

Abstract
Background & Aims:
The carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) exhibits pleotropic functions, including promoting hepatic insulin clearance. The current studies investigate the functions of its close relative, CEACAM2, with more limited tissue-specific distribution.
Methods:
A global Ceacam2 (Cc2−/−) null mouse (on 129/Sv × C57BL/6J) was generated. Females were subjected to hyperinsulinemic-euglycemic clamp, indirect calorimetry, body fat composition, and pair-feeding followed by insulin tolerance test for phenotypic characterization.
Results:
Female, but not male Cc2−/− mice exhibit obesity, which results from hyperphagia and reduced energy expenditure. Hyperphagia leads to peripheral insulin resistance, as demonstrated by pair-feeding experiments. Whereas insulin action is normal in liver, it is compromised in skeletal muscle, which exhibits incomplete fatty acid oxidation and impaired glucose uptake and disposal. The mechanism of hyperphagia in Cc2−/− mice is not well delineated, but appears to result partly from hyperinsulinemia-induced hypothalamic fatty acid synthase level and activity. Hyperinsulinemia is, in turn, caused by increased insulin secretion.
Conclusions:
Consistent with the absence of CEACAM2 from peripheral insulin target tissues and its localization to the hypothalamus, the phenotype of Cc2−/− mice identifies a novel role for CEACAM2 in the central regulation of energy balance and insulin sensitivity.
Keywords: hypothalamic obesity, hyperphagia, CEACAM2, energy balance
Introduction
The carcinoembryonic antigen-related cell adhesion molecules 1 and 2 (CEACAM1 and CEACAM2) are transmembrane glycoproteins, which are encoded by two separate genes on mouse chromosome 7.1 Sequence analysis in mouse 129Sv/Ev library revealed that these two genes are homologous, mapping to 9 exons that share the lowest homology in exons 2 (76.9%) and 6 (78.5%), with the rest sharing 87-100% homology.2
Whereas CEACAM1 is widely expressed, the CEACAM2 protein is limited to kidney, uterus and crypt epithelia of intestinal tissues. Its transcripts are also detected in brain (www.brain-map.org), spleen, testis and prostate.1-4 CEACAM2 protein is absent in peripheral insulin target tissues, such as liver, muscle and white adipose tissue. Unlike CEACAM1, CEACAM2 is not detected in the embryonic stage.2 The differential tissue-distribution and developmental expression predict that these two related proteins are not functionally redundant.
CEACAM1 has multiple roles, including cell adhesion,5 tumor suppression,6-8 angiogenesis,9 anti-inflammation,10 and hepatic insulin clearance.11, 12 Consistent with the major role for the liver in insulin extraction, CEACAM1 is predominantly expressed in this tissue and to a lower extent in kidney, the other, albeit minor, site for insulin removal. Neither CEACAM protein is expressed in skeletal muscle or adipose tissue, two active insulin signaling sites that do not contribute to insulin clearance.
The function for CEACAM2 remains elusive. While it acts as an alternative murine hepatitis virus receptor to CEACAM1,1 it does not act as a cell adhesion molecule.4 To further characterize its function, we have generated a global Ceacam2 null mouse (Cc2−/−). Consistent with the localization of CEACAM2 protein to (among other brain regions) the ventromedial nucleus of the hypothalamus (VMH), which is implicated in the neuronal control of food intake, energy balance, glucose uptake in skeletal muscle, glucose sensing, and insulin secretion13-17 female Cc2−/− mice develop hypothalamic obesity, hyperphagia, hyperinsulinemia and altered peripheral glucose disposal.
Materials and Methods
Generation of the Cc2−/− Mouse
The targeting vector was constructed by replacing Exon 1, including the translation initiation site in the Ceacam2 gene with a gene encoding Neomycin resistance (Neor) (manuscript in preparation). Briefly, embryonic stem cells (129 Sv) were transfected by electroporation (Genome Systems Inc.) and 200 clones screened by Southern blot analysis. Two clones with the appropriate homologous recombination events were obtained. Blastocyst injection (C57BL/6J) yielded three fertile chimeric offsprings that gave rise to germ line transmission. F1 heterozygous mice were bred to homozygosity by brother-sister matings. The homozygous wild-type (Cc2+/+) and mutant (Cc2−/−) progenies were genotyped by PCR, Southern and Western analyses, and backcrossed twice onto the C57BL/6 genetic background.
All animals were housed in a 12-hour dark/light cycle and fed standard chow (Teklad 2016) ad libitum. All procedures were approved by the Institutional Animal Care and Utilization Committee.
Metabolic Analysis
Following an overnight fast, mice were anesthetized with sodium pentobarbital at 1100h. Whole venous blood was drawn from the retro-orbital sinuses to measure by radioimmunoassay the serum levels of insulin, C-peptide and leptin (Linco Research, Billerica, MA), and of somatostatin (Phoenix Pharmaceuticals, Belmont, CA). Serum free fatty acids (FFA) by NEFA C kit (Wako, Wako, Richmond, VA) and serum triglycerides by Triglyceride kit (Pointe Scientific, Canton, MI). Tissue triglyceride and glycogen content were measured, as described previously.18 Tissue content of reduced glutathione were assayed using the Bioxytech GSH-400 kit (OXISResearch), as previously done.19
Glucagon levels were measured, using an RIA assay (Linco), in retro-orbital blood of mice fasted from 800 to 1400h before being injected intraperitoneally (ip) with regular human insulin (1.5 U/kg BWT Novolin R, Novo Nordisk, Denmark) for an hour.
Body Composition Measurements and Hyperinsulinemic-Euglycemic Clamp
Visceral adipose tissue was excised, weighed, and visceral adiposity expressed as percentage of total body weight. Whole body fat and lean mass were measured by 1H-magnetic resonance spectroscopy (MRS; Echo Medical Systems, Houston, TX), and a 2-hour hyperinsulinemic-euglycemic clamp was performed in awake mice (n=10-12) with primed and continuous infusion of human regular insulin (Humulin) at a rate of 2.5 mU·kg−1·min−1, as previously described.12, 18 Glucose metabolism was estimated with a continuous infusion of [3-3H]glucose (PerkinElmer Life and Analytical Sciences) for 2 hours prior to (0.05 μCi/min) and throughout the clamps (0.1 μCi/min). Tissues were removed to determine glycogen and triglyceride content.12, 18
Pair-Feeding Experiments
Based on the average daily food intake of the mice, pair-fed (PF) mice initially received 3.5 g of food per day, 0.5 g less than ad libitum (AL)-fed Cc2−/− mice. As this feeding program prevented weight gain even in AL-fed Cc2−/− mice, the amount of food was increased to 4.0 g per day in the PF-group as opposed to 6.0 g for the AL-group in the second week, at the end of which, insulin tolerance was determined.
Insulin Tolerance Test
Following overnight fast, mice were anesthetized, injected ip with regular human insulin (0.75 U/kg BWT Novolin R), and blood was removed from the tail to determine glucose levels (Accu-check, Roche).
Glucose Uptake in Isolated Muscle
As described previously,11 soleus muscle from hind-limbs of fasted mice was removed, treated with 1200 pM insulin, and incubated at 30°C at 95% O2-5%CO2 for 20 minutes in 2-deoxy-D-[l,2-3H]glucose (2-DG) (1 mM), [U-14C]mannitol (39 mM) and 0.1% BSA. Muscles were blotted and frozen in liquid nitrogen, stored at −80°C before digestion, counting and measuring intracellular 2-DG accumulation in nmol/g wet muscle.min−1.
Fatty Acid Synthase Assay
Following a 6 hour-fast, the hypothalamus was removed and fatty acid synthase (FAS) activity measured immediately in the presence of 0.1 μCi [14C] Malonyl CoA, (Perkin Elmer, Waltham, MA), 25 nmol Malonyl CoA and 500 μM NADPH, as previously described.20 FAS activity was calculated as cpm of [14C] incorporated/mg of lysates.
Indirect Calorimetry-CLAMS
Indirect calorimetry was performed at the University of Michigan Metabolomics and Obesity Center. Mice were acclimated to dummy cages for 48 hours prior to transfer to the Comprehensive Laboratory Monitoring System (CLAMS, Columbus Instruments, Columbus, OH). Animals were provided food and water ad libitum except during fasting-refeeding, when food was removed at 1800h and replaced at 0800h the next morning. Activity levels were measured with an optical beam device. VO2 and VCO2 were sampled every 10 minutes for 5 seconds and respiratory quotient (RQ) was calculated from the VCO2/VO2 ratio.
Hypothalamic Leptin Signaling
Leptin signaling and immunohistochemistry (IHC) were analyzed as previously described21 with some modifications: Mice were fasted overnight prior to receiving an ip injection of either saline (−) or 5 μg/g body weight (BWT) recombinant mouse leptin (a generous gift from Dr. A. F. Parlow at the National Hormone and Peptide Program, Los Angeles, CA). Sixty minutes after injection, mice were anesthetized and perfused with PBS followed by 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO). Brains were removed and fixed overnight in 4% paraformaldehyde, followed by dehydration in 10% sucrose solution. Brains were frozen and cut to 30 μM thickness on a microtome (Leica, Germany). Free-floating sections were incubated with α-phospho-STAT3 (Tyr705) (Cell Signaling, Beverly, MA), followed by streptavidin-conjugated anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA). Sections were incubated with DAB (Invitrogen, Carlsbad, CA), counterstained with hemotoxylin, mounted, and visualized using a Nikon microscope. Cells positive for phosphorylated STAT3 (p-STAT3) were identified as containing a brown nucleus, counted in the arcuate nucleus (ARC) of one coronal section per each mouse.
For IHC,21 the α-CEACAM2 antibody was raised against the KLH-conjugated HPLC purified peptide CNAEIVRFVTGTNKTIKGPVH (Bethyl Laboratories, Montgomery, TX). The secondary antibody used was donkey anti-rabbit AlexaFluor568 antibody (Invitrogen, Carlsbad, CA).
Ex-Vivo Palmitate Oxidation
Ex-vivo palmitate oxidation was analyzed as previously described22 with some modifications: mice were fasted overnight and anesthetized, before soleus and gastrocnemius muscle were removed, weighed, and homogenized at 20-fold dilution in buffer (10 mM Tris pH 7.2, 300 mM sucrose, 2 mM EDTA). Homogenate (1 ml) was injected via syringe into a sealed beaker at 30°C to initiate the reaction in incubation buffer (0.2 mM of [1-14C] palmitate at 0.5 uCi/ml, 100 mM sucrose, 10 mM Tris-HCl, 5 mM potassium phosphate, 80 mM potassium chloride, 1 mM magnesium chloride, 2 mM L-carnitine, 0.1 mM malic acid, 2 mM ATP, 0.05 mM coenzyme A, 1 mM dithiothreitol, 0.2 mM EDTA, and 0.5% bovine serum albumin, pH7.4). After 45 minutes, the reaction was terminated with glacial acetic acid. Completely oxidized CO2 was trapped in a well suspended above the medium filled with benzothonium hydroxide. Trapped CO2 radioactivity was measured by liquid scintillation in CytoCint (MP Biomedicals, Solon, OH).
Semi-Quantitative Real Time PCR Analysis
Total RNA was prepared using PerfectPure RNA Tissue kit (5Prime, Gaithersburg, MD) following manufacturer's instructions. cDNA was synthesized with oligo dT primers and Improm II Reverse Transcriptase (Promega, Madison, WI), using 1 μg of total RNA and primers (Table 1). cDNA was evaluated with Real-time quantitative PCR (RT-qPCR) using Fast SYBR Green PCR mix on a StepOne Plus Real Time PCR system (Applied Biosystems, Foster City, CA). Standard curves for each primer set were generated using serial 1:4 dilutions of pooled samples. The relative amounts of mRNA were calculated by comparison to the corresponding standards and normalized relative to GAPDH or 18S. Results are expressed as mean ± SEM in fold change relative to wild-type controls.
Table 1.
| Gene | Primer Sequence 5′ to 3′ | Annealing Temperature |
|---|---|---|
| Ceacam2 | F–AATATGATGAAGGGAGTCTTGGCC | 60 |
| R–AAATTGTCCAGTCAGGACCCTACG | ||
| Pparα | F–AGATCGGCCTGGCCTTCTAAACAT | 60 |
| R–AGCTTTGGGAAGAGGAAGGTGTCA | ||
| Fatp1 | F–GCAGAAGACGCAGGAAGA | 60 |
| R–GGACGTGGCTGTGTATGG | ||
| Cpt1b | F–TCCCATAAGAAACAAGACCTCC | 60 |
| R–GCTCCAGGGTTCAGAAAGTAC | ||
| Ucp3 | F–GTTTACTGACAACTTCCCCT | 55 |
| R–CTCCTGAGCCACCATCT | ||
| Pdk4 | F–CCTTTGGCTGGTTTTGGTTA | 60 |
| R–CCTGCTTGGGATACACCAGT | ||
| Fasn | F–GTGACTGGCAGGCTGCTGGG | 61 |
| R–CGCTCGGCTCGATGGCTCAG | ||
| 18S | F–TTCGAACGTCTGCCCTATCAA | 58 |
| R–ATGGTAGGCACGGCGACT | ||
| Gapdh | F–CCAGGTTGTCTCCTGCGACT | 61 |
| R–ATACCAGGAAATGAGCTTGACAAAGT |
Statistical Methods
Statistical significance was determined by two-tailed Student t-test unless otherwise indicated. Welch's correction was used if variances were significantly different. Statistical significance was set at 5%.
Results
Elevated Body and Fat Mass in Female Cc2−/− Mice
Contrary to male Cc2−/− mice which grow at a comparable body weight to their wild-type Cc2+/+ counterparts (Figure 1B.b), female mutants exhibit a larger body weight starting at 2 months of age (Figure 1B.a). This is attributed to a significant increase in both fat and lean mass (Figure 1C), as no change in body length is detected (Table 2). While the abdominal fat mass is normal in younger mice, it progressively increases to become significantly elevated in female Cc2−/− mice at ≥ 4 months of age (Figure 1D). Because of the sexual dimorphic effect of Ceacam2 mutation on body weight, we have focused on characterizing the phenotype of female mice.
Figure 1.
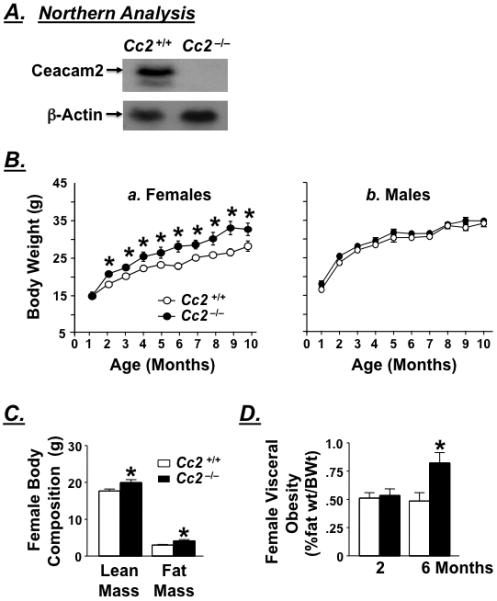
Obesity and fat distribution. (A) Northern analysis of mRNA reveals complete deletion of Ceacam2 from Cc2−/−, as compared to Cc2+/+ mice (n>3 of 2 month-old mice/group). (B) Body weight was measured over a period of 10 months in a. ad libitum-fed female and b. male wild-type (Cc2+/+, open circles) and Cc2−/− null mice (dark circles). (C) 1-H mass resonance spectroscopy analysis of fat mass and lean mass body composition in 4 month-old (n>10) female wild-type (open bar) and Cc2−/− mice (filled bars). (D) Visceral adiposity in 2-6 month-old female mice measured as %abdominal fat to body weight (n≥8/age/group). Values are mean ± SEM. *P≤0.05 versus Cc2+/+.
Table 2.
Fasting metabolic parameters of female Cc2+/+ and Cc2−/− mice
| 2-3 Months |
5-6 Months |
|||
|---|---|---|---|---|
| Cc2+/+ | Cc2−/− | Cc2+/+ | Cc2−/− | |
| Visceral Adiposity (%BWT) | 0.51 ± 0.05 | 0.54 ± 0.06 | 0.49 ± 0.07 | 0.82 ± 0.09 A |
| Body Length (cm) | NA | NA | 99.4 ± 1.28 | 102. ± 0.83 |
| Blood Glucose (mg/dl) | 90.3 ± 3.48 | 99.7 ± 6.39 | 94.2 ± 2.66 | 99.9 ± 3.68 |
| Serum Insulin (pM) | 74. ± 3.0 | 124. ± 10.2 A | 58.7 ± 4.80 | 110. ± 21.0 A |
| Serum C-Peptide (pM) | 132. ± 18.5 | 263. ± 29.5 A | 197. ± 26.0 | 453. ± 95.7 A |
| C/I Molar Ratio | 1.72 ± 0.18 | 1.91 ± 0.16 | 3.31 ± 0.33 | 5.09 ± 1.01 |
| Serum Glucagon (ng/ml) | NA | NA | 0.50 ± 0.12 | 0.57 ± 0.10 |
| Serum Somatostatin (ng/ml) | 0.84 ± 0.01 | 0.85 ± 0.01 | NA | NA |
| Serum Leptin (ng/ml) | 2.88 ± 0.18 | 2.92 ± 0.24 | 2.59 ± 0.14 | 3.19 ± 0.24 A |
| Serum Triglyceride (mg/dl) | 78.3 ± 7.51 | 56.8 ± 6.33 A | 75.5 ± 7.99 | 51.5 ± 5.65 A |
| Serum FFA (mEq/L) | 0.51 ± 0.03 | 0.55 ± 0.06 | 0.76 ± 0.09 | 0.74 ± 0.10 |
P<0.05 Cc2−/− versus Cc2+/+ mice (n≥7). Mice were fasted overnight prior to blood drawing and determination of serum metabolic parameters except for leptin and somatostatin where mice were fasted for 6 hours. For glucagon, mice were fasted for 6 hours prior to being injected with insulin for an hour and their blood removed.
Female Cc2−/− Mice are Insulin Resistant
Female Cc2−/− mice exhibit hyperinsulinemia starting at 2 months of age (Table 2). This results from elevated insulin secretion (as estimated by the ~2-fold increase in serum C-peptide levels), without a defect in insulin clearance (as assessed by normal steady state molar ratio of C-peptide/Insulin),11 (Table 2). Normal insulin clearance in Cc2−/− mice is expected given the minimal level of CEACAM2 in liver, the major site of insulin clearance, and normal levels of hepatic CEACAM1 (Figure 3C.b), a key promoter of insulin extraction.11
Figure 3.
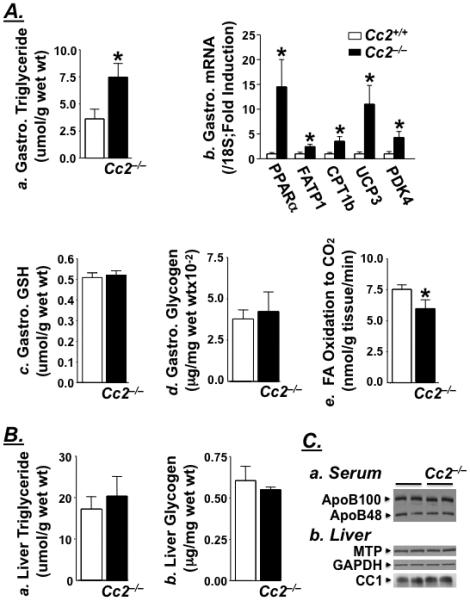
Biochemical analysis of lipid metabolism in female mice. (A) Gastrocnemius (Gastro.) muscle was isolated from female mice [Cc2+/+ (open bar) and Cc2−/− (filled bar)] to measure: a. triglyceride content (4 month-old; n=6); b. mRNA content normalized to 18S by RT-qPCR (fasted 5 month-old; n≥6); c. Reduced glutothione (GSH) levels (fasted 7 month-old; n≥6) and d. Glycogen content (4 month-old refed for 7 hours; n≥4). In e. both gastrocnemius and soleus muscle were extracted from overnight fasted 7 month-old mice (n≥5). (B) a. Hepatic triglyceride (at the end of the clamp) and b. glycogen content in 4 month-old mice refed for 7 hours (n=6). Values are expressed as mean ± SEM. *P<0.05 versus Cc2+/+. (C) Representative Western blots from ad libitum-fed 7 month-old mice (n>4) to determine: a. serum ApoB100 and ApoB48, and b. hepatic MTP and CEACAM1 (CC1) normalized to GAPDH.
Cc2−/− mutants exhibit normal basal glucose levels (Table 2, Figure 2A.a) and basal hepatic glucose production (HGP) (Figure 2A.b) by comparison to Cc2+/+ mice. Subjected to a hyperinsulinemic-euglycemic clamp, which maintains comparable glucose levels in awake 4 month-old Cc2+/+ and Cc2−/− mice (Figure 2A.a), mutant mice exhibit lower glucose infusion rate required to maintain euglycemia during clamps (Figure 2A.c). This indicates that Cc2−/− mice develop systemic insulin resistance. Insulin-mediated suppression of hepatic glucose production by insulin (Figure 2A.d) and glycogen content in muscle and liver (Figure 3A. and 3B.b, respectively) are not altered in Cc2−/− mice. However, insulin-stimulated whole body glucose turnover (Rd) (Figure 2A.e) and glycogen/lipid synthesis (Figure 2A.f) are significantly reduced in Cc2−/− mice, suggesting insulin resistance in peripheral tissues in these mice.
Figure 2.
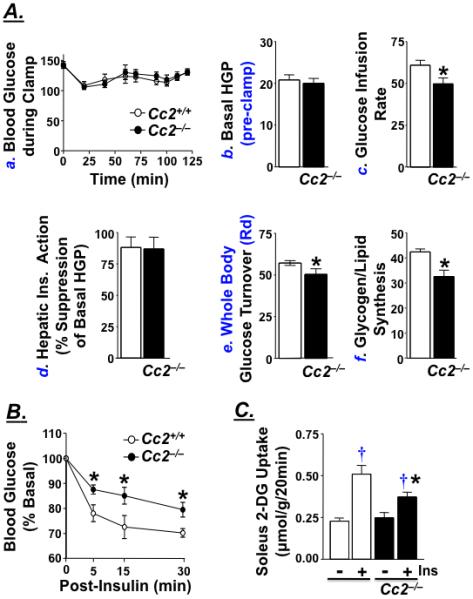
Insulin action in female mice. (A) 4 month-old awake Cc2+/+ (open bar or circle) and Cc2−/− female mice (filled bar or circle) (n≥10/group) were subjected to a 2 hour-hyperinsulinemic-euglycemic clamp to measure: a. Blood glucose levels immediately prior to (time 0) and throughout the clamp (up to 120 min); b. Basal hepatic glucose production (HGP), a measure of Ra at pre-clamp conditions; c. Steady-state glucose infusion rate during clamps; d. Hepatic insulin action, as insulin-mediated percent suppression of HGP. e. Insulin-stimulated whole body glucose turnover (Rd), and f. Insulin-stimulated whole body glycogen plus lipid synthesis. Values are all expressed in mg/kg/min, except for blood glucose levels in a, which is expressed as mg/dl. (B) 5 month-old Cc2+/+ (open circle) and Cc2−/− (filled circle) mice (n=7/group) were ip injected with insulin (0.75 U kg−1) for 0-30 minutes (min) and their tail vein blood glucose measured. (C) 2-deoxyglucose (2-DG) uptake in the absence (−) or presence (+) of insulin (Ins) was measured in soleus muscle isolated from 7 month-old mice. Values are expressed as mean ± SEM. *P<0.05 versus Cc2+/+ and †P<0.05 versus no insulin (Ins).
Glucose clearance following insulin injection is reduced in Cc2−/− mice (Figure 2B). To determine the site of altered glucose disposal, we examined 2-DG uptake in isolated soleus muscle. Whereas insulin induces glucose uptake in Cc2+/+ mice by ~125%, it only causes a 50% increase in muscle fibers from 7 (Figure 2C) and 3 month-old (not shown) Cc2−/− mice. The effect of insulin on glucose uptake in mutants is significantly lower than that in wild-type mice.
Fasting serum triglyceride levels are significantly reduced in Cc2−/− mice, starting at 2 months of age (Table 2). Given that hepatic triglyceride level is normal (Figure 3B.a), and the protein content of hepatic MTP (Figure 3C.b) and serum ApoB100/B48 is intact (Figure 3C.a), it is likely that reduction in serum triglyceride is associated with altered metabolism in extra-hepatic organs. Accordingly, the gastrocnemius muscle exhibit elevation in triglyceride content (Figure 3A.a) and potentially, fatty acid β-oxidation, as suggested by increased Pparα, Cpt1b, Ucp3 and Pdk4 mRNA levels (Figure 3A.b). Induction in Ucp3 content may contribute to the maintenance of normal antioxidant GSH level (Figure 3A.c) and protection against oxidative stress.23 While β-oxidation is likely to be increased in Cc2−/− mice, complete oxidation to CO2 is not achieved, as assessed by lower CO production in muscle isolated from Cc2−/− compared to Cc2+/+ mice (Figure 3A.e).
Female Cc2−/− Mice are hyperphagic
Because CEACAM2 is not expressed in skeletal muscle, the metabolic effects of Ceacam2 deletion are likely secondary to the genetic deficiency in other organs, in particular brain.1 Immunohistochemical analysis detects CEACAM2 protein in the ventromedial (VMH) hypothalamic nucleus (Figure 4A), as well as a number of other regions of the brain (Supplemental Figure S1), including some with a role in food reward and intake [globus pallidus (GP), ventral pallidum (VP), striatum, olfactory bulb and hippocampus].24, 25 Furthermore, brain Ceacam2 mRNA levels rise at fasting and decrease at 4 and 7 hours of refeeding (Figure 4A) when insulin surges,20 suggesting an association between CEACAM2 and reduction in food intake. This notion is supported by the observation that Cc2−/− mice are hyperphagic by 3 months of age; eating on average 20% more than their Cc2+/+ counterparts (Figure 4B), as assessed by measuring daily food intake over a one week-period. Furthermore, pair-fed (PF) Cc2−/− mutants exhibit comparable body weight (Figure 4C.a) and glucose disposal response to exogenous insulin as PF-Cc2+/+ mice (Figure 4C.b). In contrast, ad libitum (AL)-fed mice remain heavier and less insulin tolerant. This indicates that hyperphagia plays a primary role in the pathogenesis of insulin resistance in Cc2−/− mice.
Figure 4.
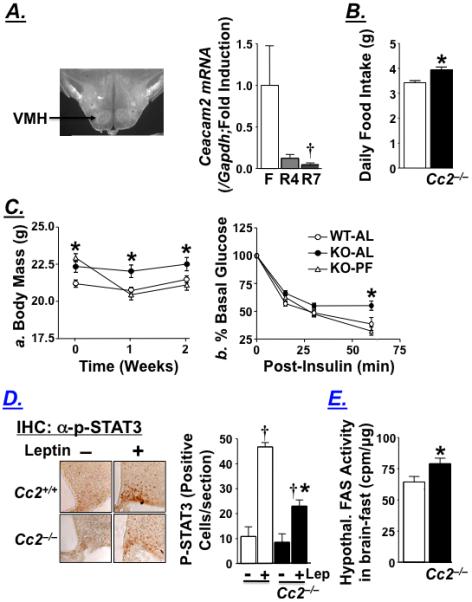
Characterization of the feeding behavior of female Cc2−/− mice. (A) Immunohistochemical analysis of CEACAM2 in the hypothalamus of wild-type mice reveals expression in the VMH (arrow), and RT-qPCR of whole brain homogenate for Ceacam2 normalized to Gapdh at fasting (F) and after being refed for 4 (R4) and 7 hours (R7) (n≥7). Values are expressed as mean ± SEM; †P<0.05 versus fasting. B) Daily food intake in 3 month-old mice measured over 7 consecutive days in Cc2+/+ (open bar) and Cc2−/− (filled bar) mice (n=8/group). (C) 3 month-old mice (n=15/group) were subjected to a pair-feeding experiment for 2 weeks and a. their body mass and b. insulin tolerance assessed. Values are mean ± SEM at each time point. *P<0.05 versus Cc2+/+. (D) Immunohistochemical analysis of coronal sections with α-phospho-STAT3 in the hypothalamic ARC of fasted 4 month-old mice (n=4/group) treated with saline or leptin (5.0 μg/g ip) for an hour. Representative sections are shown in the left panel. Brown nuclei of positive cells were counted in one section per mouse and the mean was measured as ± SEM and represented in the graph on the right. †P<0.05 versus no leptin (Lep) and *P<0.05 versus Cc2+/+. (E) Analysis of fatty acid synthase activity in the fasting hypothalamus of 3 month-old mice (n≥8). Values are expressed as mean ± SEM. *P<0.05 versus Cc2+/+.
Leptin levels do not rise significantly until about 4 months of age (Table 2). Moreover, immunohistochemical analysis of ARC coronal sections with α-phospho-STAT3 (the signal transducer and activator of transcription 3) reveals reduced hypothalamic STAT3 activation by leptin in 4 month-old Cc2−/− relative to Cc2+/+ mice (2.7-fold induction in p-STAT3 labeling by leptin in Cc2−/− mutants versus 4.3-fold in Cc2+/+ mice; P<0.05) (Figure 4D). Thus, cellular leptin resistance follows hyperphagia and insulin resistance in Cc2−/− mutants.
Consistent with elevated transcription of lipogenic enzymes in chronic hyperinsulinemia, hypothalamic FAS mRNA (Fasn) is higher in Cc2−/− than Cc2+/+ mice (not shown), in association with higher hypothalamic FAS activity (Figure 4E). Based on studies showing leptin-independent reduction of food intake and obesity by inhibiting26 or null mutating27 hypothalamic FAS activity, the data suggest that elevated FAS activity contributes to the mechanism of hyperphagia and obesity in Cc2−/− mice.
Decreased Energy Expenditure in Female Cc2−/− Mice
In concordance with lower energy expenditure in leptin resistance,28 indirect calorimetry reveals reduction in VO2 and VCO2 levels during a dark/light cycle in 6 month-old Cc2−/− mice (Figure 5A.a and 5A.b, respectively). The lower energy expenditure is not due to decreased activity, as activity-X is normal (Figure 5A.d). Subjected to a fasting/refeeding paradigm, Cc2−/− mice also exhibit a hypometabolic state during refeeding (Figure 5B.a and 5B.b) without changes in activity-X (Figure 5B.d).
Figure 5.
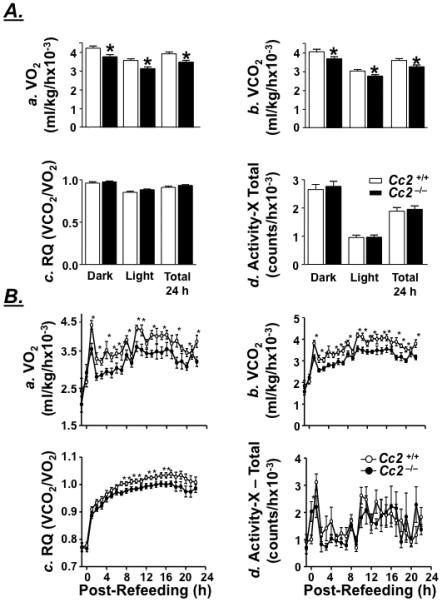
Energy expenditure and activity in female mice. 6 month-old Cc2+/+ and Cc2−/− mice (n=8) were (A) fed ad libitum and subjected to Indirect Calorimetry analysis to assess light and dark cycle a. VO2, b. VCO2, c. the respiratory quotient (VCO2/VO2), and d. total activity along the x-axis. (B) To measure energy expenditure and activity during switching from lipolytic to glycolytic metabolism, food was removed overnight and returned the next morning before measuring a. VO2, b. VCO2 and c. respiratory quotient (VCO2/VO2) by hourly average from one hour proceeding the return of food through the 22 hours following refeeding. d. Total activity along the x-axis following refeeding was also measured. Values are expressed as mean ± SEM. *P<0.05 versus Cc2+/+.
Impaired Substrate Switching Upon Refeeding in Female Cc2−/− Mice
In a normal light/dark cycle, Cc2−/− mice exhibit normal respiratory quotient (RQ) (Figure 5A.c). However, they exhibit a reduction in RQ during refeeding following an overnight fast (Figure 5B.c). This demonstrates that mutant mice do not efficiently switch from lipolytic to glycolytic metabolism when refed. Altered lipid and glucose metabolism in skeletal muscle, but not liver, suggests that impairment in metabolic switch occurs mostly in skeletal muscle.
Discussion
In contrast to males, female Cc2−/− mice exhibit elevated body weight and insulin resistance starting at 2 months of age. These metabolic abnormalities result from hyperphagia, as demonstrated by prevention of weight gain and restoration of insulin sensitivity in pair-fed mice. At about 4 months of age, the mice develop cellular leptin resistance, which could contribute to reduction in energy expenditure.29, 30 Together with hyperphagia, the hypometabolic state causes a higher weight gain in female Cc2−/− mice than in the wild type controls,31-33 and ultimately, visceral obesity at about 6 months of age.
The localization of CEACAM2 to VMH and other brain regions that are important in the regulation of food intake and body weight,16, 24, 25, 34, 35 and the finding that brain Ceacam2 expression is induced at fasting, but is markedly reduced during refeeding, provide circumstantial evidence that CEACAM2 is involved in the regulation of feeding behavior. The mechanism of this novel function of CEACAM2 awaits further explanation. Nevertheless, the observed sexual dimorphism in obesity in Cc2−/− mutants lends support to the notion that CEACAM2 is involved in the VMH regulation of food intake based on ample documentation that VMH lesions cause hyperphagia and obesity more commonly in females.16 Thus, it is likely that the hypothalamic obesity in female Cc2−/− mice is related to disturbances in the sympathetic nervous system relay from VMH, especially that it is accompanied by hyperinsulinemia and hyperphagia. Hyperphagia in Cc2−/− mice could be secondary to hyperinsulinemia and other autonomic alterations36 arising from Ceacam2 deletion. Hyperinsulinemia is caused by increased insulin secretion. Given that CEACAM2 is not expressed in β-cells (unpublished observation), and that circulating glucagon and somatostatin levels are normal (Table 2), increased insulin secretion in Cc2−/− mice could involve altered glucosensing activity37 or leptin signaling in VMH.38 The mechanism of the regulation of these processes by hypothalamic CEACAM2 remains unclear. However, hyperinsulinemia induces the expression of lipogenic enzymes, including FAS,39 which has been implicated in a leptin-independent hypothalamic pathway regulating food intake.26, 27, 40 Mice with conditional null mutation of hypothalamic FAS are hypophagic and lean.27 Thus, elevated hypothalamic FAS activity may contribute to hyperphagia and the obesity phenotype in Cc2−/− mice.
Hyperinsulinemic-euglycemic clamps and 2-deoxyglucose uptake experiments in isolated soleus muscle indicate reduction in glucose uptake without changes in hepatic glucose or lipid metabolism. This is likely secondary to “lipotoxicity”, which results from increased triglyceride accumulation41 and incomplete fatty acid oxidation in muscle.42 Moreover, Cc2−/− mice display a reduced capacity to switch from lipolytic to glycolytic metabolism when subjected to a fasting-refeeding paradigm. The sustained fatty acid oxidation state contributes to peripheral insulin resistance.43
Absence of CEACAM2 in skeletal muscle2 and its localization to VMH, which is implicated in leptin regulation of glucose uptake in skeletal muscle44 points to central dysregulation of glucose uptake and fatty acid oxidation in skeletal muscle in Cc2−/− mice. Leptin injection in VMH increases glucose uptake in skeletal muscle, but not white adipose tissue44 through activation of the sympathetic nervous system.14 Although central leptin resistance Cc2−/− mice remains to be tested,45 the development of cellular leptin resistance in addition to impairment of glucose uptake in soleus muscle, but not in adipose tissue in Cc2−/−, propose that CEACAM2 may be involved in leptin-dependent signaling pathways regulating glucose uptake in skeletal muscle. Moreover, hypothalamic FAS activity is involved in the central regulation of not only feeding but also fatty acid oxidation to CO2 in skeletal muscle.40 Thus, it is also possible that elevated hypothalamic FAS activity contributes mechanistically to the disturbed central control of peripheral glucose disposal in Cc2−/− mice.
In conclusion, our data identify a novel and an unexpected function for CEACAM2 in the central control of feeding behavior, body weight, energy balance, insulin secretion and peripheral insulin action. Further studies are required to unravel the underlying mechanisms, but it appears that this may involve both leptin-dependent and independent hypothalamic pathways.
Supplementary Material
Acknowledgements
The authors thank Mats A. Fernstrom and Jennifer Kalisz for excellent technical assistance in the generation, genotyping and maintenance of Cc2−/− null mice, in addition to carrying out routine enzymatic assays and RNA analyses. We also thank Dr. Tong Dai for his assistance in genotyping and analysis of male mice, and Dr. Nathan R. Qi from the University of Michigan Metabolomics and Obesity Center (MMOC) for his assistance in indirect calorimetry (CLAMS) analysis. We specially thank Dr. Randy J. Seeley at the University of Cincinnati for his continuous scientific discussions, and Dr. Jennifer W. Hill from the University of Toledo College of Medicine for her critical reading of the manuscript.
The authors disclose the following: This work was supported by grants from the National Institutes of Health (R01DK054254 and R01DK083850 to SM Najjar, R37DK56731 to MG Myers and R01-DK80756 and U24-DK59635 to JK Kim), United States Department of Agriculture (USDA 38903-19826) to SM Najjar, the American Diabetes Association (to J Kim and MG Myers) and the American Heart Association (to MG Myers and G Heinrich).
Abbreviations used in this paper
- CEACAM2
the carcinoembryonic antigen-related cell adhesion molecule 2
- Cc2
Ceacam2 gene
- Cc2+/+
wild type mouse
- Cc2−/−
Ceacam2 null mutant mouse
- FFA
free fatty acids
- FAS
fatty acid synthase
- PPARα
peroxisome proliferator-activated receptor α
- UCP3
uncoupling protein 3
- CPT-1
carnitine palmitoyl transferase 1
- FATP1
fatty acid transport protein 1
- PDK-4
pyruvate dehydrogenate kinase
- GSH
reduced glutathione
- 2-DG
2-deoxy-D-[l,2-3H]glucose (2-DG)
- STAT3
the signal transducer and activator of transcription 3
- RQ
respiratory quotient
- VMH
ventromedial nucleus of the hypothalamus
- ARC
arcuate nucleus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Garrett Heinrich: study concept and design; acquisition of data; analysis and interpretation of data; statistical analysis; drafting of the manuscript.
Sumona Ghosh: acquisition of data
Anthony M. DeAngelis: acquisition of data
Jill M. Schroeder-Gloeckler: acquisition of data
Payal R. Patel: acquisition of data
Tamara Castaneda: acquisition of data
Shane Jeffers: acquisition of data
Abraham D. Lee: acquisition of data
Dae Young Jung: acquisition of data
Zhiyou Zhang: acquisition of data
Darren M. Opland: acquisition of data
Martin G. Myers, Jr: acquisition of data; analysis and interpretation of data; critical revision of the manuscript for important intellectual content; obtained funding.
Jason K. Kim: acquisition of data; analysis and interpretation of data; critical revision of the manuscript for important intellectual content; statistical analysis; obtained funding.
Sonia M. Najjar: study concept and design; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; statistical analysis; obtained funding; study supervision.
Conflict of Interest: Authors declare that no conflicts of interest exist.
References
- 1.Nedellec P, Dveksler GS, Daniels E, Turbide C, Chow B, Basile AA, Holmes KV, Beauchemin N. Bgp2, a new member of the carcinoembryonic antigen-related gene family, encodes an alternative receptor for mouse hepatitis viruses. J Virol. 1994;68:4525–37. doi: 10.1128/jvi.68.7.4525-4537.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han E, Phan D, Lo P, Poy MN, Behringer R, Najjar SM, Lin SH. Differences in tissue-specific and embryonic expression of mouse Ceacam1 and Ceacam2 genes. Biochem J. 2001;355:417–23. doi: 10.1042/0264-6021:3550417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zebhauser R, Kammerer R, Eisenried A, McLellan A, Moore T, Zimmermann W. Identification of a novel group of evolutionarily conserved members within the rapidly diverging murine Cea family. Genomics. 2005;86:566–80. doi: 10.1016/j.ygeno.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Robitaille J, Izzi L, Daniels E, Zelus B, Holmes KV, Beauchemin N. Comparison of expression patterns and cell adhesion properties of the mouse biliary glycoproteins Bbgp1 and Bbgp2. Eur J Biochem. 1999;264:534–44. doi: 10.1046/j.1432-1327.1999.00660.x. [DOI] [PubMed] [Google Scholar]
- 5.Öbrink B. CEA adhesion molecules: multifunctional proteins with signal-regulatory properties. Curr. Opin. Cell Biol. 1997;9:616–626. doi: 10.1016/S0955-0674(97)80114-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang J, Simpson JF, Glackin C, Riethorf L, Wagener C, Shively JE. Expression of biliary glycoprotein (CD66a) in normal and malignant breast epithelial cells. Anticancer Res. 1998;18:3203–12. [PubMed] [Google Scholar]
- 7.Lin SH, Pu YS. Function and therapeutic implication of C-CAM cell-adhesion molecule in prostate cancer. Semin Oncol. 1999;26:227–33. [PubMed] [Google Scholar]
- 8.Laurie NA, Comegys MM, Carreiro MP, Brown JF, Flanagan DL, Brilliant KE, Hixson DC. Carcinoembryonic antigen-related cell adhesion molecule 1a-4L suppression of rat hepatocellular carcinomas. Cancer Res. 2005;65:11010–7. doi: 10.1158/0008-5472.CAN-04-2841. [DOI] [PubMed] [Google Scholar]
- 9.Ergun S, Kilik N, Ziegeler G, Hansen A, Nollau P, Gotze J, Wurmbach JH, Horst A, Weil J, M. F, Wagener C. CEA-related cell adhesion molecule 1: a potent angiogenic factor and a major effector of vascular endothelial growth factor. Mol. Cell. 2000;5:311–320. doi: 10.1016/s1097-2765(00)80426-8. [DOI] [PubMed] [Google Scholar]
- 10.Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat. Rev. Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- 11.Poy MN, Yang Y, Rezaei K, Fernstrom MA, Lee AD, Kido Y, Erickson SK, Najjar SM. CEACAM1 regulates insulin clearance in liver. Nat Genet. 2002;30:270–6. doi: 10.1038/ng840. [DOI] [PubMed] [Google Scholar]
- 12.DeAngelis AM, Heinrich G, Dai T, Bowman TA, Patel PR, Lee SJ, Hong EG, Jung DY, Assmann A, Kulkarni RN, Kim JK, Najjar SM. Carcinoembryonic antigen-related cell adhesion molecule 1: a link between insulin and lipid metabolism. Diabetes. 2008;57:2296–303. doi: 10.2337/db08-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudo M, Minokoshi Y, Shimazu T. Ventromedial hypothalamic stimulation enhances peripheral glucose uptake in anesthetized rats. Am J Physiol. 1991;261:E298–303. doi: 10.1152/ajpendo.1991.261.3.E298. [DOI] [PubMed] [Google Scholar]
- 14.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48:1706–12. doi: 10.2337/diabetes.48.9.1706. [DOI] [PubMed] [Google Scholar]
- 15.Minokoshi Y, Okano Y, Shimazu T. Regulatory mechanism of the ventromedial hypothalamus in enhancing glucose uptake in skeletal muscles. Brain Res. 1994;649:343–7. doi: 10.1016/0006-8993(94)91085-5. [DOI] [PubMed] [Google Scholar]
- 16.King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87:221–44. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Shiuchi T, Haque MS, Okamoto S, Inoue T, Kageyama H, Lee S, Toda C, Suzuki A, Bachman ES, Kim YB, Sakurai T, Yanagisawa M, Shioda S, Imoto K, Minokoshi Y. Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab. 2009;10:466–80. doi: 10.1016/j.cmet.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Park SY, Cho YR, Kim HJ, Hong EG, Higashimori T, Lee SJ, Goldberg IJ, Shulman GI, Najjar SM, Kim JK. Mechanism of glucose intolerance in mice with dominant negative mutation of CEACAM1. Am. J. Physiol. Endocrinol. Metab. 2006;291:E517–E524. doi: 10.1152/ajpendo.00077.2006. [DOI] [PubMed] [Google Scholar]
- 19.Lee SJ, Heinrich G, Fedorova L, Al-Share QY, Ledford KJ, Fernstrom MA, McInerney MF, Erickson SK, Gatto-Weis C, Najjar SM. Development of nonalcoholic steatohepatitis in insulin-resistant liver-specific S503A carcinoembryonic antigen-related cell adhesion molecule 1 mutant mice. Gastroenterology. 2008;135:2084–95. doi: 10.1053/j.gastro.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Najjar SM, Yang Y, Fernstrom MA, Lee SJ, Deangelis AM, Rjaily GA, Al-Share QY, Dai T, Miller TA, Ratnam S, Ruch RJ, Smith S, Lin SH, Beauchemin N, Oyarce AM. Insulin acutely decreases hepatic fatty acid synthase activity. Cell Metab. 2005;2:43–53. doi: 10.1016/j.cmet.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Munzberg H, Jobst EE, Bates SH, Jones J, Villanueva E, Leshan R, Bjornholm M, Elmquist J, Sleeman M, Cowley MA, Myers MG., Jr Appropriate inhibition of orexigenic hypothalamic arcuate nucleus neurons independently of leptin receptor/STAT3 signaling. J Neurosci. 2007;27:69–74. doi: 10.1523/JNEUROSCI.3168-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JY, Koves TR, Yu GS, Gulick T, Cortright RN, Dohm GL, Muoio DM. Evidence of a malonyl-CoA-insensitive carnitine palmitoyltransferase I activity in red skeletal muscle. Am J Physiol Endocrinol Metab. 2002;282:E1014–22. doi: 10.1152/ajpendo.00233.2001. [DOI] [PubMed] [Google Scholar]
- 23.Lowell BB, Bachman ES. Beta-Adrenergic receptors, diet-induced thermogenesis, and obesity. J Biol Chem. 2003;278:29385–8. doi: 10.1074/jbc.R300011200. [DOI] [PubMed] [Google Scholar]
- 24.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–95. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 25.Cota D, Barrera JG, Seeley RJ. Leptin in energy balance and reward: two faces of the same coin? Neuron. 2006;51:678–80. doi: 10.1016/j.neuron.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 27.Chakravarthy MV, Zhu Y, Lopez M, Yin L, Wozniak DF, Coleman T, Hu Z, Wolfgang M, Vidal-Puig A, Lane MD, Semenkovich CF. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J Clin Invest. 2007;117:2539–52. doi: 10.1172/JCI31183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, Collins F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–3. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 29.Rosenbaum M, Leibel RL. Leptin: a molecule integrating somatic energy stores, energy expenditure and fertility. Trends Endocrinol Metab. 1998;9:117–24. doi: 10.1016/s1043-2760(98)00028-9. [DOI] [PubMed] [Google Scholar]
- 30.Badman MK, Flier JS. The adipocyte as an active participant in energy balance and metabolism. Gastroenterology. 2007;132:2103–15. doi: 10.1053/j.gastro.2007.03.058. [DOI] [PubMed] [Google Scholar]
- 31.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 32.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–32. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 35.Ahima RS, Lazar MA. Adipokines and the peripheral and neural control of energy balance. Mol Endocrinol. 2008;22:1023–31. doi: 10.1210/me.2007-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powley TL. The ventromedial hypothalamic syndrome, satiety, and a cephalic phase hypothesis. Psychol Rev. 1977;84:89–126. [PubMed] [Google Scholar]
- 37.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes. 2004;53:2521–8. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- 38.Hinoi E, Gao N, Jung DY, Yadav V, Yoshizawa T, Kajimura D, Myers MG, Jr., Chua SC, Jr., Wang Q, Kim JK, Kaestner KH, Karsenty G. An Osteoblast-dependent mechanism contributes to the leptin regulation of insulin secretion. Ann N Y Acad Sci. 2009;1173(Suppl 1):E20–30. doi: 10.1111/j.1749-6632.2009.05061.x. [DOI] [PubMed] [Google Scholar]
- 39.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cha SH, Hu Z, Chohnan S, Lane MD. Inhibition of hypothalamic fatty acid synthase triggers rapid activation of fatty acid oxidation in skeletal muscle. Proc Natl Acad Sci U S A. 2005;102:14557–62. doi: 10.1073/pnas.0507300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H, Knaub LA, Jensen DR, Young Jung D, Hong EG, Ko HJ, Coates AM, Goldberg IJ, de la Houssaye BA, Janssen RC, McCurdy CE, Rahman SM, Soo Choi C, Shulman GI, Kim JK, Friedman JE, Eckel RH. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes. 2009;58:116–24. doi: 10.2337/db07-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 43.Muoio DM, Koves TR. Skeletal muscle adaptation to fatty acid depends on coordinated actions of the PPARs and PGC1 alpha: implications for metabolic disease. Appl Physiol Nutr Metab. 2007;32:874–83. doi: 10.1139/H07-083. [DOI] [PubMed] [Google Scholar]
- 44.Toda C, Shiuchi T, Lee S, Yamato-Esaki M, Fujino Y, Suzuki A, Okamoto S, Minokoshi Y. Distinct effects of leptin and a melanocortin receptor agonist injected into medial hypothalamic nuclei on glucose uptake in peripheral tissues. Diabetes. 2009 doi: 10.2337/db09-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scarpace PJ, Zhang Y. Leptin resistance: a prediposing factor for diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2009;296:R493–500. doi: 10.1152/ajpregu.90669.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.