dursun H, Cimsit M. A new treatment modality for fibromyalgia syndrome: hyperbaric oxygen therapy. J Int Med Res. 2004;32:263–267. doi: 10.1177/147323000403200305. [DOI] [PubMed] [Google Scholar]
- 33.Zelinski LM, Ohgami Y, Chung E, Shirachi DY, Quock RM. A prolonged NO-dependent, opioid-mediated antinociceptive effect of hyperbaric oxygen in mice. J Pain. 2009;10:167–172. doi: 10.1016/j.jpain.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]